

Retinitis Pigmentosa Masquerades: Case Series and Review of the Literature

Abstract
1. Introduction
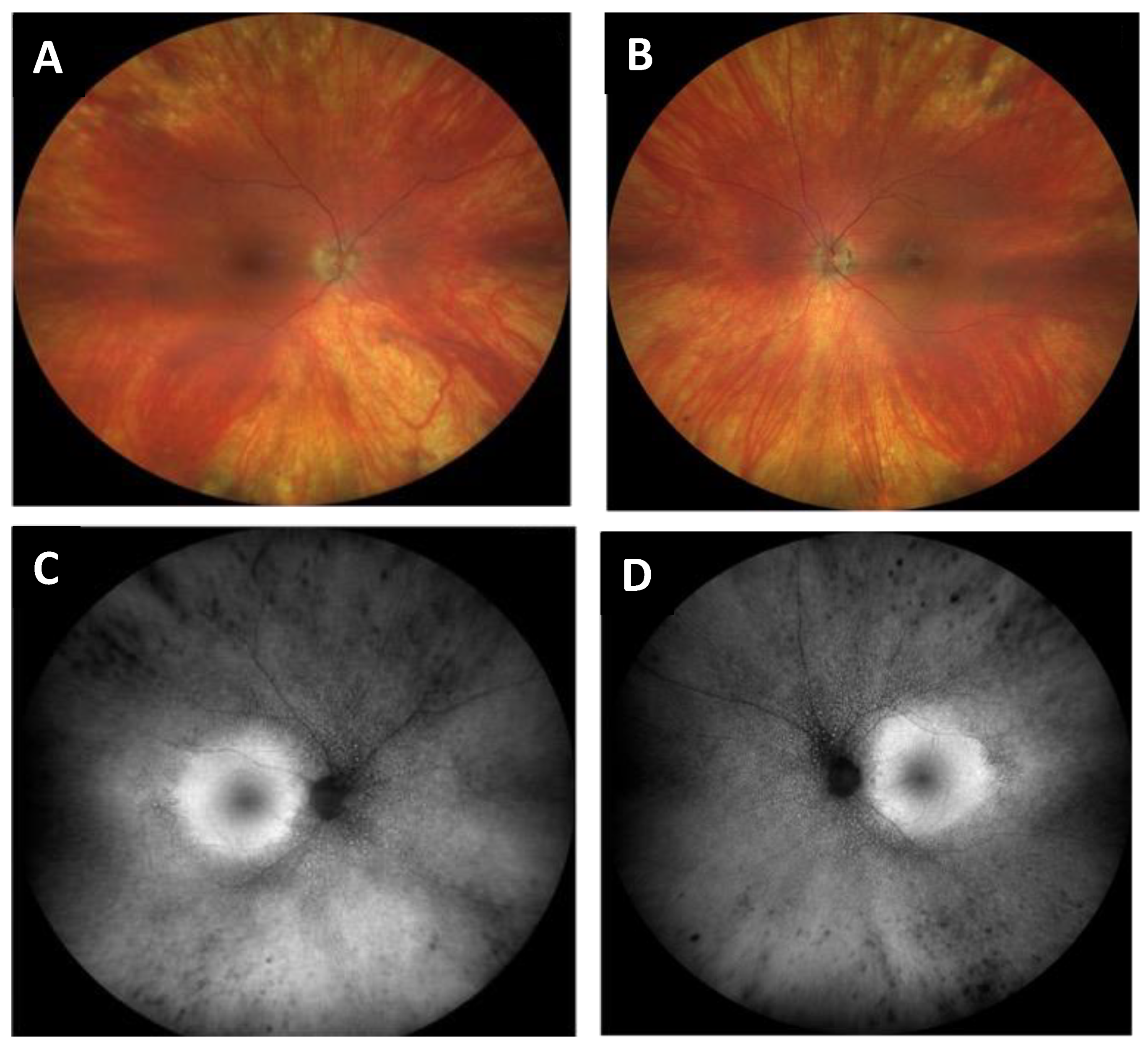
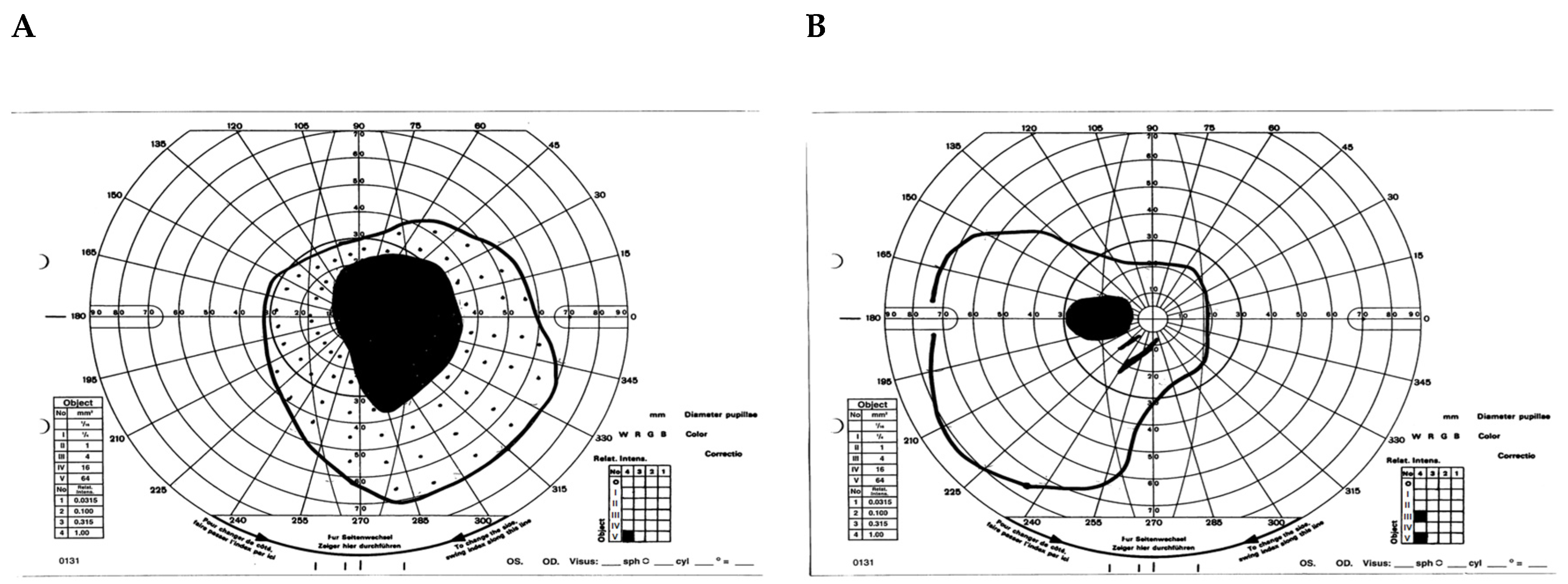
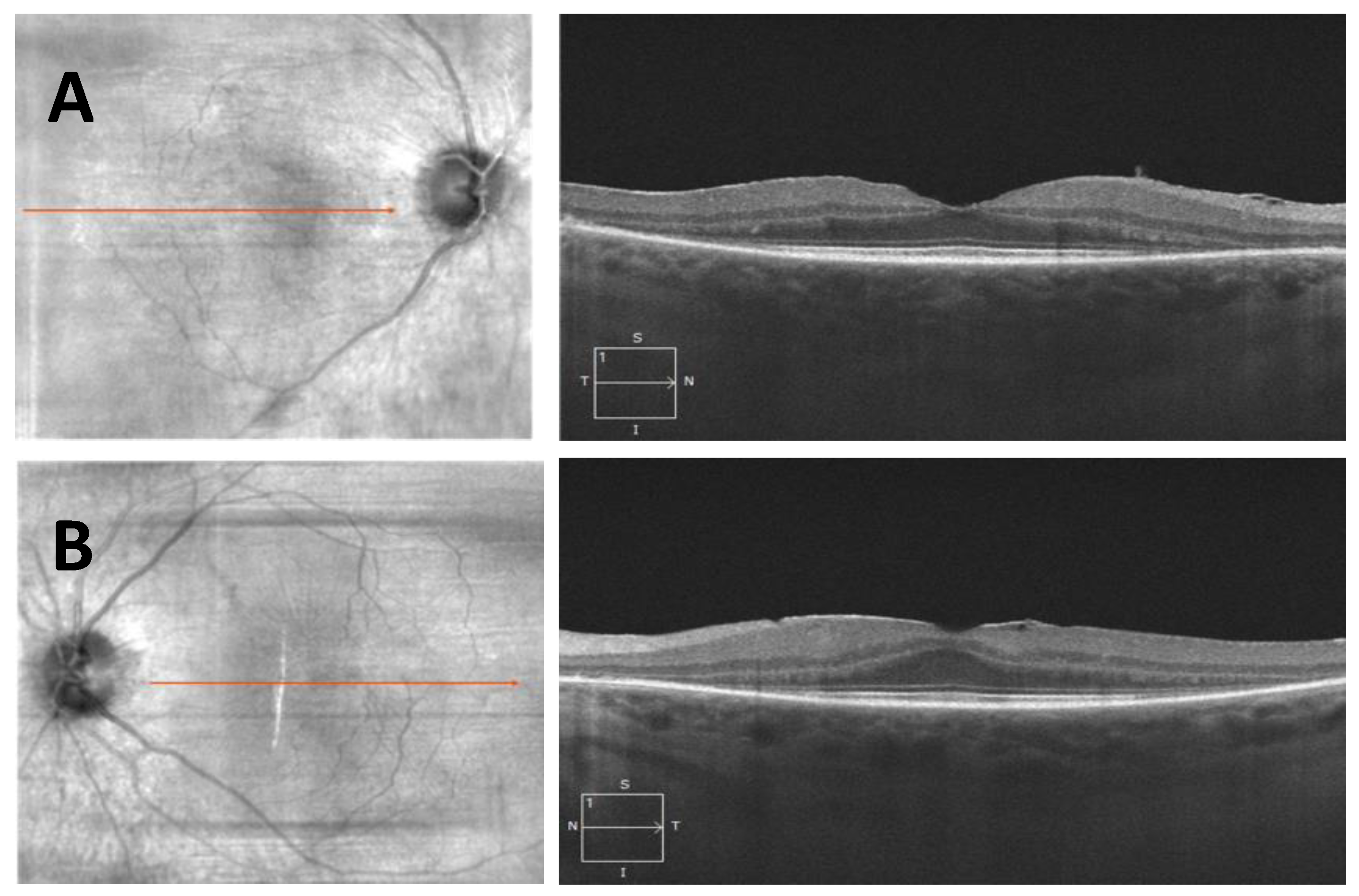
- Case 1:
- Case 2:
2. Review of RP Masquerades
3. Metabolic: Vitamin A deficiency
4. Drug-Induced
5. Chorioretinal Infections
6. Noninfectious Inflammatory Diseases
7. Autoimmune and Paraneoplastic Retinopathies
8. Miscellaneous
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muirhead, F. Adrien van Trigt, and the first published ophthalmoscopic images (De Speculo Oculi, 1853). J. Med. Biogr. 2020, 29, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Donders, F.C. Beiträge zur pathologischen Anatomie des Auges. Arch. Ophthalmol. 1857, 3, 139–165. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- McClard, C.K.; Pollalis, D.; Jamshidi, F.; Kingsley, R.; Lee, S.Y. Utility of No-Charge Panel Genetic Testing for Inherited Retinal Diseases in a Real-World Clinical Setting. J. Vitr. Dis. 2022, 6, 351–357. [Google Scholar] [CrossRef]
- Berson, E.L. Retinitis Pigmentosa the Friedenwald Lecture. Investig. Ophthalmol. Vis. Sci. 1993, 34, 1659–1676. [Google Scholar]
- Gregory-Evans, K.; Pennesi, M.E.; Weleber, R.G. Retinitis Pigmentosa and Allied Disorders. In Retina, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Strong, S.; Liew, G.; Michaelides, M. Retinitis pigmentosa-associated cystoid macular oedema: Pathogenesis and avenues of intervention. Br. J. Ophthalmol. 2017, 101, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Chae, T.; Foroozan, R. Vitamin A deficiency in patients with a remote history of intestinal surgery. Br. J. Ophthalmol. 2006, 90, 955–956. [Google Scholar] [CrossRef] [PubMed]
- Slater, G.H.; Ren, C.J.; Siegel, N.; Williams, T.; Barr, D.; Wolfe, B.; Dolan, K.; Fielding, G.A. Serum fat-soluble vitamin deficiency andabnormal calcium metabolism after malabsorptivebariatric surgery. J. Gastrointest. Surg. 2004, 8, 48–55, discussion 54–55. [Google Scholar] [CrossRef]
- Gouras, P.; Carr, R.E.; Gunkel, R.D. Retinitis pigmentosa in abetalipoproteinemia: Effects of vitamin A. Investig. Ophthalmol. Vis. Sci. 1971, 10, 784–793. [Google Scholar] [PubMed]
- Marmor, M.F. A Randomized Trial of Vitamin A and Vitamin E Supplementation for Retinitis Pigmentosa. Arch. Ophthalmol. 1993, 111, 1460. [Google Scholar] [CrossRef]
- Jorge, A.; Ung, C.; Young, L.H.; Melles, R.B.; Choi, H.K. Hydroxychloroquine retinopathy—Implications of research advances for rheumatology care. Nat. Rev. Rheumatol. 2018, 14, 693–703. [Google Scholar] [CrossRef]
- Ricketts, R.; Christoforidis, J.; Loizos, T.; Chang, S. Optical coherence tomography findings of quinine poisoning. Clin. Ophthalmol. 2011, 5, 75–80. [Google Scholar] [CrossRef]
