

Reversing the Inflammatory Process—25 Years of Tumor Necrosis Factor-α Inhibitors


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
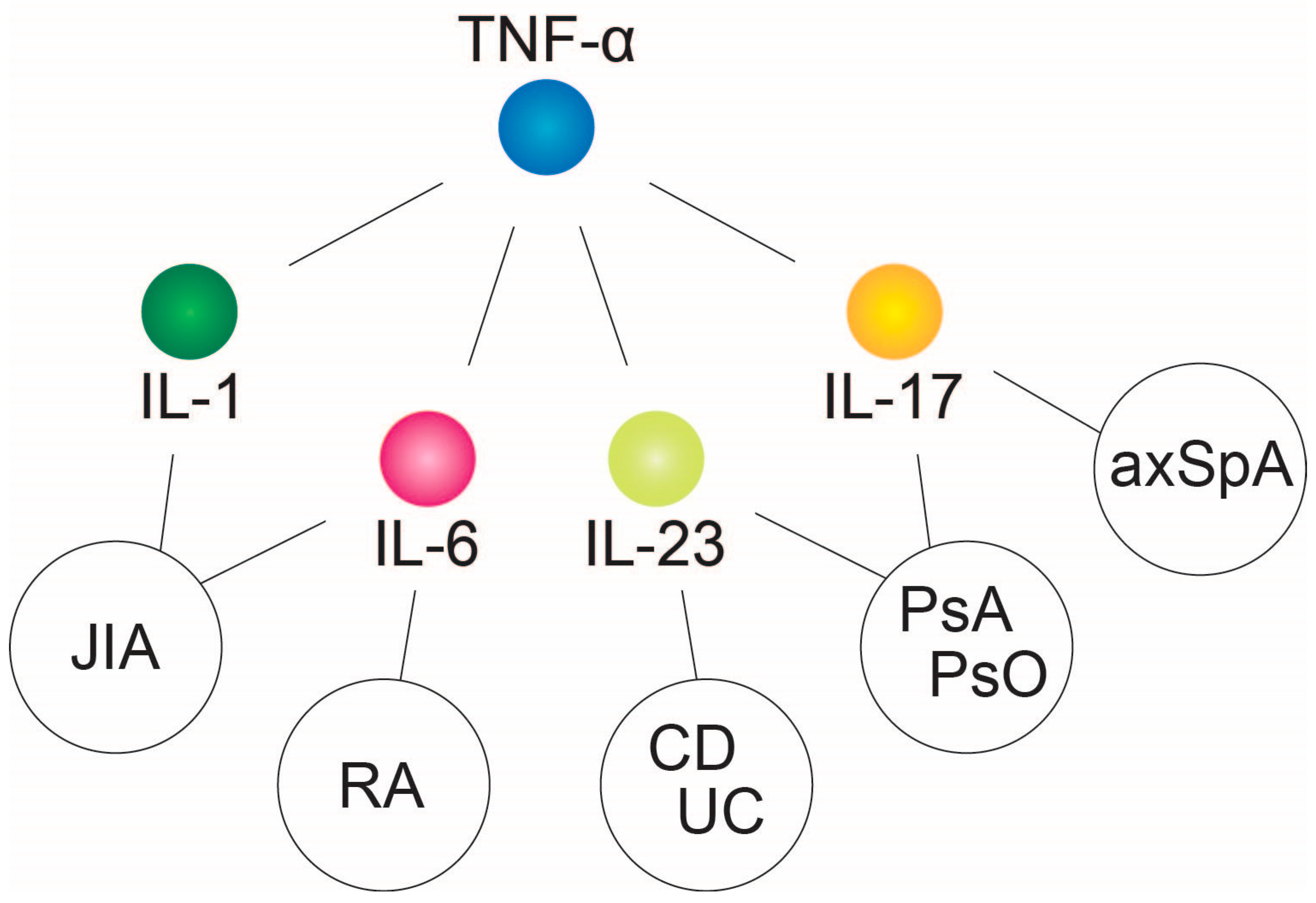
2. Tumor Necrosis Factor-α—Central Downstream Mediator of Chronic Inflammatory Response
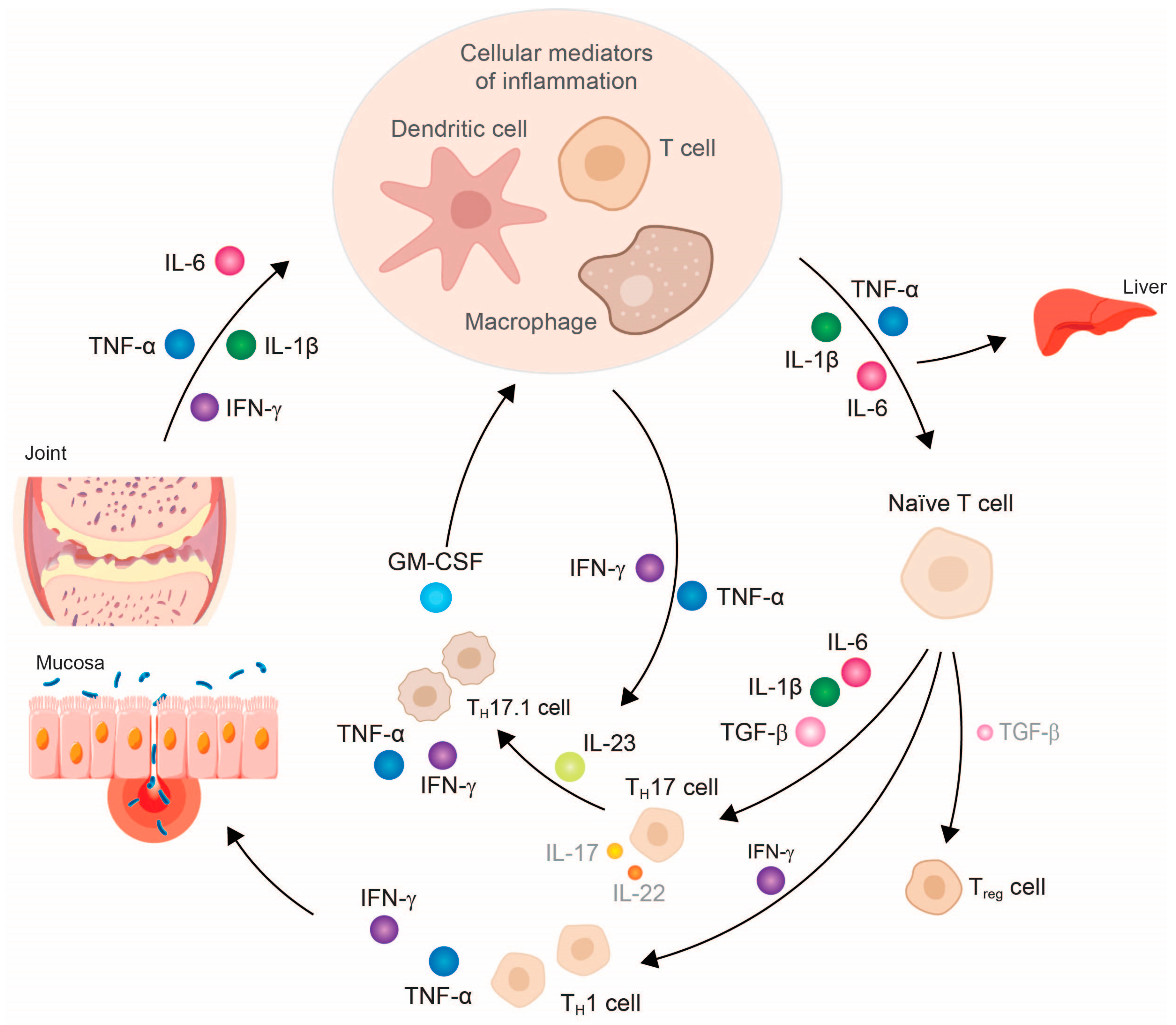
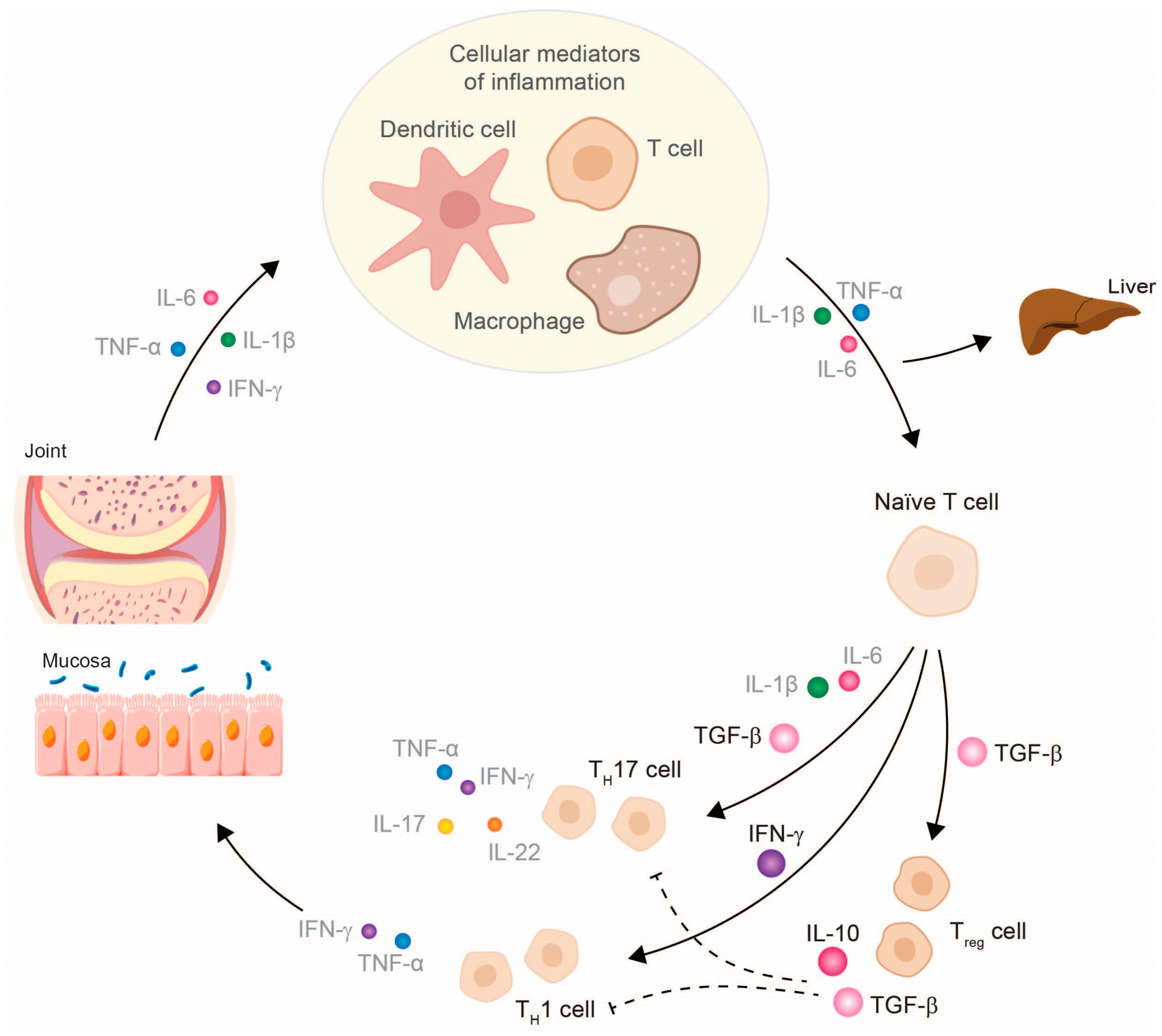
3. TNF-α Inhibitor Therapy Restores Cytokine Balance and Normalizes Organ Function
4. TNF-α Inhibitor Therapy and Its Clinical Impact
5. Future Directions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGonagle, D.; Stockwin, L.; Isaacs, J.; Emery, P. An enthesitis based model for the pathogenesis of spondyloarthropathy. additive effects of microbial adjuvant and biomechanical factors at disease sites. J. Rheumatol. 2001, 28, 2155–2159. [Google Scholar] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Germolec, D.R.; Shipkowski, K.A.; Frawley, R.P.; Evans, E. Markers of inflammation. Methods Mol. Biol. 2018, 1803, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulou, E.; Pircalabioru, G.G.; Bezirtzoglou, E. The role of cytochromes P450 in infection. Front. Immunol. 2018, 9, 89. [Google Scholar] [CrossRef]
- Szekanecz, Z.; McInnes, I.B.; Schett, G.; Szamosi, S.; Benkő, S.; Szűcs, G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat. Rev. Rheumatol. 2021, 17, 585–595. [Google Scholar] [CrossRef]
- Toubi, E.; Vadasz, Z. Innate immune-responses and their role in driving autoimmunity. Autoimmun. Rev. 2019, 18, 306–311. [Google Scholar] [CrossRef]
- Foster, J.R. The functions of cytokines and their uses in toxicology. Int. J. Exp. Pathol. 2001, 82, 171–192. [Google Scholar] [CrossRef]
- Cicchese, J.M.; Evans, S.; Hult, C.; Joslyn, L.R.; Wessler, T.; Millar, J.A.; Marino, S.; Cilfone, N.A.; Mattila, J.T.; Linderman, J.J.; et al. Dynamic balance of pro- and anti-inflammatory signals controls disease and limits pathology. Immunol. Rev. 2018, 285, 147–167. [Google Scholar] [CrossRef]
- Schett, G.; Elewaut, D.; McInnes, I.B.; Dayer, J.M.; Neurath, M.F. How cytokine networks fuel inflammation: Toward a cytokine-based disease taxonomy. Nat. Med. 2013, 19, 822–824. [Google Scholar] [CrossRef]
- Schett, G.; McInnes, I.B.; Neurath, M.F. Reframing immune-mediated inflammatory diseases through signature cytokine hubs. N. Engl. J. Med. 2021, 385, 628–639. [Google Scholar] [CrossRef]
- Ohkura, N.; Kitagawa, Y.; Sakaguchi, S. Development and maintenance of regulatory T cells. Immunity 2013, 38, 414–423. [Google Scholar] [CrossRef] [PubMed]
- van Hamburg, J.P.; Tas, S.W. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J. Autoimmun. 2018, 87, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Pesce, B.; Ribeiro, C.H.; Larrondo, M.; Ramos, V.; Soto, L.; Catalán, D.; Aguillón, J.C. TNF-α affects signature cytokines of Th1 and Th17 T cell subsets through differential actions on TNFR1 and TNFR2. Int. J. Mol. Sci. 2022, 23, 9306. [Google Scholar] [CrossRef]
