
Association of ADP-Induced Whole-Blood Platelet Aggregation with Serum Low-Density Lipoprotein Cholesterol in Patients with Coronary Artery Disease When Receiving Maintenance Ticagrelor-Based Dual Antiplatelet Therapy



Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Procedure
2.3. Statistical Analysis
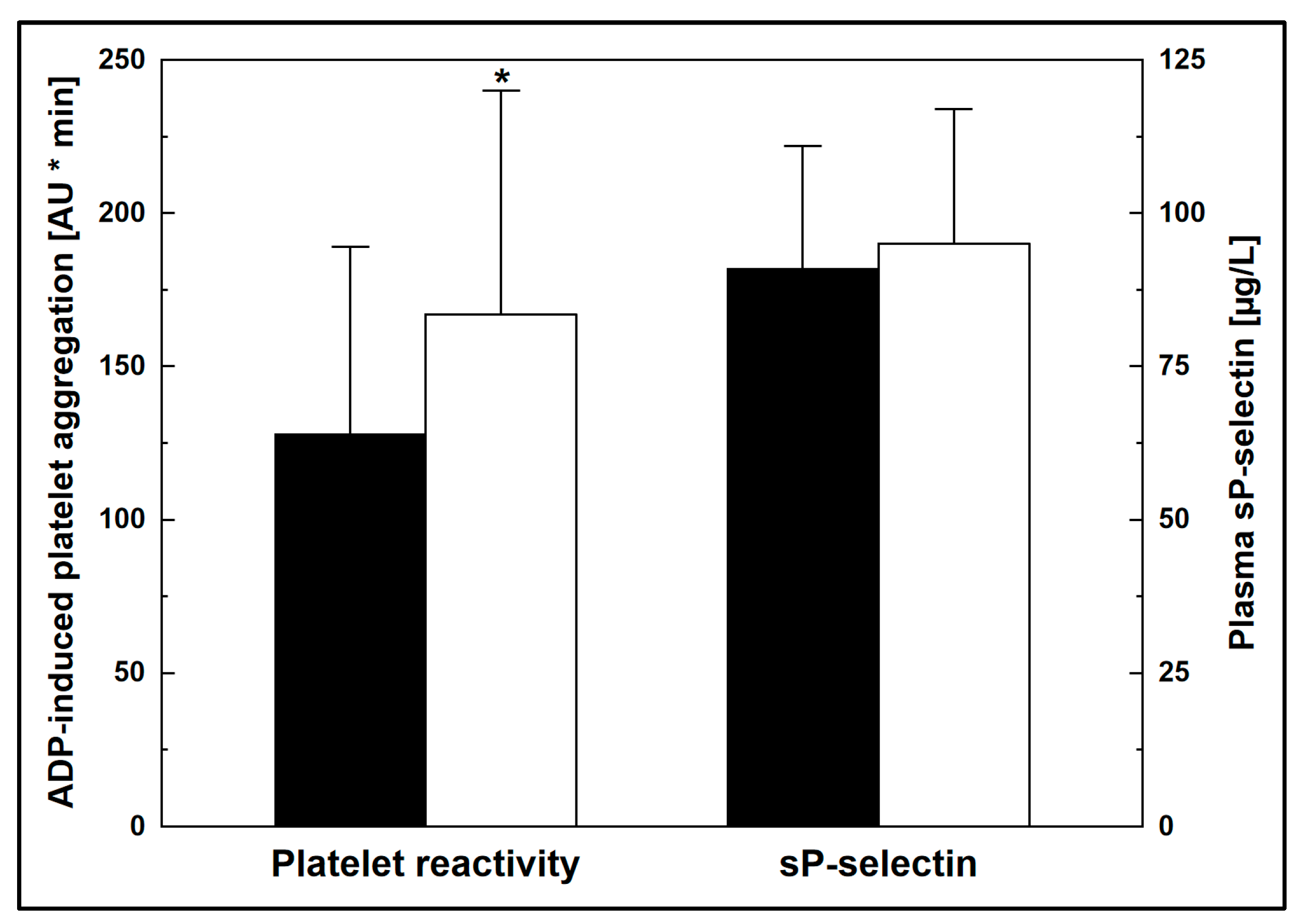
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Angiolillo, D.J.; Bernardo, E.; Sabaté, M.; Jimenez-Quevedo, P.; Costa, M.A.; Palazuelos, J.; Hernández-Antolin, R.; Moreno, R.; Escaned, J.; Alfonso, F.; et al. Impact of platelet reactivity on cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J. Am. Coll. Cardiol. 2007, 50, 1541–1547. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Antonino, M.J.; Bliden, K.P.; Dichiara, J.; Suarez, T.A.; Singla, A.; Tantry, U.S. Platelet reactivity to adenosine diphosphate and long-term ischemic event occurrence following percutaneous coronary intervention: A potential antiplatelet therapeutic target. Platelets 2008, 19, 595–604. [Google Scholar] [CrossRef]
- Price, M.J.; Endemann, S.; Gollapudi, R.R.; Valencia, R.; Stinis, C.T.; Levisay, J.P.; Ernst, A.; Sawhney, N.S.; Schatz, R.A.; Teirstein, P.S. Prognostic significance of post-clopidogrel platelet reactivity assessed by a point-of-care assay on thrombotic events after drug-eluting stent implantation. Eur. Heart J. 2008, 29, 992–1000. [Google Scholar] [CrossRef]
- Mega, J.L.; Close, S.L.; Wiviott, S.D.; Shen, L.; Hockett, R.D.; Brandt, J.T.; Walker, J.R.; Antman, E.M.; Macias, W.; Braunwald, E.; et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N. Engl. J. Med. 2009, 360, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Stone, G.W.; Witzenbichler, B.; Weisz, G.; Rinaldi, M.J.; Neumann, F.J.; Metzger, D.C.; Henry, T.D.; Cox, D.A.; Duffy, P.L.; Mazzaferri, E.; et al. Platelet reactivity and clinical outcomes after coronary artery implantation of drug-eluting stents (ADAPT-DES): A prospective multicentre registry study. Lancet 2013, 382, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Aradi, D.; Kirtane, A.; Bonello, L.; Gurbel, P.A.; Tantry, U.S.; Huber, K.; Freynhofer, M.K.; ten Berg, J.; Janssen, P.; Angiolillo, D.J.; et al. Bleeding and stent thrombosis on P2Y12-inhibitors: Collaborative analysis on the role of platelet reactivity for risk stratification after percutaneous coronary intervention. Eur. Heart J. 2015, 36, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Sibbing, D.; Aradi, D.; Alexopoulos, D.; Ten Berg, J.; Bhatt, D.L.; Bonello, L.; Collet, J.P.; Cuisset, T.; Franchi, F.; Gross, L.; et al. Updated Expert Consensus Statement on Platelet Function and Genetic Testing for Guiding P2Y12 Receptor Inhibitor Treatment in Percutaneous Coronary Intervention. JACC Cardiovasc. Interv. 2019, 12, 1521–1537. [Google Scholar] [CrossRef]
- Tantry, U.S.; Bonello, L.; Aradi, D.; Price, M.J.; Jeong, Y.H.; Angiolillo, D.J.; Stone, G.W.; Curzen, N.; Geisler, T.; Ten Berg, J.; et al. Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J. Am. Coll. Cardiol. 2013, 62, 2261–2273. [Google Scholar] [CrossRef]