- Yam, J.C.S.; Kwok, A.K.H. Ocular toxicity of hydroxychloroquine. Hong Kong Med. J. 2006, 12, 294–304. [Google Scholar] [PubMed]
- Wolfe, F.; Marmor, M.F. Rates and predictors of hydroxychloroquine retinal toxicity in patients with rheumatoid arthritis and systemic lupus erythematosus. Arthritis Care Res. 2010, 62, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.A.; Marmor, M.F. ERG and other discriminators between advanced hydroxychloroquine retinopathy and retinitis pigmentosa. Doc. Ophthalmol. 2017, 132, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Richa, S.; Yazbek, J.-C. Ocular Adverse Effects of Common Psychotropic Agents: A review. CNS Drugs 2010, 24, 501–526. [Google Scholar] [CrossRef]
- Siddall, J.R. Ocular toxic changes associated with chlorpromazine and thioridazine. Can. J. Ophthalmol. 1966, 1, 190–198. [Google Scholar]
- Hagopian, V.; Stratton, D.B.; Busiek, R.D. Five Cases of Pigmentary Retinopathy Associated with Thioridazine Administration. Am. J. Psychiatry 1966, 123, 97–100. [Google Scholar] [CrossRef]
- Marmor, M.F. Is thioridazine retinopathy progressive? Relationship of pigmentary changes to visual function. Br. J. Ophthalmol. 1990, 74, 739–742. [Google Scholar] [CrossRef]
- Meredith, T.A.; Aaberg, T.M.; Willerson, W.D. Progressive Chorioretinopathy After Receiving Thioridazine. Arch. Ophthalmol. 1978, 96, 1172–1176. [Google Scholar] [CrossRef]
- Chao, J.R.; Khurana, R.N.; Fawzi, A.A.; Reddy, H.S.; Rao, N.A.; Chao, J.R.; Khurana, R.N.; Fawzi, A.A.; Reddy, H.S.; Rao, N.A. Syphilis: Reemergence of an Old Adversary. Ophthalmology 2006, 113, 2074–2079. [Google Scholar] [CrossRef]
- Klein, A.; Fischer, N.; Goldstein, M.; Shulman, S.; Habot-Wilner, Z. The great imitator on the rise: Ocular and optic nerve manifestations in patients with newly diagnosed syphilis. Acta Ophthalmol. 2019, 97, e641–e647. [Google Scholar] [CrossRef]
- Paiva, A.d.C.M.; de Britto, V.S.; Criado, G.G.; Simões, K.M.P.; Motta, M.M.d.S. Pseudoretinitis pigmentosa due to syphilis: A case report and literature review. Rev. Bras. Oftalmol. 2021, 80, e0025. [Google Scholar] [CrossRef]
- Lee, S.Y.; Cheng, V.; Rodger, D.; Rao, N. Clinical and laboratory characteristics of ocular syphilis: A new face in the era of HIV co-infection. J. Ophthalmic Inflamm. Infect. 2015, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Du, Q.; Ma, F.; Lu, Y.; Wang, M.; Li, X. Characteristics of syphilitic uveitis in northern China. BMC Ophthalmol. 2017, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Margo, C.E.; Hamed, L.M. Ocular syphilis. Surv. Ophthalmol. 1992, 37, 203–220. [Google Scholar] [CrossRef]
