
Etiologies of Acute Bronchiolitis in Children at Risk for Asthma, with Emphasis on the Human Rhinovirus Genotyping Protocol
 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Design and Population
2.2. Data and Specimens’ Collection and Follow-Up
2.3. Nucleic Acid Extraction from Samples
2.4. Respiratory Viral Detection
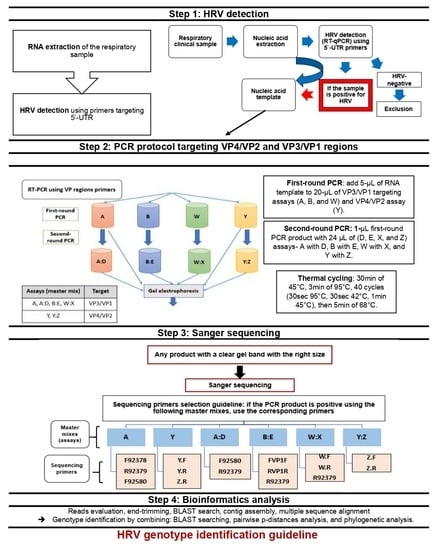
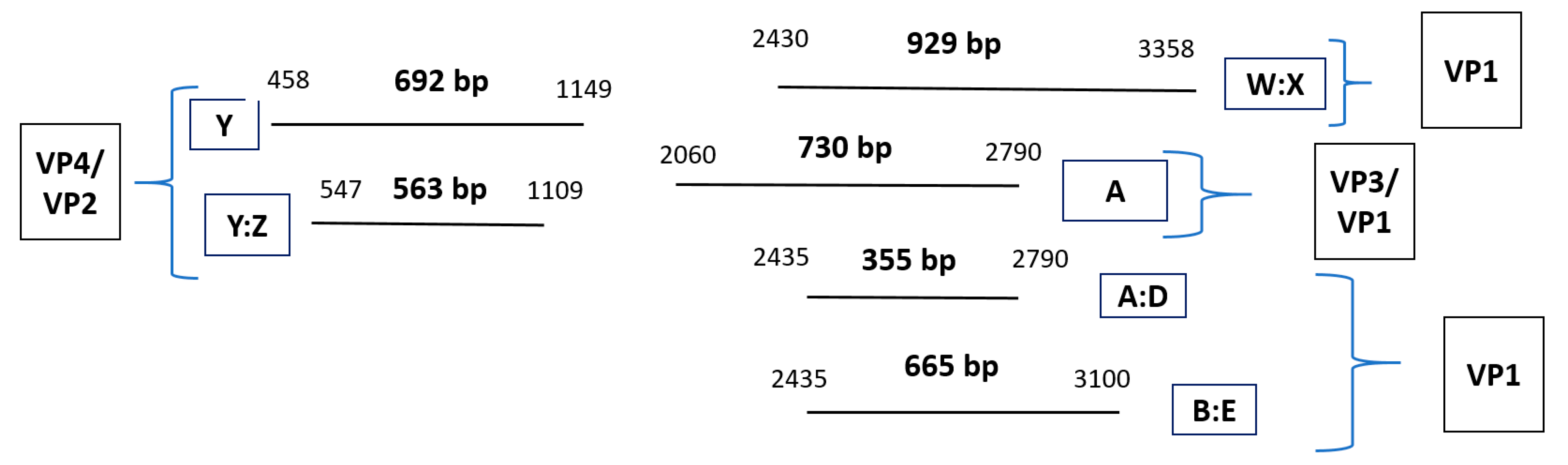
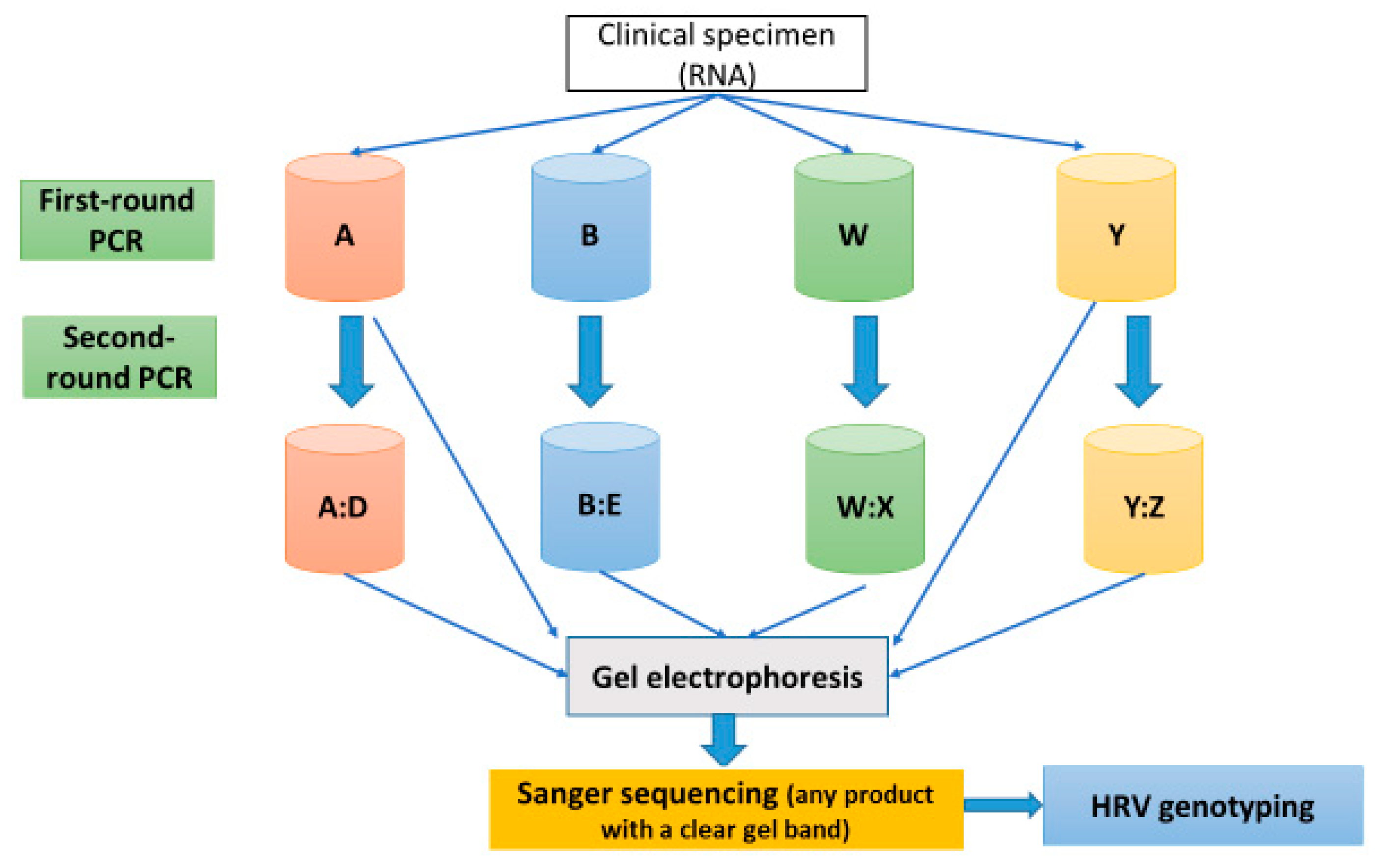
2.5. PCR Protocol for HRV-Positive Samples Targeting VP4/VP2 and VP3/VP1 Regions
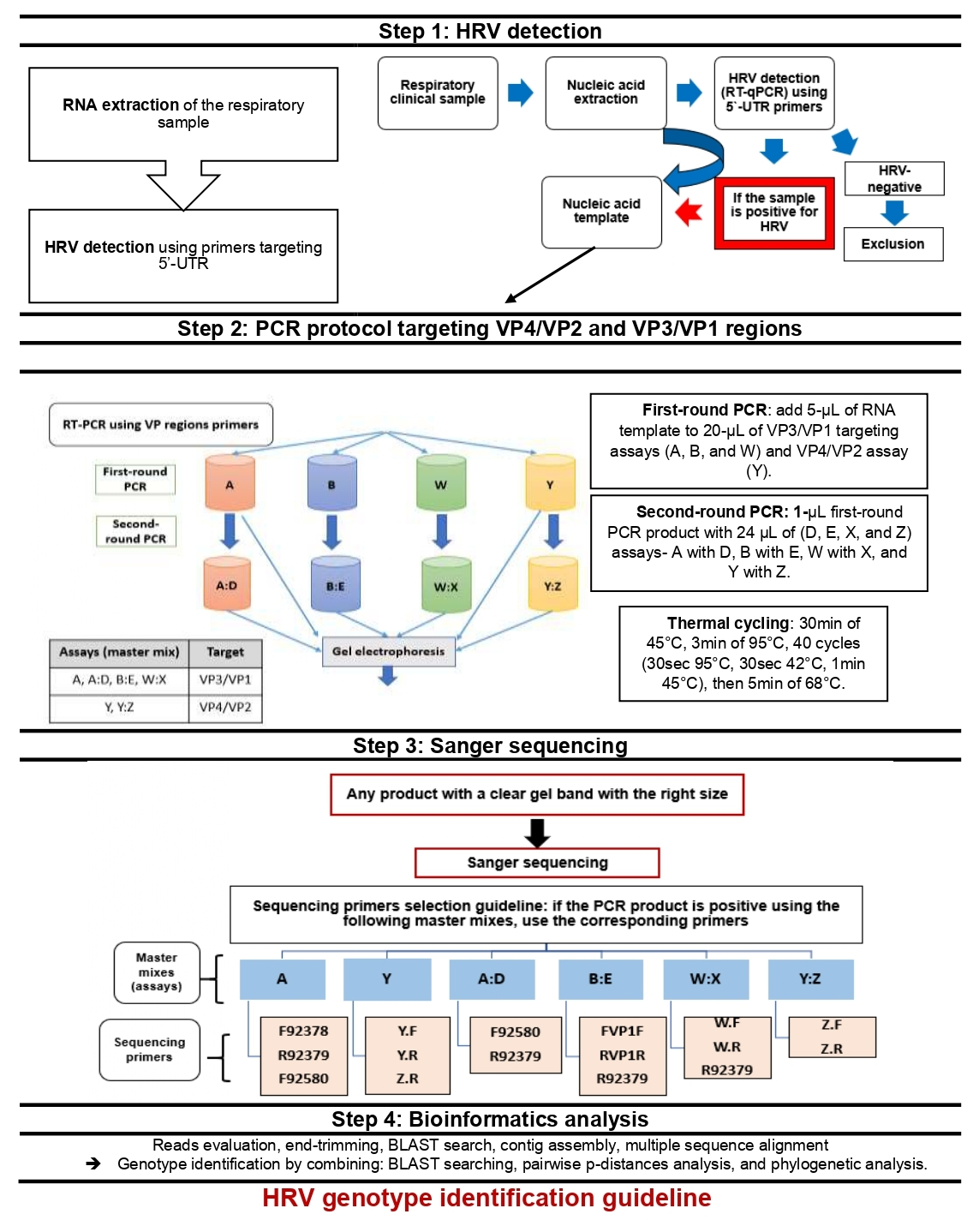
2.5.1. PCR Primers for HRV VP Regions
2.5.2. Pre-PCR and PCR Protocol
2.5.3. Variations of the Standard PCR Technique (Nested and Semi-Nested PCR)
2.5.4. Agarose Gel Electrophoresis
2.5.5. PCR Product Clean-Up Procedure
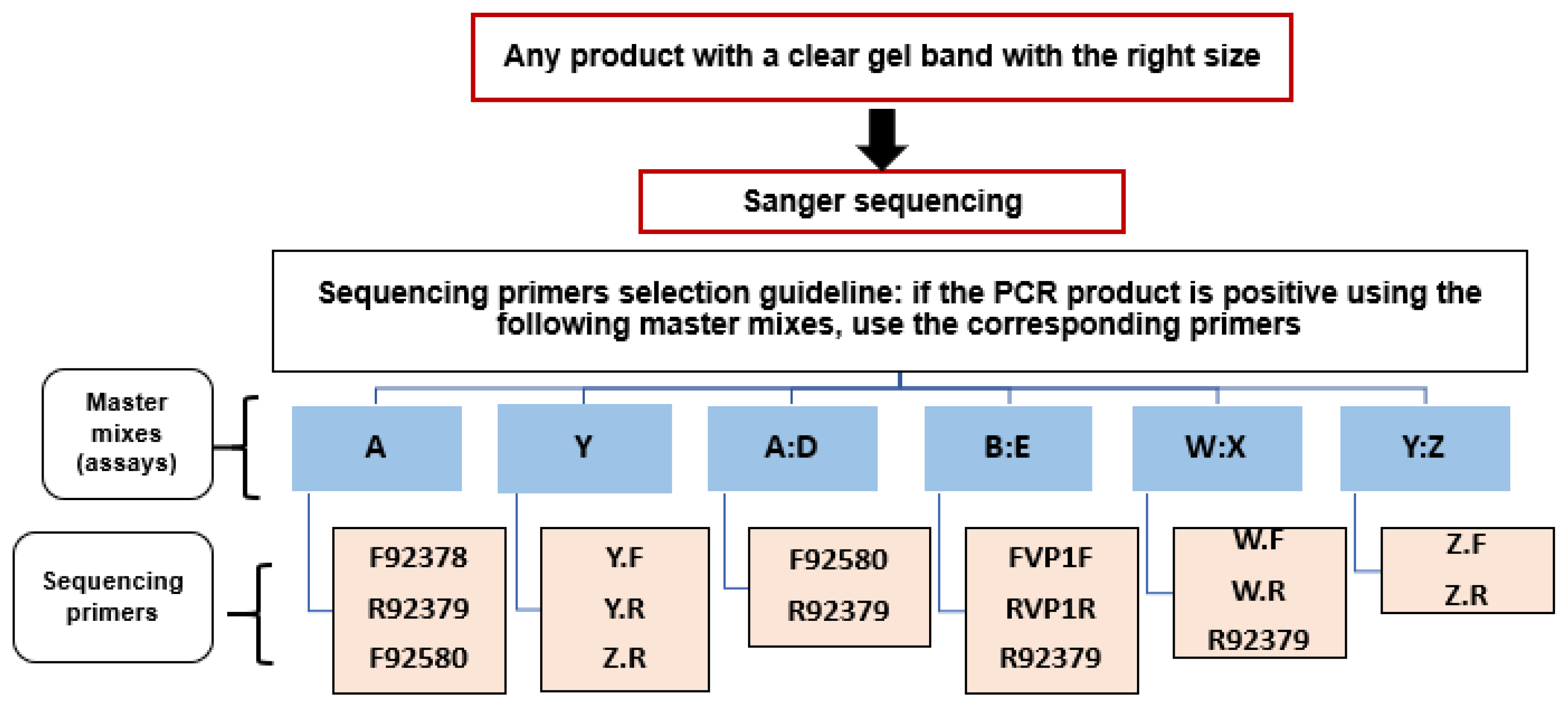
2.6. Sanger Sequencing Workflow
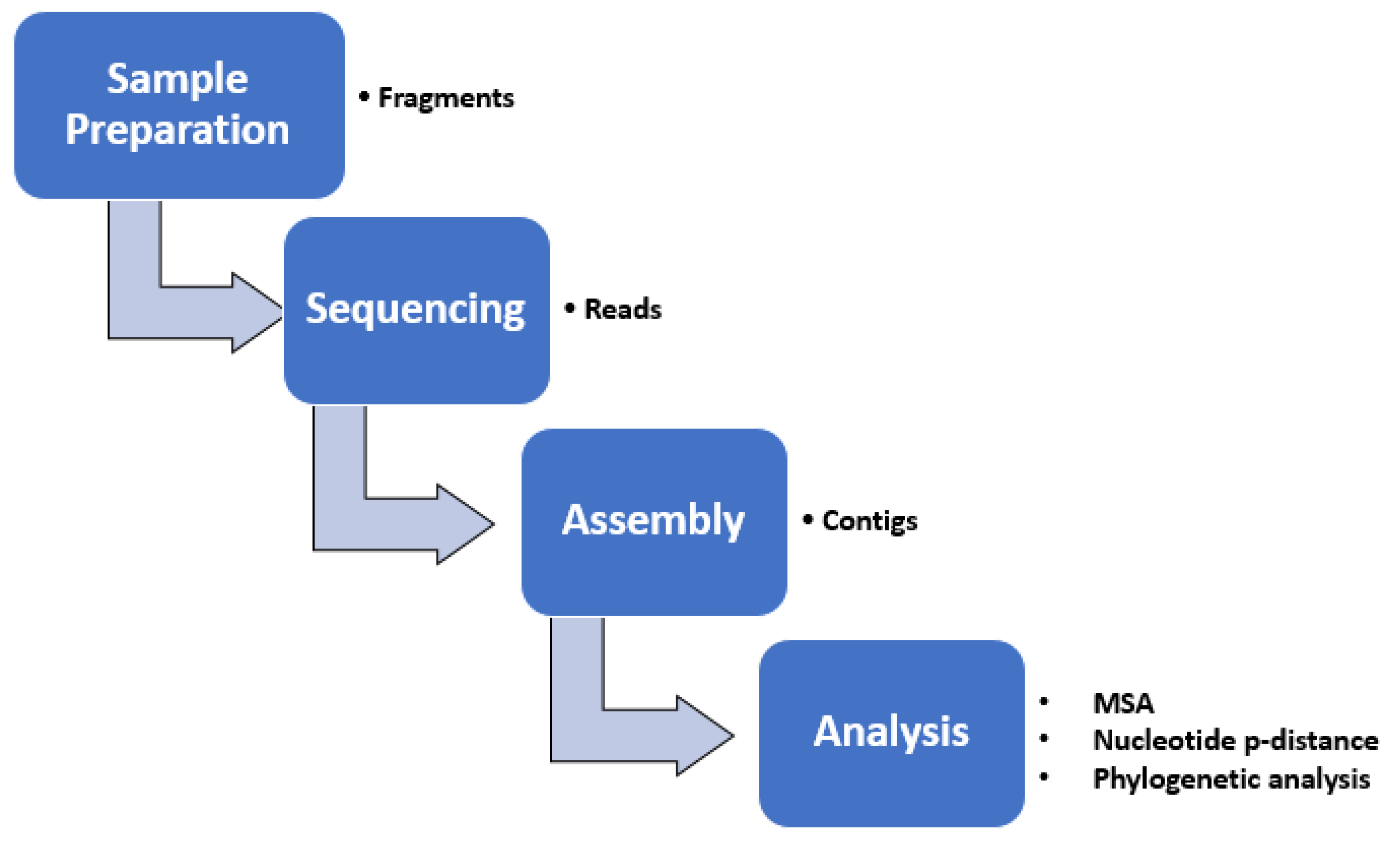
2.7. Bioinformatic Methods
2.7.1. Sequence Alignment and Database Searching
2.7.2. Assembly of VP Region Sequences
2.7.3. Multiple Sequence Alignment, Nucleotide p-Distances, and Phylogenetic Analysis
2.7.4. Recombination Analysis within the VP4/VP2 and VP3/VP1 Regions
3. Results
3.1. Viral Detection
3.2. Real-Time qPCR for the Template Samples
3.3. HRV Detection Rate of First- and Second-Round PCR Assays Using Gel Electrophoresis
3.4. Sequencing and Sequence Analysis
3.5. Pairwise Nucleotide p-Distances of HRVs
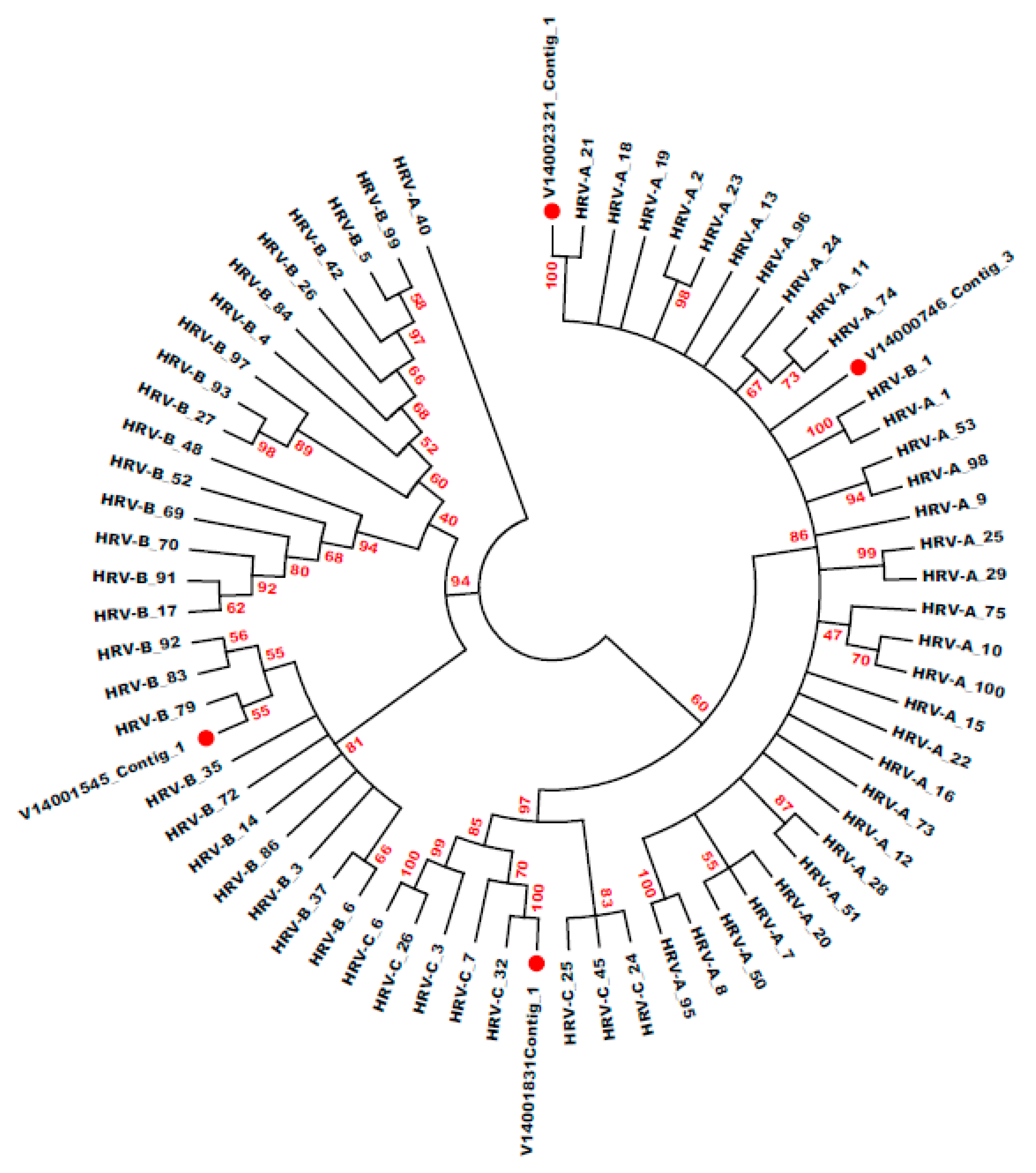
3.6. HRV Genotype Identification and Phylogenetic Analysis
3.7. Recombination Analysis within the VP4/VP2 and VP3/VP1 Regions
4. Discussion
4.1. Development of a Method for the Amplification of the VP Region of HRV
4.2. Proposed Criteria for HRV Genotyping
4.3. The Importance and Implications of HRV Genotyping
4.4. Limitations and Future Research