- Chan, M.V.; Knowles, R.B.; Lundberg, M.H.; Tucker, A.T.; Mohamed, N.A.; Kirkby, N.S.; Armstrong, P.C.; Mitchell, J.A.; Warner, T.D. P2Y12 receptor blockade synergizes strongly with nitric oxide and prostacyclin to inhibit platelet activation. Br. J. Clin. Pharmacol. 2016, 81, 621–633. [Google Scholar] [CrossRef]
- Knowles, R.B.; Warner, T.D. Anti-platelet drugs and their necessary interaction with endothelial mediators and platelet cyclic nucleotides for therapeutic efficacy. Pharmacol. Ther. 2019, 193, 83–90. [Google Scholar] [CrossRef]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. The anti-aggregating properties of vascular endothelium: Interactions between prostacyclin and nitric oxide. Br. J. Pharmacol. 1987, 92, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Lecchi, A. Inhibition of the platelet P2Y12 receptor for adenosine diphosphate potentiates the antiplatelet effect of prostacyclin. J. Thromb. Haemost. 2007, 5, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Kirkby, N.S.; Lundberg, M.H.; Chan, M.V.; Vojnovic, I.; Solomon, A.B.; Emerson, M.; Mitchell, J.A.; Warner, T.D. Blockade of the purinergic P2Y12 receptor greatly increases the platelet inhibitory actions of nitric oxide. Proc. Natl. Acad. Sci. USA 2013, 110, 15782–15787. [Google Scholar] [CrossRef] [PubMed]
- Chyrchel, B.; Kruszelnicka, O.; Surdacki, A. Endothelial biomarkers and platelet reactivity on ticagrelor versus clopidogrel in patients after acute coronary syndrome with and without concomitant type 2 diabetes: A preliminary observational study. Cardiovasc. Diabetol. 2022, 21, 249. [Google Scholar] [CrossRef]
- Nagy, Z.; Smolenski, A. Cyclic nucleotide-dependent inhibitory signaling interweaves with activating pathways to determine platelet responses. Res. Pract. Thromb. Haemost. 2018, 2, 558–571. [Google Scholar] [CrossRef]
- Sibbing, D.; Braun, S.; Jawansky, S.; Vogt, W.; Mehilli, J.; Schömig, A.; Kastrati, A.; von Beckerath, N. Assessment of ADP-induced platelet aggregation with light transmission aggregometry and multiple electrode platelet aggregometry before and after clopidogrel treatment. Thromb. Haemost. 2008, 99, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Chyrchel, B.; Surdacki, A.; Chyrchel, M.; Dudek, D.; Dubiel, J.S. Separate dosing of clopidogrel and omeprazole may improve platelet inhibition on dual antiplatelet therapy. Int. J. Cardiol. 2011, 149, 124–125. [Google Scholar] [CrossRef]
- Verdoia, M.; Pergolini, P.; Rolla, R.; Nardin, M.; Schaffer, A.; Barbieri, L.; Marino, P.; Bellomo, G.; Suryapranata, H.; De Luca, G. Advanced age and high-residual platelet reactivity in patients receiving dual antiplatelet therapy with clopidogrel or ticagrelor. J. Thromb. Haemost. 2016, 14, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Sartori, C.; Pergolini, P.; Nardin, M.; Rolla, R.; Barbieri, L.; Schaffer, A.; Marino, P.; Bellomo, G.; Suryapranata, H.; et al. Prevalence and predictors of high-on treatment platelet reactivity with ticagrelor in ACS patients undergoing stent implantation. Vascul. Pharmacol. 2016, 77, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Nardin, M.; Verdoia, M.; Sartori, C.; Pergolini, P.; Rolla, R.; Barbieri, L.; Schaffer, A.; Bellomo, G.; Suryapranata, H.; De Luca, G.; et al. Diabetes mellitus, glucose control parameters and platelet reactivity in ticagrelor treated patients. Thromb. Res. 2016, 143, 45–49. [Google Scholar] [CrossRef]