- Luo, J.; Jing, D.; Kozak, I.; Huiming, Z.; Siying, C.; Yezhen, Y.; Xin, Q.; Luosheng, T.; Adelman, R.A.; Forster, S.H. Prevalence of Ocular Manifestations of HIV/AIDS in the Highly Active Antiretroviral Therapy (HAART) Era: A Different Spectrum in Central South China. Ophthalmic Epidemiol. 2013, 20, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Heiden, D.; Saranchuk, P.; Keenan, J.D.; Ford, N.; Lowinger, A.; Yen, M.; McCune, J.; Rao, N.A. Eye examination for early diagnosis of disseminated tuberculosis in patients with AIDS. Lancet Infect. Dis. 2016, 16, 493–499. [Google Scholar] [CrossRef]
- Gupta, V.; Gupta, A.; Rao, N.A. Intraocular Tuberculosis—An Update. Surv. Ophthalmol. 2007, 52, 561–587. [Google Scholar] [CrossRef] [PubMed]
- Melo, I.M.; Gomes, R.C.F.; Yung, A.A. Unique case of presumed ocular tuberculosis presenting as bilateral pseudoretinitis pigmentosa. Am. J. Ophthalmol. Case Rep. 2022, 26, 101412. [Google Scholar] [CrossRef] [PubMed]
- Winward, K.E.; Smith, J.L.; Culbertson, W.W.; Paris-Hamelin, A. Ocular Lyme Borreliosis. Am. J. Ophthalmol. 1989, 108, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Winward, K.E.; Nicholson, D.F.; Albert, D.W. Retinal vasculitis in Lyme borreliosis. J. Clin. Neuroophthalmol. 1991, 11, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Schechter, S.L. Lyme disease associated with optic neuropathy. Am. J. Med. 1986, 81, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, C.W.; Osterholm, M.T.; MacDonald, K.L.; White, K.E. An Interlaboratory Study of Antibody to Borrelia burgdorferi. J. Infect. Dis. 1987, 155, 1325–1327. [Google Scholar] [CrossRef] [PubMed]
- Karma, A.; Pirttila, T.A.; Viljanen, M.K.; Lahde, Y.E.; Raitta, C.M. Secondary retinitis pigmentosa and cerebral demyelination in Lyme borreliosis. Br. J. Ophthalmol. 1993, 77, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Gasch, A.T.; Smith, J.A.; Whitcup, S.M. Birdshot retinochoroidopathy. Br. J. Ophthalmol. 1999, 83, 241–249. [Google Scholar] [CrossRef]
- Pleyer, U.; Schlüter, D.; Mänz, M. Ocular Toxoplasmosis: Recent Aspects of Pathophysiology and Clinical Implications. Ophthalmic Res. 2014, 52, 116–123. [Google Scholar] [CrossRef]
- Delair, E.; Latkany, P.; Noble, A.G.; Rabiah, P.; McLeod, R.; Brézin, A. Clinical Manifestations of Ocular Toxoplasmosis. Ocul. Immunol. Inflamm. 2011, 19, 91–102. [Google Scholar] [CrossRef]
- Smith, J.R.; Cunningham, E.T., Jr. Atypical presentations of ocular toxoplasmosis. Curr. Opin. Ophthalmol. 2002, 13, 387–392. [Google Scholar] [CrossRef]
- Silveira, C.; Belfort, R., Jr.; Nussenblatt, R.; Farah, M.; Takahashi, W.; Imamura, P.; Burnier, M., Jr. Unilateral Pigmentary Retinopathy Associated with Ocular Toxoplasmosis. Am. J. Ophthalmol. 1989, 107, 682–684. [Google Scholar] [CrossRef]
- Karska-Basta, I.; Romanowska-Dixon, B.; Pojda-Wilczek, D.; Mackiewicz, N. Ocular Toxoplasmosis Associated with Unilateral Pigmentary Retinopathy That May Mimic Retinitis Pigmentosa: Diagnostic Dilemmas. Medicina 2021, 57, 892. [Google Scholar] [CrossRef] [PubMed]