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smyth, R.L.; Openshaw, P.J.M. Bronchiolitis. Lancet 2006, 368, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Esposito, S. Emerging role of Mycoplasma pneumoniae and Chlamydia pneumoniae in paediatric respiratorytract infections. Lancet Infect. Dis. 2001, 1, 334–344. [Google Scholar] [CrossRef]
- van Woensel, J.; van Aalderen, W.; Kimpen, J.L.L. Viral lower respiratory tract infection in infants and young children. BMJ 2003, 327, 36–40. [Google Scholar] [CrossRef] [Green Version]
- Jartti, T.; Lehtinen, P.; Vuorinen, T.; Österback, R.; van den Hoogen, B.; Osterhaus, A.D.; Ruuskanen, O. Respiratory picornaviruses and respiratory syncytial virus as causative agents of acute expiratory wheezing in children. Emerg. Infect. Dis. 2004, 10, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Manoha, C.; Espinosa, S.; Aho, S.-L.; Huet, F.; Pothier, P. Epidemiological and clinical features of hMPV, RSV and RVs infections in young children. J. Clin. Virol. 2007, 38, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Jacques, J.; Bouscambert-Duchamp, M.; Moret, H.; Carquin, J.; Brodard, V.; Lina, B.; Motte, J.; Andréoletti, L. Association of respiratory picornaviruses with acute bronchiolitis in French infants. J. Clin. Virol. 2006, 35, 463–466. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Moustaki, M.; Tsolia, M.; Bossios, A.; Astra, E.; Prezerakou, A.; Gourgiotis, D.; Kafetzis, D. Association of Rhinovirus infection with increased disease severity in acute bronchiolitis. Am. J. Respir. Crit. Med. 2002, 165, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Korppi, M.; Kotaniemi-Syrjänen, A.; Waris, M.; Vainionpää, R.; Reijonen, T. Rhinovirus-associated wheezing in infancy: Comparison with respiratory syncytial virus bronchiolitis. Pediatr. Infect. Dis. J. 2004, 23, 995–999. [Google Scholar] [CrossRef]