- Verdoia, M.; Nardin, M.; Gioscia, R.; Negro, F.; Marcolongo, M.; Suryapranata, H.; Kedhi, E.; De Luca, G.; on behalf of the Novara Atherosclerosis Study Group (NAS). Higher neutrophil-to-lymphocyte ratio (NLR) increases the risk of suboptimal platelet inhibition and major cardiovascular ischemic events among ACS patients receiving dual antiplatelet therapy with ticagrelor. Vascul. Pharmacol. 2020, 132, 106765. [Google Scholar] [CrossRef]
- Verdoia, M.; Rolla, R.; Pergolini, P.; Gioscia, R.; Nardin, M.; Negro, F.; Viglione, F.; Suryapranata, H.; Kedhi, E.; De Luca, G.; et al. Low hemoglobin predicts high-platelet reactivity and major cardiovascular ischemic events at long-term follow-up among ACS patients receiving dual antiplatelet therapy with ticagrelor. Catheter Cardiovasc. Interv. 2021, 98, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Pergolini, P.; Rolla, R.; Sartori, C.; Nardin, M.; Schaffer, A.; Barbieri, L.; Daffara, V.; Marino, P.; Bellomo, G.; et al. Vitamin D levels and high-residual platelet reactivity in patients receiving dual antiplatelet therapy with clopidogrel or ticagrelor. Platelets 2016, 27, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Rolla, R.; Negro, F.; Tonon, F.; Pergolini, P.; Nardin, M.; Marcolongo, M.; De Luca, G.; on behalf of the Novara Atherosclerosis Study Group (NAS). Homocysteine levels and platelet reactivity in coronary artery disease patients treated with ticagrelor. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 292–299. [Google Scholar] [CrossRef]
- Barbieri, L.; Pergolini, P.; Verdoia, M.; Rolla, R.; Nardin, M.; Marino, P.; Bellomo, G.; Suryapranata, H.; De Luca, G.; on behalf of the Novara Atherosclerosis Study Group (NAS). Platelet reactivity in patients with impaired renal function receiving dual antiplatelet therapy with clopidogrel or ticagrelor. Vascul. Pharmacol. 2016, 79, 11–15. [Google Scholar] [CrossRef]
- Barbieri, L.; Verdoia, M.; Pergolini, P.; Nardin, M.; Rolla, R.; Marino, P.; Bellomo, G.; Suryapranata, H.; De Luca, G.; on behalf of the Novara Atherosclerosis Study Group (NAS). Uric acid and high-residual platelet reactivity in patients treated with clopidogrel or ticagrelor. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 352–358. [Google Scholar] [CrossRef]
- Nardin, M.; Verdoia, M.; Sartori, C.; Pergolini, P.; Rolla, R.; Barbieri, L.; Schaffer, A.; Marino, P.; Bellomo, G.; Suryapranata, H.; et al. Body Mass Index and Platelet Reactivity During Dual Antiplatelet Therapy With Clopidogrel or Ticagrelor. J. Cardiovasc. Pharmacol. 2015, 66, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, D.; Xanthopoulou, I.; Storey, R.F.; Bliden, K.P.; Tantry, U.S.; Angiolillo, D.J.; Gurbel, P.A. Platelet reactivity during ticagrelor maintenance therapy: A patient-level data meta-analysis. Am. Heart J. 2014, 168, 530–536. [Google Scholar] [CrossRef]
- Ndrepepa, G.; Holdenrieder, S.; Bernlochner, I.; Kastrati, A. Influence of body size on platelet response to ticagrelor and prasugrel in patients with acute coronary syndromes. Clin. Res. Cardiol. 2022, 111, 838–842. [Google Scholar] [CrossRef]