- Givens, K.T.; Lee, D.A.; Jones, T.; Ilstrup, D.M. Congenital rubella syndrome: Ophthalmic manifestations and associated systemic disorders. Br. J. Ophthalmol. 1993, 77, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Boniuk, M.; Zimmerman, L.E. Ocular Pathology in the Rubella Syndrome. Arch. Ophthalmol. 1967, 77, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, D.M.; Wan, S.L.; Chew, A.L.; Chelva, E.; Tang, I.; Mackey, D.A.; Chen, F.K. Fundus Autofluorescence in Rubella Retinopathy: Correlation with Photoreceptor Structure and Function. Retina 2017, 37, 124–134. [Google Scholar] [CrossRef]
- Kuppermann, B.D.; Petty, J.G.; Richman, D.D.; Mathews, W.C.; Fullerton, S.C.; Rickman, L.S.; Freeman, W.R. Correlation Between CD4+ Counts and Prevalence of Cytomegalovirus Retinitis and Human Immunodeficiency Virus--related Noninfectious Retinal Vasculopathy in Patients with Acquired Immunodeficiency Syndrome. Am. J. Ophthalmol. 1993, 115, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Recchia, F.M.; Lalezary, M.; Kim, S.J. Treatment of Congenital Cytomegalovirus Retinitis with Intravitreous Ganciclovir. Arch. Ophthalmol. 2012, 130, 525–527. [Google Scholar] [CrossRef][Green Version]
- Coats, D.K.; Demmler, G.J.; Paysse, E.A.; Du, L.T.; Libby, C. Ophthalmologic findings in children with congenital cytomegalovirus infection. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2000, 4, 110–116. [Google Scholar] [CrossRef]
- Bagga, B.; Kate, A.; Joseph, J.; Dave, V.P. Herpes simplex infection of the eye: An introduction. Community Eye Health 2020, 33, 68–70. [Google Scholar]
- Ames, P.; Agarwal-Sinha, S. Congenital neonatal herpes simplex retinitis. Can. J. Ophthalmol. 2019, 54, e102–e105. [Google Scholar] [CrossRef]
- Lambert, S.R.; Taylor, D.; Kriss, A.; Holzel, H.; Heard, S. Ocular Manifestations of the Congenital Varicella Syndrome. Arch. Ophthalmol. 1989, 107, 52–56. [Google Scholar] [CrossRef]
- Banker, A. Posterior segment manifestations of human immunodeficiency virus/acquired immune deficiency syndrome. Indian J. Ophthalmol. 2008, 56, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Minos, E.; Barry, R.J.; Southworth, S.; Folkard, A.; Murray, P.I.; Duker, J.S.; Keane, P.A.; Denniston, A.K. Birdshot chorioretinopathy: Current knowledge and new concepts in pathophysiology, diagnosis, monitoring and treatment. Orphanet J. Rare Dis. 2016, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Willermain, F.; Greiner, K.; Forrester, J.V. Atypical end-stage birdshot retinochoroidopathy. Ocul. Immunol. Inflamm. 2003, 11, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.-K.; Buggage, R.R.; Nussenblatt, R.B. Serpiginous Choroiditis. Surv. Ophthalmol. 2005, 50, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.D. Acute zonal occult outer retinopathy. Donders Lecture: The Netherlands Ophthalmological Society, Maastricht, Holland, 19 June 1992. J. Clin. Neuroophthalmol. 1993, 13, 79–97. [Google Scholar] [PubMed]