- Aeberli, D.; Seitz, M.; Jüni, P.; Villiger, P.M. Increase of peripheral CXCR3 positive T lymphocytes upon treatment of RA patients with TNF-alpha inhibitors. Rheumatology 2005, 44, 172–175. [Google Scholar] [CrossRef][Green Version]
- Bjarnadóttir, U.; Einarsdóttir, H.K.; Stefánsdóttir, E.; Helgason, E.A.; Jónasdóttir, D.; Gudmundsson, S.; Gudbjornsson, B.; Ludviksson, B.R. Resolution of Th/Tc17-driven inflammation during anti-TNFα treatment of rheumatoid arthritis reveals a unique immune biomarker profiling pattern. Scand. J. Immunol. 2022, 95, e13116. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Munoz, F.; Dominguez-Lopez, A.; Yamamoto-Furusho, J.K. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 4280–4288. [Google Scholar] [CrossRef] [PubMed]
- de Jong, L.M.; Jiskoot, W.; Swen, J.J.; Manson, M.L. Distinct effects of inflammation on cytochrome P450 regulation and drug metabolism: Lessons from experimental models and a potential role for pharmacogenetics. Genes 2020, 11, 1509. [Google Scholar] [CrossRef]
- Lenoir, C.; Rollason, V.; Desmeules, J.A.; Samer, C.F. Influence of inflammation on cytochromes P450 activity in adults: A systematic review of the literature. Front. Pharmacol. 2021, 12, 733935. [Google Scholar] [CrossRef]
- Stanke-Labesque, F.; Gautier-Veyret, E.; Chhun, S.; Guilhaumou, R.; French Society of Pharmacology and Therapeutics. Inflammation is a major regulator of drug metabolizing enzymes and transporters: Consequences for the personalization of drug treatment. Pharmacol. Ther. 2020, 215, 107627. [Google Scholar] [CrossRef]
- Wollmann, B.M.; Syversen, S.W.; Vistnes, M.; Lie, E.; Mehus, L.L.; Molden, E. Associations between cytokine levels and CYP3A4 phenotype in patients with rheumatoid arthritis. Drug Metab. Dispos. 2018, 46, 1384–1389. [Google Scholar] [CrossRef]
- Tiegs, G.; Horst, A.K. TNF in the liver: Targeting a central player in inflammation. Semin. Immunopathol. 2022, 44, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Parlati, L.; Régnier, M.; Guillou, H.; Postic, C. New targets for NAFLD. JHEP Rep. 2021, 3, 100346. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Tamiya, G.; Ando, S.; Ohsumi, K.; Chiyo, T.; Mizutani, A.; Kitamura, N.; Toda, K.; Kaneko, T.; Horie, Y.; et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 2006, 55, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Charles, P.; Elliott, M.J.; Davis, D.; Potter, A.; Kalden, J.R.; Antoni, C.; Breedveld, F.C.; Smolen, J.S.; Eberl, G.; deWoody, K.; et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J. Immunol. 1999, 163, 1521–1528. [Google Scholar] [CrossRef]