- Calvo, C.; García-García, M.L.; Blanco, C.; Pozo, F.; Flecha, I.C.; Pérez-Breña, P. Role of rhinovirus in hospitalized infants with respiratory tract infections in Spain. Pediatr. Infect. Dis. J. 2007, 26, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Pitrez, P.; Stein, R.T.; Stuermer, L.; Macedo, I.S.; Schmitt, V.M.; Jones, M.H.; Arruda, E. Rhinovirus and acute bronchiolitis in young infants. J. Pediatr. 2005, 81, 417–420. [Google Scholar] [CrossRef]
- Kusel, M.M.; de Klerk, N.H.; Kebadze, T.; Vohma, V.; Holt, P.G.; Johnston, S.L.; Sly, P. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J. Allergy Clin. Immunol. 2007, 119, 1105–1110. [Google Scholar] [CrossRef]
- Lemanske, R.F., Jr.; Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Li, Z.; Shult, P.A.; Kirk, C.J.; Reisdorf, E.; Roberg, K.A.; Anderson, E. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J. Allergy Clin. Immunol. 2005, 116, 571–577. [Google Scholar] [CrossRef]
- Singh, A.M.; Moore, P.E.; Gern, J.E.; Lemanske, R.F., Jr.; Hartert, T.V. Bronchiolitis to asthma: A review and call for studies of gene-virus interactions in asthma causation. Am. J. Respir. Crit. Care Med. 2007, 175, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Roberg, K.A.; Anderson, E.L.; Pappas, T.E.; Printz, M.C.; Lee, W.M.; Shult, P.A.; Reisdorf, E.; et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008, 178, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Bosis, S.; Esposito, S.; Niesters, H.G.; Zuccotti, G.V.; Marseglia, G.; Lanari, M.; Zuin, G.; Pelucchi, C.; Osterhaus, A.D.; Principi, N. Role of respiratory pathogens in infants hospitalized for a first episode of wheezing and their impact on recurrences. Clin. Microbiol. Infect. 2008, 14, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Kotaniemi-Syrjänen, A.; Vainionpää, R.; Reijonen, T.M.; Waris, M.; Korhonen, K.; Korppi, M. Rhinovirus-induced wheezing in infancy—The first sign of childhood asthma? J. Allergy Clin. Immunol. 2003, 111, 66–71. [Google Scholar] [CrossRef]