- Alexopoulos, D.; Vogiatzi, C.; Stavrou, K.; Vlassopoulou, N.; Perperis, A.; Pentara, I.; Xanthopoulou, I. Diabetes mellitus and platelet reactivity in patients under prasugrel or ticagrelor treatment: An observational study. Cardiovasc. Diabetol. 2015, 14, 68. [Google Scholar] [CrossRef]
- Verdoia, M.; Pergolini, P.; Nardin, M.; Rolla, R.; Barbieri, L.; Marino, P.; Carriero, A.; Suryapranata, H.; De Luca, G.; on behalf of the Novara Atherosclerosis Study Group (NAS). Prevalence and predictors of high-on treatment platelet reactivity during prasugrel treatment in patients with acute coronary syndrome undergoing stent implantation. J. Cardiol. 2019, 73, 198–203. [Google Scholar] [CrossRef]
- Golukhova, E.Z.; Grigoryan, M.V.; Ryabinina, M.N.; Bulaeva, N.I.; Serebruany, V.L. Body Mass Index and Plasma P-Selectin before Coronary Stenting Predict High Residual Platelet Reactivity at 6 Months on Dual Antiplatelet Therapy. Cardiology 2018, 139, 132–136. [Google Scholar] [CrossRef]
- Jäger, B.; Piackova, E.; Haller, P.M.; Andric, T.; Kahl, B.; Christ, G.; Geppert, A.; Wojta, J.; Huber, K. Increased platelet reactivity in dyslipidemic patients with coronary artery disease on dual anti-platelet therapy. Arch. Med. Sci. 2019, 15, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Buring, J.E.; Rifai, N. Soluble P-selectin and the risk of future cardiovascular events. Circulation 2001, 103, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D.; Goode, G.K.; Miller, J.P.; McCollum, C.N. Soluble P-selectin in hyperlipidaemia with and without symptomatic vascular disease: Relationship with von Willebrand factor. Blood Coagul. Fibrinolysis 1997, 8, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D.; Faragher, E.B.; McCollum, C.N. Increased soluble P-selectin following myocardial infarction: A new marker for the progression of atherosclerosis. Blood Coagul. Fibrinolysis 1997, 8, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Tscharre, M.; Vogel, B.; Tentzeris, I.; Freynhofer, M.K.; Rohla, M.; Wojta, J.; Weiss, T.W.; Ay, C.; Huber, K.; Farhan, S. Prognostic Impact of Soluble P-Selectin on Long-Term Adverse Cardiovascular Outcomes in Patients Undergoing Percutaneous Coronary Intervention. Thromb. Haemost. 2019, 119, 340–347. [Google Scholar] [CrossRef]
- Davì, G.; Romano, M.; Mezzetti, A.; Procopio, A.; Iacobelli, S.; Antidormi, T.; Bucciarelli, T.; Alessandrini, P.; Cuccurullo, F.; Bittolo Bon, G. Increased levels of soluble P-selectin in hypercholesterolemic patients. Circulation 1998, 97, 953–957. [Google Scholar] [CrossRef]
- Asadifar, M.; Bakhti, M.; Habibi-Rezaei, M.; Moosavi-Movahedi, A.A.; Tabatabi, M.R.; Ahmadinejad, M.; Badlou, B.A. Platelet Aggregation Increased by Advanced Glycated Hemoglobin. J. Blood Disord. Transfus. 2015, 6, 4. [Google Scholar] [CrossRef]
- Nusca, A.; Tuccinardi, D.; Proscia, C.; Melfi, R.; Manfrini, S.; Nicolucci, A.; Ceriello, A.; Pozzilli, P.; Ussia, G.P.; Grigion, F.; et al. Incremental role of glycaemic variability over HbA1c in identifying type 2 diabetic patients with high platelet reactivity undergoing percutaneous coronary intervention. Cardiovasc. Diabetol. 2019, 18, 147. [Google Scholar] [CrossRef]