- Shu, W.; Pang, Z.; Xu, C.; Lin, J.; Li, G.; Wu, W.; Sun, S.; Li, J.; Li, X.; Liu, Z. Anti-TNF-α monoclonal antibody therapy improves anemia through downregulating hepatocyte hepcidin expression in inflammatory bowel disease. Mediat. Inflamm. 2019, 2019, 4038619. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Carter, P.; Bruzelius, M.; Vithayathil, M.; Kar, S.; Mason, A.M.; Lin, A.; Burgess, S.; Larsson, S.C. Effects of tumour necrosis factor on cardiovascular disease and cancer: A two-sample Mendelian randomization study. EBiomedicine 2020, 59, 102956. [Google Scholar] [CrossRef]
- Sinh, P.; Cross, R. Cardiovascular risk assessment and impact of medications on cardiovascular disease in inflammatory bowel disease. Inflamm. Bowel Dis. 2021, 27, 1107–1115. [Google Scholar] [CrossRef]
- Bystrom, J.; Clanchy, F.I.; Taher, T.E.; Mangat, P.; Jawad, A.S.; Williams, R.O.; Mageed, R.A. TNFα in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine 2018, 101, 4–13. [Google Scholar] [CrossRef]
- Sauzullo, I.; Scrivo, R.; Sessa, P.; Mengoni, F.; Vullo, V.; Valesini, G.; Mastroianni, C.M. Changes in T cell effector functions over an 8-year period with TNF antagonists in patients with chronic inflammatory rheumatic diseases. Sci. Rep. 2018, 8, 7881. [Google Scholar] [CrossRef]
- Wen, H.; Chen, D.; Lu, J.; Jiao, Z.; Chen, B.; Zhang, B.; Ye, C.; Liu, L. Probable drug interaction between etanercept and cyclosporine resulting in clinically unexpected low trough concentrations: First case report. Front. Pharmacol. 2020, 11, 939. [Google Scholar] [CrossRef] [PubMed]
- Schnell, A.; Schwarz, B.; Wahlbuhl, M.; Allabauer, I.; Hess, M.; Weber, S.; Werner, F.; Schmidt, H.; Rechenauer, T.; Siebenlist, G.; et al. Distribution and cytokine profile of peripheral B cell subsets is perturbed in pediatric IBD and partially restored during a successful IFX therapy. Inflamm. Bowel Dis. 2021, 27, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Defendenti, C.; Atzeni, F.; Malandrin, S.; Ardizzone, S.; Almasio, P.L.; Saibeni, S.; Bezzio, C.; Bollani, S.; Salerno, R.; Declich, P.; et al. Anti-tumour necrosis factor-α antibodies and B cell homeostasis in human inflammatory bowel diseases. Int. Immunopharmacol. 2018, 54, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Vermeire, S.; Bullens, D.; Ferrante, M.; Van Steen, K.; Noman, M.; Bossuyt, X.; Rutgeerts, P.; Ceuppens, J.L.; Van Assche, G. Anti-tumor necrosis factor therapy restores peripheral blood B-cell subsets and CD40 expression in inflammatory bowel diseases. Inflamm. Bowel Dis. 2015, 21, 2787–2796. [Google Scholar] [CrossRef]
- Ringheanu, M.; Daum, F.; Markowitz, J.; Levine, J.; Katz, S.; Lin, X.; Silver, J. Effects of infliximab on apoptosis and reverse signaling of monocytes from healthy individuals and patients with Crohn’s disease. Inflamm. Bowel Dis. 2004, 10, 801–810. [Google Scholar] [CrossRef]
- Agnholt, J.; Kaltoft, K. Infliximab downregulates interferon-gamma production in activated gut T-lymphocytes from patients with Crohn’s disease. Cytokine 2001, 15, 212–222. [Google Scholar] [CrossRef]