- Hamparian, V.; Colonno, R.; Cooney, M.; Dick, E.; Gwaltney Jr, J.; Hughes, J.; Jordan Jr, W.; Kapikian, A.; Mogabgab, W.; Monto, A. A collaborative report: Rhinoviruses--extension of the numbering system from 89 to 100. Virology 1987, 159, 191–192. [Google Scholar] [PubMed]
- Alsayed, A.R.; Abed, A.; Khader, H.A.; Al-Shdifat, L.M.; Hasoun, L.; Al-Rshaidat, M.M.; Alkhatib, M.; Zihlif, M. Molecular Accounting and Profiling of Human Respiratory Microbial Communities: Toward Precision Medicine by Targeting the Respiratory Microbiome for Disease Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 4086. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-M.; Kiesner, C.; Pappas, T.; Lee, I.; Grindle, K.; Jartti, T.; Jakiela, B.; Lemanske Jr, R.F.; Shult, P.A.; Gern, J.E. A diverse group of previously unrecognized human rhinoviruses are common causes of respiratory illnesses in infants. PLoS ONE 2007, 2, e966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiang, D.; Kalra, I.; Yagi, S.; Louie, J.K.; Boushey, H.; Boothby, J.; Schnurr, D.P. Assay for 5′ noncoding region analysis of all human rhinovirus prototype strains. J. Clin. Microbiol. 2008, 46, 3736–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Holloway, B.; Dare, R.K.; Kuypers, J.; Yagi, S.; Williams, J.V.; Hall, C.B.; Erdman, D.D. Real-time reverse transcription-PCR assay for comprehensive detection of human rhinoviruses. J. Clin. Microbiol. 2008, 46, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Tapparel, C.; Cordey, S.; Van Belle, S.; Turin, L.; Lee, W.