- Gresele, P.; Guglielmini, G.; De Angelis, M.; Ciferri, S.; Ciofetta, M.; Falcinelli, E.; Lalli, C.; Ciabattoni, G.; Davì, G.; Bolli, G.B. Acute, short-term hyperglycemia enhances shear stress-induced platelet activation in patients with type II diabetes mellitus. J. Am. Coll. Cardiol. 2003, 41, 1013–1020. [Google Scholar] [CrossRef]
- Notarbartolo, A.; Davì, G.; Averna, M.; Barbagallo, C.M.; Ganci, A.; Giammarresi, C.; La Placa, F.P.; Patrono, C. Inhibition of thromboxane biosynthesis and platelet function by simvastatin in type IIa hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Morotti, A.; Barale, C.; Melchionda, E.; Russo, I. Platelet Redox Imbalance in Hypercholesterolemia: A Big Problem for a Small Cell. Int. J. Mol. Sci. 2022, 23, 11446. [Google Scholar] [CrossRef]
- Roberts, W.; Magwenzi, S.; Aburima, A.; Naseem, K.M. Thrombospondin-1 induces platelet activation through CD36-dependent inhibition of the cAMP/protein kinase A signaling cascade. Blood 2010, 116, 4297–4306. [Google Scholar] [CrossRef]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E.; Loscalzo, J.; Barnard, M.R.; Alpert, C.; Keaney, J.F.; Michelson, A.D. Nitric oxide released from activated platelets inhibits platelet recruitment. J. Clin. Invest. 1997, 100, 350–356. [Google Scholar] [CrossRef]
- Ikeda, H.; Takajo, Y.; Murohara, T.; Ichiki, K.; Adachi, H.; Haramaki, N.; Katoh, A.; Imaizumi, T. Platelet-derived nitric oxide and coronary risk factors. Hypertension 2000, 35, 904–907. [Google Scholar] [CrossRef]
- Katoh, A.; Ikeda, H.; Takajo, Y.; Haramaki, N.; Murohara, T.; Shintani, S.; Kanaya, S.; Yokoyama, S.; Ueno, T.; Honma, T.; et al. Coexistence of impairment of endothelium-derived nitric oxide and platelet-derived nitric oxide in patients with coronary risk factors. Circ. J. 2002, 66, 837–840. [Google Scholar] [CrossRef]
- Barale, C.; Frascaroli, C.; Senkeev, R.; Cavalot, F.; Russo, I. Simvastatin Effects on Inflammation and Platelet Activation Markers in Hypercholesterolemia. Biomed. Res. Int. 2018, 2018, 6508709. [Google Scholar] [CrossRef]
- Willoughby, S.R.; Stewart, S.; Holmes, A.S.; Chirkov, Y.Y.; Horowitz, J.D. Platelet nitric oxide responsiveness: A novel prognostic marker in acute coronary syndromes. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Russo, I. Influence of Cardiometabolic Risk Factors on Platelet Function. Int. J. Mol. Sci. 2020, 21, 623. [Google Scholar] [CrossRef]
- Nagy, B., Jr.; Jin, J.; Ashby, B.; Reilly, M.P.; Kunapuli, S.P. Contribution of the P2Y12 receptor-mediated pathway to platelet hyperreactivity in hypercholesterolemia. J. Thromb. Haemost. 2011, 9, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Söderbäck, U.; Sollevi, A.; Fredholm, B.B. The disappearance of adenosine from blood and platelet suspension in relation to the platelet cyclic AMP content. Acta Physiol. Scand. 1987, 129, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Mullershausen, F.; Russwurm, M.; Thompson, W.J.; Liu, L.; Koesling, D.; Friebe, A. Rapid nitric oxide-induced desensitization of the cGMP response is caused by increased activity of phosphodiesterase type 5 paralleled by phosphorylation of the enzyme. J. Cell Biol. 2001, 155, 271–278. [Google Scholar] [CrossRef]