- Holleran, G.; Lopetuso, L.; Petito, V.; Graziani, C.; Ianiro, G.; McNamara, D.; Gasbarrini, A.; Scaldaferri, F. The innate and adaptive immune system as targets for biologic therapies in inflammatory bowel disease. Int. J. Mol. Sci. 2017, 18, 2020. [Google Scholar] [CrossRef]
- Karsulovic, C.; Tempio, F.; Lopez, M.; Guerrero, J.; Goecke, A. In vitro phenotype induction of circulating monocytes: CD16 and CD163 analysis. J. Inflamm. Res. 2021, 14, 191–198. [Google Scholar] [CrossRef]
- Nadkarni, S.; Mauri, C.; Ehrenstein, M.R. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J. Exp. Med. 2007, 204, 33–39. [Google Scholar] [CrossRef]
- McGovern, J.L.; Nguyen, D.X.; Notley, C.A.; Mauri, C.; Isenberg, D.A.; Ehrenstein, M.R. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheum. 2012, 64, 3129–3138. [Google Scholar] [CrossRef]
- Bankó, Z.; Pozsgay, J.; Gáti, T.; Rojkovich, B.; Ujfalussy, I.; Sármay, G. Regulatory B cells in rheumatoid arthritis: Alterations in patients receiving anti-TNF therapy. Clin. Immunol. 2017, 184, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Flores-Borja, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci. Transl. Med. 2013, 5, 173ra123. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, B.; Jiang, Z.; Jiang, Y. Reduced numbers of regulatory B cells are negatively correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin. Rheumatol. 2014, 33, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Salomon, S.; Guignant, C.; Morel, P.; Flahaut, G.; Brault, C.; Gourguechon, C.; Fardellone, P.; Marolleau, J.P.; Gubler, B.; Goeb, V. Th17 and CD24(hi)CD27(+) regulatory B lymphocytes are biomarkers of response to biologics in rheumatoid arthritis. Arthritis. Res. Ther. 2017, 19, 33. [Google Scholar] [CrossRef]
- Menegatti, S.; Guillemot, V.; Latis, E.; Yahia-Cherbal, H.; Mittermüller, D.; Rouilly, V.; Mascia, E.; Rosine, N.; Koturan, S.; Millot, G.A.; et al. Immune response profiling of patients with spondyloarthritis reveals signalling networks mediating TNF-blocker function in vivo. Ann. Rheum. Dis. 2021, 80, 475–486. [Google Scholar] [CrossRef]
- Jongsma, M.M.E.; Aardoom, M.A.; Cozijnsen, M.A.; van Pieterson, M.; de Meij, T.; Groeneweg, M.; Norbruis, O.F.; Wolters, V.M.; van Wering, H.M.; Hojsak, I.; et al. First-line treatment with infliximab versus conventional treatment in children with newly diagnosed moderate-to-severe Crohn’s disease: An open-label multicentre randomised controlled trial. Gut 2022, 71, 34–42. [Google Scholar] [CrossRef]
- Payen, E.; Neuraz, A.; Zenzeri, L.; Talbotec, C.; Abi Nader, E.; Chatenoud, L.; Chhun, S.; Goulet, O.; Ruemmele, F.M.; Pigneur, B. Adalimumab therapy in pediatric Crohn disease: A 2-year follow-up comparing “top-down” and “step-up” strategies. J. Pediatr. Gastroenterol. Nutr. 2023, 76, 166–173. [Google Scholar] [CrossRef]