-M.; Regamey, N.; Meylan, P.; Mühlemann, K.; Gobbini, F.; Kaiser, L. New molecular detection tools adapted to emerging rhinoviruses and enteroviruses. J. Clin. Microbiol. 2009, 47, 1742–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gama, R.E.; Horsnell, P.R.; Hughes, P.J.; North, C.; Bruce, C.B.; al-Nakib, W.; Stanway, G. Amplification of rhinovirus specific nucleic acids from clinical samples using the polymerase chain reaction. J. Med. Virol. 1989, 28, 73–77. [Google Scholar] [CrossRef]
- Ireland, D.C.; Kent, J.; Nicholson, K.G. Improved detection of rhinoviruses in nasal and throat swabs by seminested RT-PCR. J. Med. Virol. 1993, 40, 96–101. [Google Scholar] [CrossRef]
- Andeweg, A.C.; Bestebroer, T.M.; Huybreghs, M.; Kimman, T.G.; de Jong, J.C. Improved detection of rhinoviruses in clinical samples by using a newly developed nested reverse transcription-PCR assay. J. Clin. Microbiol. 1999, 37, 524–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faux, C.E.; Arden, K.E.; Lambert, S.B.; Nissen, M.D.; Nolan, T.M.; Chang, A.B.; Sloots, T.P.; Mackay, I.M. Usefulness of published PCR primers in detecting human rhinovirus infection. Emerg. Infect. Dis. 2011, 17, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, Y.A.; Gern, J.E. Clinical and molecular features of human rhinovirus C. Microbes Infect. 2012, 14, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Wang, W.; Bessaud, M.; Ren, P.; Sheng, J.; Yan, H.; Zhang, J.; Lin, X.; Wang, Y.; Delpeyroux, F. Evidence of recombination and genetic diversity in human rhinoviruses in children with acute respiratory infection. PLoS ONE 2009, 4, e6355. [Google Scholar] [CrossRef]
- McIntyre, C.L.; Leitch, E.C.M.; Savolainen-Kopra, C.; Hovi, T.; Simmonds, P. Analysis of genetic diversity and sites of recombination in human rhinovirus species C. J. Virol. 2010, 84, 10297–10310. [Google Scholar] [CrossRef] [Green Version]