- Mo, E.; Amin, H.; Bianco, I.H.; Garthwaite, J. Kinetics of a cellular nitric oxide/cGMP/phosphodiester-rase-5 pathway. J. Biol. Chem. 2004, 279, 26149–26158. [Google Scholar] [CrossRef]
- Nolte, C.; Eigenthaler, M.; Schanzenbächer, P.; Walter, U. Endothelial cell-dependent phosphorylation of a platelet protein mediated by cAMP- and cGMP-elevating factors. J. Biol. Chem. 1991, 266, 14808–14812. [Google Scholar] [CrossRef]
- Johnston-Cox, H.A.; Ravid, K. Adenosine and blood platelets. Purinergic Signal. 2011, 7, 357–365. [Google Scholar] [CrossRef]
- Muller, O.; Hamilos, M.; Bartunek, J.; Ulrichts, H.; Mangiacapra, F.; Holz, J.B.; Ntalianis, A.; Trana, C.; Dierickx, K.; Vercruysse, K.; et al. Relation of endothelial function to residual platelet reactivity after clopidogrel in patients with stable angina pectoris undergoing percutaneous coronary intervention. Am. J. Cardiol. 2010, 105, 333–338. [Google Scholar] [CrossRef]
- Ohman, J.; Kudira, R.; Albinsson, S.; Olde, B.; Erlinge, D. Ticagrelor induces adenosine triphosphate release from human red blood cells. Biochem. Biophys. Res. Commun. 2012, 418, 754–758. [Google Scholar] [CrossRef]
- van Giezen, J.J.; Sidaway, J.; Glaves, P.; Kirk, I.; Björkman, J.A. Ticagrelor inhibits adenosine uptake in vitro and enhances adenosine-mediated hyperemia responses in a canine model. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Smits, P.; Williams, S.B.; Lipson, D.E.; Banitt, P.; Rongen, G.A.; Creager, M.A. Endothelial release of nitric oxide contributes to the vasodilator effect of adenosine in humans. Circulation 1995, 92, 2135–2141. [Google Scholar] [CrossRef]
- Cattaneo, M.; Schulz, R.; Nylander, S. Adenosine-mediated effects of ticagrelor: Evidence and potential clinical relevance. J. Am. Coll. Cardiol. 2014, 63, 2503–2509. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Jeong, Y.H.; Tantry, U.S. The Dogged Search for Cryptic Effects of Ticagrelor: Wishful Thinking or Real Benefits Beyond P2Y12 Inhibition? Circulation 2016, 134, 1720–1723. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Shamburek, R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis 2009, 203, 325–330. [Google Scholar] [CrossRef]
- Zinellu, A.; Mangoni, A.A. A systematic review and meta-analysis of the effect of statin treatment on sVCAM-1 and sICAM-1. Expert Rev. Clin. Pharmacol. 2022, 15, 601–620. [Google Scholar] [CrossRef]
- Song, K.; Jin, X.; Kim, M.H.; Li, J.X.; Jin, C.D.; Yuan, S.L.; Song, Z.Y.; Jin, E.Z.; Lee, K.M.; Lim, K.H.; et al. Differences in Optimal Platelet Reactivity after Potent P2Y12 Inhibitor Treatment in Acute Coronary Syndrome Patients Undergoing Percutaneous Coronary Intervention. J. Clin. Med. 2022, 11, 2480. [Google Scholar] [CrossRef]
- Yang, Q.; Sun, D.; Pei, C.; Zeng, Y.; Wang, Z.; Li, Z.; Hao, Y.; Song, X.; Li, Y.; Liu, G.; et al. LDL cholesterol levels and in-hospital bleeding in patients on high-intensity antithrombotic therapy: Findings from the CCC-ACS project. Eur. Heart J. 2021, 42, 3175–3186. [Google Scholar] [CrossRef]