- Jongsma, M.M.E.; Costes, L.M.M.; Tindemans, I.; Cozijnsen, M.A.; Raatgreep, R.H.C.; van Pieterson, M.; Li, Y.; Escher, J.C.; de Ridder, L.; Samsom, J.N. Serum immune profiling in pediatric Crohn’s disease demonstrates stronger immune modulation with first-line infliximab than conventional therapy and pre-treatment profiles predict clinical response to both treatments. J. Crohns Colitis 2023, jjad049. [Google Scholar] [CrossRef]
- Claßen, M.; de Laffolie, J.; Claßen, M.; Schnell, A.; Sohrabi, K.; Hoerning, A. Significant advantages for first line treatment with TNF-alpha inhibitors in pediatric patients with inflammatory bowel disease—Data from the multicenter CEDATA-GPGE registry study. Front. Pediatr. 2022, 10, 903677. [Google Scholar] [CrossRef]
- de Laffolie, J.; Zimmer, K.P.; Sohrabi, K.; Hauer, A.C. Early immune suppression in children and adolescents with Crohn’s disease—Data from the CEDATA GPGE registry. Dtsch. Arztebl. Int. 2021, 118, 421–422. [Google Scholar] [CrossRef]
- Walters, T.D.; Kim, M.O.; Denson, L.A.; Griffiths, A.M.; Dubinsky, M.; Markowitz, J.; Baldassano, R.; Crandall, W.; Rosh, J.; Pfefferkorn, M.; et al. Increased effectiveness of early therapy with anti-tumor necrosis factor-α vs an immunomodulator in children with Crohn’s disease. Gastroenterology 2014, 146, 383–391. [Google Scholar] [CrossRef] [PubMed]
- England, B.R.; Thiele, G.M.; Anderson, D.R.; Mikuls, T.R. Increased cardiovascular risk in rheumatoid arthritis: Mechanisms and implications. BMJ 2018, 361, k1036. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Li, Y.; Luo, W.W.; Cheng, X.; Xiang, H.R.; Zhang, Q.Z.; He, J.; Peng, W.X. The risk of adverse effects of TNF-α inhibitors in patients with rheumatoid arthritis: A network meta-analysis. Front. Immunol. 2022, 13, 814429. [Google Scholar] [CrossRef]
- Conti, F.; Atzeni, F.; Massaro, L.; Gerardi, M.C.; Gremese, E.; Passiu, G.; Carletto, A.; Malavolta, N.; Foti, R.; Ramonda, R.; et al. The influence of comorbidities on the efficacy of tumour necrosis factor inhibitors, and the effect of tumour necrosis factor inhibitors on comorbidities in rheumatoid arthritis: Report from a National Consensus Conference. Rheumatology 2018, 57, vii11–vii22. [Google Scholar] [CrossRef] [PubMed]
- Olivera, P.A.; Zuily, S.; Kotze, P.G.; Regnault, V.; Al Awadhi, S.; Bossuyt, P.; Gearry, R.B.; Ghosh, S.; Kobayashi, T.; Lacolley, P.; et al. International consensus on the prevention of venous and arterial thrombotic events in patients with inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 857–873. [Google Scholar] [CrossRef]
- Bundesinstitut für Arzneimittel und Medizinprodukte. Januskinase-Inhibitoren: Behandlung von Entzündungskrankheiten. Available online: https://www.bfarm.de/SharedDocs/Risikoinformationen/Pharmakovigilanz/DE/RV_STP/g-l/januskinase.html;jsessionid=BD9413EADD5BFB59861897542E2169D0.internet281?nn=471274 (accessed on 15 May 2023).