- Savolainen, C.; Mulders, M.N.; Hovi, T. Phylogenetic analysis of rhinovirus isolates collected during successive epidemic seasons. Virus Res. 2002, 85, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Ledford, R.M.; Patel, N.R.; Demenczuk, T.M.; Watanyar, A.; Herbertz, T.; Collett, M.S.; Pevear, D.C. VP1 sequencing of all human rhinovirus serotypes: Insights into genus phylogeny and susceptibility to antiviral capsid-binding compounds. J. Virol. 2004, 78, 3663–3674. [Google Scholar] [CrossRef] [Green Version]
- Laine, P.; Savolainen, C.; Blomqvist, S.; Hovi, T. Phylogenetic analysis of human rhinovirus capsid protein VP1 and 2A protease coding sequences confirms shared genus-like relationships with human enteroviruses. J. Gen. Virol. 2005, 86, 697–706. [Google Scholar] [CrossRef]
- Wisdom, A.; Leitch, E.M.; Gaunt, E.; Harvala, H.; Simmonds, P. Screening respiratory samples for detection of human rhinoviruses (HRVs) and enteroviruses: Comprehensive VP4-VP2 typing reveals high incidence and genetic diversity of HRV species C. J. Clin. Microbiol. 2009, 47, 3958–3967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberste, M.S.; Maher, K.; Pallansch, M.A. Molecular phylogeny and proposed classification of the simian picornaviruses. J. Virol. 2002, 76, 1244–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberste, M.S.; Maher, K.; Nix, W.A.; Michele, S.M.; Uddin, M.; Schnurr, D.; al-Busaidy, S.; Akoua-Koffi, C.; Pallansch, M.A. Molecular identification of 13 new enterovirus types, EV79–88, EV97, and EV100–101, members of the species Human Enterovirus B. Virus Res. 2007, 128, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Michele, S.M.; Maher, K.; Schnurr, D.; Cisterna, D.; Junttila, N.; Uddin, M.; Chomel, J.-J.; Lau, C.-S.; Ridha, W. Molecular identification and characterization of two proposed new enterovirus serotypes, EV74 and EV75. J. Gen. Virol. 2004, 85, 3205–3212. [Google Scholar] [CrossRef]