- Gass, J.; Agarwal, A.; Scott, I.U. Acute zonal occult outer retinopathy: A long-term follow-up study. Am. J. Ophthalmol. 2002, 134, 329–339. [Google Scholar] [CrossRef]
- Monson, D.M.; Smith, J.R. Acute Zonal Occult Outer Retinopathy. Surv. Ophthalmol. 2011, 56, 23–35. [Google Scholar] [CrossRef]
- Duncker, T.; Lee, W.M.; Jiang, F.E.; Ramachandran, R.B.; Hood, D.C.; Tsang, S.H.; Sparrow, J.R.; Greenstein, V.C. Acute Zonal Occult Outer Retinopathy: Structural and Functional Analysis Across the Transition Zone Between Healthy and Diseased Retina. Retina 2018, 38, 118–127. [Google Scholar] [CrossRef]
- Francis, P.J.; Marinescu, A.; Fitzke, F.W.; Bird, A.C.; Holder, G.E. Acute zonal occult outer retinopathy: Towards a set of diagnostic criteria. Br. J. Ophthalmol. 2005, 89, 70–73. [Google Scholar] [CrossRef]
- Mrejen, S.; Khan, S.; Gallego-Pinazo, R.; Jampol, L.M.; Yannuzzi, L.A. Acute Zonal Occult Outer Retinopathy: A classification based on multimodal imaging. JAMA Ophthalmol 2014, 132, 1089–1098. [Google Scholar] [CrossRef]
- Fujiwara, T.; Imamura, Y.; Giovinazzo, V.J.; Spaide, R.F. Fundus Autofluorescence and Optical Coherence Tomographic Findings in Acute Zonal Occult Outer Retinopathy. Retina 2010, 30, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Matsou, A.; Tsaousis, K.T. Management of chronic ocular sarcoidosis: Challenges and solutions. Clin. Ophthalmol. 2018, 12, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Dana, M.-R.; Merayo-Lloves, J.; Schaumberg, D.A.; Foster, C.S. Prognosticators for Visual Outcome in Sarcoid Uveitis. Ophthalmology 1996, 103, 1846–1853. [Google Scholar] [CrossRef] [PubMed]
- Pasadhika, S.; Rosenbaum, J.T. Ocular Sarcoidosis. Clin. Chest Med. 2015, 36, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.B.; Rao, P.K. Ocular manifestations of systemic lupus erythematosus. Curr. Opin. Ophthalmol. 2008, 19, 512–518. [Google Scholar] [CrossRef]
- Stafford-Brady, F.J.; Urowitz, M.B.; Gladman, D.D.; Easterbrook, M. Lupus retinopathy. Arthritis Rheum. 1988, 31, 1105–1110. [Google Scholar] [CrossRef]
- Lamba, N.; Lee, S.; Chaudhry, H.; Foster, C.S. A review of the ocular manifestations of rheumatoid arthritis. Cogent Med. 2016, 3, 1243771. [Google Scholar] [CrossRef]
- Heckenlively, J.R.; Ferreyra, H.A. Autoimmune retinopathy: A review and summary. Semin. Immunopathol. 2008, 30, 127–134. [Google Scholar] [CrossRef]
- Grewal, D.S.; Fishman, G.A.; Jampol, L.M. Autoimmune Retinopathy and Antiretinal Antibodies: A review. Retina 2014, 34, 827–845. [Google Scholar] [CrossRef]