- Hochholzer, W.; Wiviott, S.D.; Antman, E.M.; Contant, C.F.; Guo, J.; Giugliano, R.P.; Dalby, A.J.; Montalescot, G.; Braunwald, E. Predictors of bleeding and time dependence of association of bleeding with mortality: Insights from the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel--Thrombolysis in Myocardial Infarction 38 (TRITON-TIMI 38). Circulation 2011, 123, 2681–2689. [Google Scholar] [CrossRef]
- Iijima, R.; Ndrepepa, G.; Mehilli, J.; Byrne, R.A.; Schulz, S.; Neumann, F.J.; Richardt, G.; Berger, P.B.; Schömig, A.; Kastrati, A. Profile of bleeding and ischaemic complications with bivalirudin and unfractionated heparin after percutaneous coronary intervention. Eur. Heart J. 2009, 30, 290–296. [Google Scholar] [CrossRef]
- Aygun, S.; Tokgozoglu, L. Comparison of Current International Guidelines for the Management of Dyslipidemia. J. Clin Med. 2022, 11, 7249. [Google Scholar] [CrossRef] [PubMed]
- Claassens, D.M.; Sibbing, D. De-Escalation of Antiplatelet Treatment in Patients with Myocardial Infarction Who Underwent Percutaneous Coronary Intervention: A Review of the Current Literature. J. Clin. Med. 2020, 9, 2983. [Google Scholar] [CrossRef]
- Sibbing, D.; Aradi, D.; Jacobshagen, C.; Gross, L.; Trenk, D.; Geisler, T.; Orban, M.; Hadamitzky, M.; Merkely, B.; Kiss, R.G.; et al. Guided de-escalation of antiplatelet treatment in patients with acute coronary syndrome undergoing percutaneous coronary intervention (TROPICAL-ACS): A randomised, open-label, multicentre trial. Lancet 2017, 390, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Rizas, K.D.; Park, K.W.; Chung, J.; van den Broek, W.; Claassens, D.M.F.; Choo, E.H.; Aradi, D.; Massberg, S.; Hwang, D.; et al. Dual antiplatelet therapy de-escalation in acute coronary syndrome: An individual patient meta-analysis. Eur. Heart J. 2023, 44, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Gurevitz, C.; Auriel, E.; Elis, A.; Kornowski, R. The Association between Low Levels of Low Density Lipoprotein Cholesterol and Intracerebral Hemorrhage: Cause for Concern? J. Clin. Med. 2022, 11, 536. [Google Scholar] [CrossRef]
- Blann, A.D.; Nadar, S.K.; Lip, G.Y. The adhesion molecule P-selectin and cardiovascular disease. Eur. Heart J. 2003, 24, 2166–2179. [Google Scholar] [CrossRef]
- Siller-Matula, J.M.; Delle-Karth, G.; Lang, I.M.; Neunteufl, T.; Kozinski, M.; Kubica, J.; Maurer, G.; Linkowska, K.; Grzybowski, T.; Huber, K.; et al. Phenotyping vs. genotyping for prediction of clopidogrel efficacy and safety: The PEGASUS-PCI study. J. Thromb. Haemost. 2012, 10, 529–542. [Google Scholar] [CrossRef]
- Adamski, P.; Barańska, M.; Ostrowska, M.; Kuliczkowski, W.; Buszko, K.; Kościelska-Kasprzak, K.; Karolko, B.; Mysiak, A.; Kubica, J. Diurnal Variability of Platelet Aggregation in Patients with Myocardial Infarction Treated with Prasugrel and Ticagrelor. J. Clin. Med. 2022, 11, 1124. [Google Scholar] [CrossRef]
- Zinellu, A.; Mangoni, A.A. Systematic Review and Meta-Analysis of the Effect of Statins on Circulating E-Selectin, L-Selectin, and P-Selectin. Biomedicines 2021, 9, 1707. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Variable | LDL-c | p | |
|---|---|---|---|
| Below-Median n = 31 | Over-Median n = 31 | ||
| Platelet reactivity, AU * min | 128 ± 61 | 167 ± 73 | <0.05 |