- Brown, B.A.; Maher, K.; Flemister, M.R.; Naraghi-Arani, P.; Uddin, M.; Oberste, M.S.; Pallansch, M.A. Resolving ambiguities in genetic typing of human enterovirus species C clinical isolates and identification of enterovirus 96, 99 and 102. J. Gen. Virol. 2009, 90, 1713–1723. [Google Scholar] [CrossRef]
- Smura, T.P.; Junttila, N.; Blomqvist, S.; Norder, H.; Kaijalainen, S.; Paananen, A.; Magnius, L.O.; Hovi, T.; Roivainen, M. Enterovirus 94, a proposed new serotype in human enterovirus species D. J. Gen. Virol. 2007, 88, 849–858. [Google Scholar] [CrossRef]
- Bouslama, L.; Nasri, D.; Chollet, L.; Belguith, K.; Bourlet, T.; Aouni, M.; Pozzetto, B.; Pillet, S. Natural recombination event within the capsid genomic region leading to a chimeric strain of human enterovirus B. J. Virol. 2007, 81, 8944–8952. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhu, S.; Yan, D.; Liu, G.; Bai, R.; Wang, D.; Chen, L.; Zhu, H.; An, H.; Kew, O. Natural type 3/type 2 intertypic vaccine-related poliovirus recombinants with the first crossover sites within the VP1 capsid coding region. PLoS ONE 2010, 5, e15300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arola, A.; Santti, J.; Ruuskanen, O.; Halonen, P.; Hyypiä, T. Identification of enteroviruses in clinical specimens by competitive PCR followed by genetic typing using sequence analysis. J. Clin. Microbiol. 1996, 34, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberste, M.S.; Maher, K.; Pallansch, M.A. Molecular phylogeny of all human enterovirus serotypes based on comparison of sequences at the 5′ end of the region encoding VP2. Virus Res. 1998, 58, 35–43. [Google Scholar] [CrossRef]