- Adamus, G. Autoantibody targets and their cancer relationship in the pathogenicity of paraneoplastic retinopathy. Autoimmun. Rev. 2009, 8, 410–414. [Google Scholar] [CrossRef]
- Eltabbakh, G.H.; Hoogerland, D.L.; Kay, M.C. Paraneoplastic Retinopathy Associated with Uterine Sarcoma. Gynecol. Oncol. 1995, 58, 120–123. [Google Scholar] [CrossRef]
- Weleber, R.G.; Watzke, R.C.; Shults, W.T.; Trzupek, K.M.; Heckenlively, J.R.; Egan, R.A.; Adamus, G. Clinical and Electrophysiologic Characterization of Paraneoplastic and Autoimmune Retinopathies Associated with Antienolase Antibodies. Am. J. Ophthalmol. 2005, 139, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.L.; Beebe, J.D.; Nepple, K.G.; Zakharia, Y.; Mullins, R.F.; Flamme-Wiese, M.J.; Thurtell, M.J.; Han, I.C. Autoimmune retinopathy and optic neuropathy associated with enolase-positive renal oncocytoma. Am. J. Ophthalmol. Case Rep. 2018, 12, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ferreyra, H.A.; Jayasundera, T.; Khan, N.W.; He, S.; Lu, Y.; Heckenlively, J.R. Management of Autoimmune Retinopathies with Immunosuppression. Arch. Ophthalmol. 2009, 127, 390–397. [Google Scholar] [CrossRef]
- Fortenbach, C.R.; Whitmore, S.S.; Thurtell, M.J.; Sohn, E.H.; Critser, D.B.; Stone, E.M.; Folk, J.C.; Han, I.C.; Boyce, T.M. Retinal Sublayer Analysis in Autoimmune Retinopathy and Identification of New Optical Coherence Tomography Phenotypes. Ocul. Immunol. Inflamm. 2023, 1–8. [Google Scholar] [CrossRef]
- Wiley, L.A.; Binkley, E.M.; DeLuca, A.P.; Workalemahu, G.; Tatro, N.J.; Luse, M.A.; Kennedy, E.L.; Folk, J.C.; Scheetz, T.E.; Ballas, Z.K.; et al. Autoimmune Retinopathy Mimicking Heritable Retinal Degeneration in a Patient with Common Variable Immune Deficiency. Retin. Cases Brief Rep. 2022, 16, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Woo, S.J.; Park, K.H.; Lee, B.R. Retinal Pigment Epithelium Sequelae Caused by Blunt Ocular Trauma: Incidence, Visual Outcome, and Associated Factors. Sci. Rep. 2017, 7, 14184. [Google Scholar] [CrossRef]
- Chao, H.; Chen, Y.; Liu, J.; Lee, S.; Lee, F.; Chang, Y.; Yeh, P.; Pan, W.; Chi, C.; Liu, T.; et al. Iron-generated hydroxyl radicals kill retinal cells in vivo: Effect of ferulic acid. Hum. Exp. Toxicol. 2008, 27, 327–339. [Google Scholar] [CrossRef]
- Rosenthal, A.R.; Marmor, M.F.; Leuenberger, P.; Hopkins, J.L. Chalcosis: A Study of Natural History. Ophthalmology 1979, 86, 1956–1969. [Google Scholar] [CrossRef]
- Neubauer, H. The Montgomery Lecture, 1979. Ocular metallosis. Trans. Ophthalmol. Soc. U. K. 1979, 99, 502–510. [Google Scholar]
- Kuhn, F.; Witherspoon, C.; Skalka, H.; Morris, R. Improvement of Siderotic ERG. Eur. J. Ophthalmol. 1992, 2, 44–45. [Google Scholar] [CrossRef]