- Hakimiana, S.; Kheder, J.; Arum, S.; Cave, D.R.; Hyatt, B. Re-evaluating osteoporosis and fracture risk in Crohn’s disease patients in the era of TNF-alpha inhibitors. Scand. J. Gastroenterol. 2018, 53, 168–172. [Google Scholar] [CrossRef]
- Torre-Alonso, J.C.; Carmona, L.; Moreno, M.; Galíndez, E.; Babío, J.; Zarco, P.; Linares, L.; Collantes-Estevez, E.; Barrial, M.F.; Hermosa, J.C.; et al. Identification and management of comorbidity in psoriatic arthritis: Evidence- and expert-based recommendations from a multidisciplinary panel from Spain. Rheumatol. Int. 2017, 37, 1239–1248. [Google Scholar] [CrossRef]
- Loza, E.; Lajas, C.; Andreu, J.L.; Balsa, A.; González-Álvaro, I.; Illera, O.; Jover, J.; Mateo, I.; Orte, J.; Rivera, J.; et al. Consensus statement on a framework for the management of comorbidity and extra-articular manifestations in rheumatoid arthritis. Rheumatol. Int. 2015, 35, 445–458. [Google Scholar] [CrossRef]
- Claßen, M.; Hoerning, A. Current role of monoclonal antibody therapy in pediatric IBD: A special focus on therapeutic drug monitoring and treat-to-target strategies. Children 2023, 10, 634. [Google Scholar] [CrossRef]
- Leone, G.M.; Mangano, K.; Petralia, M.C.; Nicoletti, F.; Fagone, P. Past, Present and (Foreseeable) Future of Biological Anti-TNF Alpha Therapy. J. Clin. Med. 2023, 12, 1630. [Google Scholar] [CrossRef]
- Luchetti, M.M.; Benfaremo, D.; Gabrielli, A. Biologics in Inflammatory and Immunomediated Arthritis. Curr. Pharm. Biotechnol. 2017, 18, 989–1007. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, G.; Klein, A.; Ganser, G.; Ruehlmann, M.J.; Dressler, F.; Hospach, A.; Minden, K.; Trauzeddel, R.; Foeldvari, I.; Kuemmerle-Deschner, J.; et al. Long-term safety and effectiveness of etanercept in JIA: An 18-year experience from the BiKeR registry. Arthritis Res. Ther. 2020, 22, 258. [Google Scholar] [CrossRef] [PubMed]
- Filippini, M.; Bazzani, C.; Atzeni, F.; Sarzi Puttini, P.; Marchesoni, A.; Favalli, E.G.; Caporali, R.; Cavagna, L.; Gorla, R. Effects of anti-TNF alpha drugs on disability in patients with rheumatoid arthritis: Long-term real-life data from the Lorhen Registry. BioMed Res. Int. 2014, 2014, 416892. [Google Scholar] [CrossRef] [PubMed]
- Karadag, O.; Dalkilic, E.; Ayan, G.; Kucuksahin, O.; Kasifoglu, T.; Yilmaz, N.; Koca, S.S.; Yazisiz, V.; Erten, P.T.; Sayarlioglu, M.; et al. Real-world data on change in work productivity, activity impairment, and quality of life in patients with psoriatic arthritis under anti-TNF therapy: A postmarketing, noninterventional, observational study. Clin. Rheumatol. 2022, 41, 85–94. [Google Scholar] [CrossRef]
- van der Heijde, D.; Gladman, D.D.; Kavanaugh, A.; Mease, P.J. Assessing structural damage progression in psoriatic arthritis and its role as an outcome in research. Arthritis Res. Ther. 2020, 22, 18. [Google Scholar] [CrossRef]
- Schnitzler, F.; Seitz, T.; Tillack-Schreiber, C.; Lange, S.; Waggershauser, C.; Ochsenkuhn, T. Early start of infliximab in Crohn’s Disease increases rates of endoscopic remission and decreases stenosis formation: Experiences from a single center cohort. Crohns Colitis 360 2021, 3, otab060. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Arkkila, P.; Armuzzi, A.; Danese, S.; Guardiola, J.; Jahnsen, J.; Lees, C.; Louis, E.; Lukáš, M.; Reinisch, W.; et al. Comparative efficacy and safety of infliximab and vedolizumab therapy in patients with inflammatory bowel disease: A systematic review and meta-analysis. BMC Gastroenterol. 2022, 22, 291. [Google Scholar] [CrossRef]
- Genovese, M.C.; Bathon, J.M.; Fleischmann, R.M.; Moreland, L.W.; Martin, R.W.; Whitmore, J.B.; Tsuji, W.H.; Leff, J.A. Longterm safety, efficacy, and radiographic outcome with etanercept treatment in patients with early rheumatoid arthritis. J. Rheumatol. 2005, 32, 1232–1242. [Google Scholar]
- Marquez-Megias, S.; Nalda-Molina, R.; Sanz-Valero, J.; Más-Serrano, P.; Diaz-Gonzalez, M.; Candela-Boix, M.R.; Ramon-Lopez, A. Cost-Effectiveness of Therapeutic Drug Monitoring of Anti-TNF Therapy in Inflammatory Bowel Disease: A Systematic Review. Pharmaceutics 2022, 14, 1009. [Google Scholar] [CrossRef]