| sP-selectin, µg/L | 91 ± 20 | 95 ± 22 | n.s. |
| Clinical and biochemical characteristics | |||
| Age, years | 63 ± 10 | 65 ± 10 | n.s. |
| Men/Women, n (%) | 24/7 (77/23) | 22/9 (71/29) | n.s. |
| T2DM, n (%) | 14 (45) | 16 (52) | n.s. |
| HbA1c a, % | 7.2 ± 1.0 | 7.4 ± 1.0 | n.s. |
| BMI, kg/m2 | 30.0 ± 3.9 | 29.7 ± 3.6. | n.s. |
| HDL-c, mmol/L | 1.0 ± 0.3 | 1.1 ± 0.3 | n.s. |
| TG, mmol/L | 1.5 ± 0.8 | 1.7 ± 0.9 | n.s. |
| Arterial hypertension, n (%) | 25 (81) | 28 (90) | n.s. |
| Current smoking, n (%) | 9 (29) | 8 (26) | n.s. |
| LVEF, % | 54 ± 10 | 49 ± 9 | n.s. |
| Multivessel coronary artery disease, n (%) | 19 (61) | 23 (74) | n.s. |
| eGFR, mL/min per 1.73 m2 | 79 ± 19 | 77 ± 18 | n.s. |
| CRP, mg/L | 2.2 [1.3–3.5] | 2.1 [1.2–3.2] | n.s. |
| Hb, g/dL | 13.2 ± 1.9 | 13.0 ± 1.8 | n.s. |
| Platelet count, 103/µL | 229 ± 71 | 238 ± 75 | n.s. |
| Drugs beyond DAPT, ACEI/ARB, high-intensity statin and PPI | |||
| β-blockers, n (%) | 29 (94) | 28 (90) | n.s. |
| Diuretics, n (%) | 10 (32) | 11 (35) | n.s. |
| Calcium channel blockers, n (%) | 8 (26) | 12 (39) | n.s. |
| Metformin, n (% of diabetic subjects) | 13 (93) | 15 (94) | n.s. |
| Sulfonyloureas, n (% of diabetic subjects) | 3 (21) | 4 (25) | n.s. |
| Insulin, n (% of diabetic subjects) | 5 (36) | 3 (19) | n.s. |
| HDL-c, mmol/L | p | TG, mmol/L | p | HbA1c a, % | p | BMI, kg/m2 | p | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <1.0 | >1.0 | <1.6 | >1.6 | <7.3 | >7.3 | <29.9 | >29.9 | |||||
| Platelet reactivity, AU * min | 152 ± 65 | 144 ± 60 | n.s. | 145 ± 63 | 150 ± 64 | n.s. | 147 ± 63 | 149 ± 64 | n.s. | 153 ± 66 | 143 ± 62 | n.s. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chyrchel, B.; Kruszelnicka, O.; Wieczorek-Surdacka, E.; Surdacki, A. Association of ADP-Induced Whole-Blood Platelet Aggregation with Serum Low-Density Lipoprotein Cholesterol in Patients with Coronary Artery Disease When Receiving Maintenance Ticagrelor-Based Dual Antiplatelet Therapy. J. Clin. Med. 2023, 12, 4530. https://doi.org/10.3390/jcm12134530
Chyrchel B, Kruszelnicka O, Wieczorek-Surdacka E, Surdacki A. Association of ADP-Induced Whole-Blood Platelet Aggregation with Serum Low-Density Lipoprotein Cholesterol in Patients with Coronary Artery Disease When Receiving Maintenance Ticagrelor-Based Dual Antiplatelet Therapy. Journal of Clinical Medicine. 2023; 12(13):4530. https://doi.org/10.3390/jcm12134530
Chicago/Turabian StyleChyrchel, Bernadeta, Olga Kruszelnicka, Ewa Wieczorek-Surdacka, and Andrzej Surdacki. 2023. "Association of ADP-Induced Whole-Blood Platelet Aggregation with Serum Low-Density Lipoprotein Cholesterol in Patients with Coronary Artery Disease When Receiving Maintenance Ticagrelor-Based Dual Antiplatelet Therapy" Journal of Clinical Medicine 12, no. 13: 4530. https://doi.org/10.3390/jcm12134530
APA StyleChyrchel, B., Kruszelnicka, O., Wieczorek-Surdacka, E., & Surdacki, A. (2023). Association of ADP-Induced Whole-Blood Platelet Aggregation with Serum Low-Density Lipoprotein Cholesterol in Patients with Coronary Artery Disease When Receiving Maintenance Ticagrelor-Based Dual Antiplatelet Therapy. Journal of Clinical Medicine, 12(13), 4530. https://doi.org/10.3390/jcm12134530