- Dayan, M.R.; Cottrell, D.G.; Mitchell, K.W. Reversible retinal toxicity associated with retained intravitreal copper foreign body in the absence of intraocular inflammation. Acta Ophthalmol. Scand. 1999, 77, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Kellner, U.; Lai, T.Y.; Melles, R.B.; Mieler, W.F. Recommendations on Screening for Chloroquine and Hydroxychloroquine Retinopathy (2016 Revision). Ophthalmology 2016, 123, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.E. Goodman and Gilman’s the Pharmacological Basis of Therapeutics: 6th Ed. Edited by Alfred Goodman Gilman, Louis S. Goodman, and Alfred Gilman. Macmillan, 866 Third Ave., New York, NY 10022. 1980. 1843pp. 18 × 26cm. Price $45.00. J. Pharm. Sci. 1981, 70, 581. [Google Scholar] [CrossRef]
- Boland, M.; Roper, S.B.; Henry, J. Complications of Quinine Poisoning. Lancet 1985, 325, 384–385. [Google Scholar] [CrossRef] [PubMed]
- Vela, J.I.; Marcantonio, I.; Díaz-Cascajosa, J.; Crespí, J.; Buil, J.A. Progression of retinal pigmentation mimicking unilateral retinitis pigmentosa after bilateral pars planitis: A case report. BMC Ophthalmol. 2018, 18, 242. [Google Scholar] [CrossRef]
- Tugal-Tutkun, I.; Ozdal, P.C.; Berker, N. Pars planitis: Epidemiology, clinical characteristics, management and visual prognosis. J. Ophthalmic Vis. Res. 2015, 10, 469–480. [Google Scholar] [CrossRef]
- Khanamiri, H.N.; Rao, N.A. Serpiginous Choroiditis and Infectious Multifocal Serpiginoid Choroiditis. Surv. Ophthalmol. 2013, 58, 203–232. [Google Scholar] [CrossRef]
- Weiss, H.; Annesley, W.H.; Shields, J.A.; Tomer, T.; Christopherson, K. The Clinical Course of Serpiginous Choroidopathy. Am. J. Ophthalmol. 1979, 87, 133–142. [Google Scholar] [CrossRef]
- Adamus, G.; Amundson, D.; Seigel, G.; Machnicki, M. Anti-Enolase-α Autoantibodies in Cancer-Associated Retinopathy: Epitope Mapping and Cytotoxicity on Retinal Cells. J. Autoimmun. 1998, 11, 671–677. [Google Scholar] [CrossRef]
- Polans, A.S.; Witkowska, D.; Haley, T.L.; Amundson, D.; Baizer, L.; Adamus, G. Recoverin, a photoreceptor-specific calcium-binding protein, is expressed by the tumor of a patient with cancer-associated retinopathy. Proc. Natl. Acad. Sci. USA 1995, 92, 9176–9180. [Google Scholar] [CrossRef]
- Goldstein, S.M.; Syed, N.A.; Milam, A.H.; Maguire, A.M.; Lawton, T.J.; Nichols, C.W. Cancer-associated retinopathy. Arch. Ophthalmol. 1999, 117, 1641–1645. [Google Scholar] [CrossRef]
- Adamus, G.; Amundson, D.; MacKay, C.; Gouras, P. Long-term persistence of antirecoverin antibodies in endometrial cancer-associated retinopathy. Arch. Ophthalmol. 1998, 116, 251–253. [Google Scholar] [PubMed]