- Martelli, L.; Olivera, P.; Roblin, X.; Attar, A.; Peyrin-Biroulet, L. Cost-effectiveness of drug monitoring of anti-TNF therapy in inflammatory bowel disease and rheumatoid arthritis: A systematic review. J. Gastroenterol. 2017. [Google Scholar] [CrossRef]
- Lauper, K.; Iudici, M.; Mongin, D.; Bergstra, S.A.; Choquette, D.; Codreanu, C.; Cordtz, R.; De Cock, D.; Dreyer, L.; Elkayam, O.; et al. Effectiveness of TNF-inhibitors, abatacept, IL6-inhibitors and JAK-inhibitors in 31,846 patients with rheumatoid arthritis in 19 registers from the ‘JAK-pot’ collaboration. Ann. Rheum. Dis. 2022, 81, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Shu, J.; Shao, W.; Zhou, Z.; Guo, H.; Wang, J. Efficacy and safety of IL inhibitors, TNF-α inhibitors, and JAK inhibitors in patients with ankylosing spondylitis: A systematic review and Bayesian network meta-analysis. Ann. Transl. Med. 2023, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Baraliakos, X.; Ostergaard, M.; Poddubnyy, D.; van der Heijde, D.; Deodhar, A.; Machado, P.M.; Navarro-Compán, V.; Hermann, K.G.; Kishimoto, M.; Lee, E.Y.; et al. Effect of secukinumab versus adalimumab biosimilar on radiographic progression in patients with radiographic axial spondyloarthritis: A randomized phase IIIb study. In Proceedings of the American College of Rheumatology Convergence 2022, Philadelphia, PA, USA, 10–14 November 2022. [Google Scholar]
- Narula, N.; Wong, E.C.L.; Dulai, P.S.; Marshall, J.K.; Jairath, V.; Reinisch, W. Comparative effectiveness of biologics for endoscopic healing of the ileum and colon in Crohn’s Disease. Am. J. Gastroenterol. 2022, 117, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Schäfers, M.; Sommer, C.; Geis, C.; Hagenacker, T.; Vandenabeele, P.; Sorkin, L.S. Selective stimulation of either tumor necrosis factor receptor differentially induces pain behavior in vivo and ectopic activity in sensory neurons in vitro. Neuroscience 2008, 157, 414–423. [Google Scholar] [CrossRef]
- Hess, A.; Axmann, R.; Rech, J.; Finzel, S.; Heindl, C.; Kreitz, S.; Sergeeva, M.; Saake, M.; Garcia, M.; Kollias, G.; et al. Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc. Natl. Acad. Sci. USA 2011, 108, 3731–3736. [Google Scholar] [CrossRef]
- Hess, A.; Roesch, J.; Saake, M.; Sergeeva, M.; Hirschmann, S.; Neumann, H.; Dörfler, A.; Neurath, M.F.; Atreya, R. Functional brain imaging reveals rapid blockade of abdominal pain response upon anti-TNF therapy in Crohn’s Disease. Gastroenterology 2015, 149, 864–866. [Google Scholar] [CrossRef][Green Version]
- Cavanagh, J.; Paterson, C.; McLean, J.; Pimlott, S.; McDonald, M.; Patterson, J.; Wyper, D.; McInnes, I. Tumour necrosis factor blockade mediates altered serotonin transporter availability in rheumatoid arthritis: A clinical, proof-of-concept study. Ann. Rheum. Dis. 2010, 69, 1251–1252. [Google Scholar] [CrossRef]
- Beltagy, A.; Aghamajidi, A.; Trespidi, L.; Ossola, W.; Meroni, P.L. Biologics During Pregnancy and Breastfeeding Among Women With Rheumatic Diseases: Safety Clinical Evidence on the Road. Front. Pharmacol. 2021, 12, 621247. [Google Scholar] [CrossRef]
- Mahadevan, U.; Wolf, D.C.; Dubinsky, M.; Cortot, A.; Lee, S.D.; Siegel, C.A.; Ullman, T.; Glover, S.; Valentine, J.F.; Rubin, D.T.; et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2013, 11, 286–292. [Google Scholar] [CrossRef]
- Zundler, S.; Günther, C.; Kremer, A.E.; Zaiss, M.M.; Rothhammer, V.; Neurath, M.F. Gut immune cell trafficking: Inter-organ communication and immune-mediated inflammation. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 50–64. [Google Scholar] [CrossRef]
- Li, S.J.; Perez-Chada, L.M.; Merola, J.F. TNF Inhibitor-Induced Psoriasis: Proposed Algorithm for Treatment and Management. J. Psoriasis Psoriatic Arthritis 2019, 4, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, N.; Bessone, F. Hepatotoxicity Induced by Biological Agents: Clinical Features and Current Controversies. J. Clin. Transl. Hepatol. 2022, 10, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Bouhuys, M.; Lexmond, W.S.; van Rheenen, P.F. De-Escalation of Anti-Tumor Necrosis Factor Alpha Agents and Reduction in Adverse Effects: A Systematic Review. Biomedicines 2022, 10, 1034. [Google Scholar] [CrossRef] [PubMed]