- Marsiglia, M.; Duncker, T.; Peiretti, E.; Brodie, S.E.; Tsang, S.H. Unilateral Retinitis Pigmentosa: A Proposal of Genetic Pathogenic Mechanisms. Eur. J. Ophthalmol. 2012, 22, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Potsidis, E.; Berson, E.L.; Sandberg, M.A. Disease Course of Patients with Unilateral Pigmentary Retinopathy. Investig. Opthalmol. Vis. Sci. 2011, 52, 9244–9249. [Google Scholar] [CrossRef] [PubMed]
- Farrell, D.F. Unilateral retinitis pigmentosa and cone-rod dystrophy. Clin. Ophthalmol. 2009, 3, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.M.; Michelson, G.; Juenemann, A.G. Unilateral retinitis pigmentosa: 30 years follow-up. BMJ Case Rep. 2014, 2014, bcr2013202236. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distinguishing Features | ||
|---|---|---|
| Metabolic | Vitamin A deficiency [8,9,10,11] |
|
| Drug-Induced | Quinolines (quinine, chloroquine, hydroxychloroquine) [12,13,14,15,16] |
|
| Phenothiazines (thioridazine, chlorpromazine) [17,18,19,20,21] | ||
| Didanosine | ||
| Chorioretinal infections | Bacterial: Treponema pallidum [22,23,24,25,26,27,28], Tuberculosis [29,30,31], Borrelia burgdorferi [32,33,34,35,36,37] |
|
| Parasitic: Toxoplasma [38,39,40,41,42], Diffuse Unilateral Subacute Neuroretinitis (DUSN) | ||
| Viral (Rubella [43,44,45], CMV [46,47,48], HSV [49,50], VZV [51], HIV [28,52], Measles) | ||
| Noninfectious Inflammatory diseases | Pars planitis |
|
| Birdshot and serpiginous chorioretinopathy [37,53,54,55] | ||
| Acute zonal occult outer retinopathy (AZOOR) [53,56,57,58,59,60,61,62] | ||
| Systemic sarcoidosis [63,64,65], lupus [66,67], and rheumatoid arthritis [68]) | ||
| Autoimmune and paraneoplastic retinopathies | Cancer-associated retinopathy (CAR) [69,70,71,72,73,74] |
|
| Melanoma-associated retinopathy (MAR) [71] | ||
| Non-neoplastic autoimmune retinopathy [69,70,75,76,77] | ||
| Miscellaneous | Trauma [78] |
|
| Chronic retinal detachment | ||
| Metallic foreign bodies (siderosis bulbi or chalcosis) [79,80,81,82,83] | ||
| Central serous chorioretinopathy (CSCR) | ||
| Laser scars | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thenappan, A.; Nanda, A.; Lee, C.S.; Lee, S.Y. Retinitis Pigmentosa Masquerades: Case Series and Review of the Literature. J. Clin. Med. 2023, 12, 5620. https://doi.org/10.3390/jcm12175620
Thenappan A, Nanda A, Lee CS, Lee SY. Retinitis Pigmentosa Masquerades: Case Series and Review of the Literature. Journal of Clinical Medicine. 2023; 12(17):5620. https://doi.org/10.3390/jcm12175620
Chicago/Turabian StyleThenappan, Abinaya, Arjun Nanda, Chang Sup Lee, and Sun Young Lee. 2023. "Retinitis Pigmentosa Masquerades: Case Series and Review of the Literature" Journal of Clinical Medicine 12, no. 17: 5620. https://doi.org/10.3390/jcm12175620
APA StyleThenappan, A., Nanda, A., Lee, C. S., & Lee, S. Y. (2023). Retinitis Pigmentosa Masquerades: Case Series and Review of the Literature. Journal of Clinical Medicine, 12(17), 5620. https://doi.org/10.3390/jcm12175620