- Taskilar, K.; Hagen, M.; Kleyer, A.; Simon, D.; Reiser, M.; Hueber, A.J.; Manger, B.; Englbrecht, M.; Finzel, S.; Tony, H.-P.; et al. Treatment tapering and stopping in patients with rheumatoid arthritis in stable remission (RETRO): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Rheumatol. 2021, 3, 767–777. [Google Scholar] [CrossRef]
- Stephan, M.; Tascilar, K.; Yalcin-Mutlu, M.; Hagen, M.; Haschka, J.; Reiser, M.; Hartmann, F.; Kleyer, A.; Hueber, A.J.; Manger, B.; et al. Physical Function of RA patients Tapering Treatment-A Post Hoc Analysis of the Randomized Controlled RETRO Trial. J. Clin. Med. 2023, 12, 3723. [Google Scholar] [CrossRef]
- Little, D.H.W.; Tabatabavakili, S.; Shaffer, S.R.; Nguyen, G.C.; Weizman, A.V.; Targownik, L.E. Effectiveness of Dose De-escalation of Biologic Therapy in Inflammatory Bowel Disease: A Systematic Review. Am. J. Gastroenterol. 2020, 115, 1768–1774. [Google Scholar] [CrossRef]
- Zhang, B.; Gulati, A.; Alipour, O.; Shao, L. Relapse From Deep Remission After Therapeutic De-escalation in Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. J. Crohns Colitis 2020, 14, 1413–1423. [Google Scholar] [CrossRef]
- Michielsens, C.A.J.; Boers, N.; Broeder, N.D.; Wenink, M.H.; van der Maas, A.; Mahler, E.A.M.; Mulder, M.L.M.; van der Heijde, D.; Hoogen, F.H.J.V.D.; Verhoef, L.M.; et al. Dose reduction and withdrawal strategy for TNF-inhibitors in psoriatic arthritis and axial spondyloarthritis: Design of a pragmatic open-label, randomised, non-inferiority trial. Trials 2020, 21, 90. [Google Scholar] [CrossRef]
- Reenaers, C.; Mary, J.Y.; Nachury, M.; Bouhnik, Y.; Laharie, D.; Allez, M.; Fumery, M.; Amiot, A.; Savoye, G.; Altwegg, R.; et al. Outcomes 7 years after infliximab withdrawal for patients with Crohn’s Disease in sustained remission. Clin. Gastroenterol. Hepatol. 2018, 16, 234–243.e232. [Google Scholar] [CrossRef]
- Gomes, C.F.; Colombel, J.-F.; Torres, J. De-escalation of Therapy in Inflammatory Bowel Disease. Curr. Gastroenterol. Rep. 2018, 20, 35. [Google Scholar] [CrossRef]
- Louis, E.; Resche-Rigon, M.; Laharie, D.; Satsangi, J.; Ding, N.; Siegmund, B.; D’Haens, G.; Picon, L.; Bossuyt, P.; Vuitton, L.; et al. Withdrawal of infliximab or concomitant immunosuppressant therapy in patients with Crohn’s disease on combination therapy (SPARE): A multicentre, open-label, randomised controlled trial. Lancet Gastroenterol. Hepatol. 2023, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Goncalves, J.; Quinn, M.; Benedetti, F.; Lee, J.Y. Era of biosimilars in rheumatology: Reshaping the healthcare environment. RMD Open 2019, 5, e000900. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Jung, J.-Y.; Suh, C.-H. Real-world observational study of biosimilars in inflammatory arthritis treatment: A systematic literature review. Expert Opin. Biol. Ther. 2020, 21, 57–73. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muth, K.N.; Rech, J.; Losch, F.O.; Hoerning, A. Reversing the Inflammatory Process—25 Years of Tumor Necrosis Factor-α Inhibitors. J. Clin. Med. 2023, 12, 5039. https://doi.org/10.3390/jcm12155039
Muth KN, Rech J, Losch FO, Hoerning A. Reversing the Inflammatory Process—25 Years of Tumor Necrosis Factor-α Inhibitors. Journal of Clinical Medicine. 2023; 12(15):5039. https://doi.org/10.3390/jcm12155039
Chicago/Turabian StyleMuth, Katharina N., Juergen Rech, Florian O. Losch, and André Hoerning. 2023. "Reversing the Inflammatory Process—25 Years of Tumor Necrosis Factor-α Inhibitors" Journal of Clinical Medicine 12, no. 15: 5039. https://doi.org/10.3390/jcm12155039
APA StyleMuth, K. N., Rech, J., Losch, F. O., & Hoerning, A. (2023). Reversing the Inflammatory Process—25 Years of Tumor Necrosis Factor-α Inhibitors. Journal of Clinical Medicine, 12(15), 5039. https://doi.org/10.3390/jcm12155039