- Oberste, M.S.; Maher, K.; Flemister, M.R.; Marchetti, G.; Kilpatrick, D.R.; Pallansch, M.A. Comparison of classic and molecular approaches for the identification of untypeable enteroviruses. J. Clin. Microbiol. 2000, 38, 1170–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Flemister, M.R.; Brown, B.A.; Pallansch, M.A. Typing of human enteroviruses by partial sequencing of VP1. J. Clin. Microbiol. 1999, 37, 1288–1293. [Google Scholar] [CrossRef] [Green Version]
- Alsayed, A.R.; Hasoun, L.; Khader, H.A.; Abu-Samak, M.S.; Al-Shdifat, L.M.; Al-Shammari, B.; Maqbali, M.A. Co-infection of COVID-19 patients with atypical bacteria: A study based in Jordan. Pharm. Pract. 2023, 21, 1–5. [Google Scholar] [CrossRef]
- Alsayed, A.R.; Abed, A.; Jarrar, Y.B.; Alshammari, F.; Alshammari, B.; Basheti, I.A.; Zihlif, M. Alteration of the Respiratory Microbiome in Hospitalized Patients with Asthma–COPD Overlap during and after an Exacerbation. J. Clin. Med. 2023, 12, 2118. [Google Scholar] [CrossRef]
- Alsayed, A.; Al-Doori, A.; Al-Dulaimi, A.; Alnaseri, A.; Abuhashish, J.; Aliasin, K.; Alfayoumi, I. Influences of bovine colostrum on nasal swab microbiome and viral upper respiratory tract infections–A case report. Respir. Med. Case Rep. 2020, 31, 101189. [Google Scholar] [CrossRef]
- Al-Dulaimi, A.; Alsayed, A.R.; Maqbali, M.A.; Zihlif, M. Investigating the human rhinovirus co-infection in patients with asthma exacerbations and COVID-19. Pharm. Pr. 2022, 20, 2665. [Google Scholar] [CrossRef]
- Alsayed, A.R.; Talib, W.; Al-Dulaimi, A.; Daoud, S.; Al Maqbali, M. The first detection of Pneumocystis jirovecii in asthmatic patients post-COVID-19 in Jordan. Bosn. J. Basic Med. Sci. 2022, 22, 784–790. [Google Scholar] [CrossRef]
- Tapparel, C.; Junier, T.; Gerlach, D.; Van Belle, S.; Turin, L.; Cordey, S.; Mühlemann, K.; Regamey, N.; Aubert, J.-D.; Soccal, P.M. New respiratory enterovirus and recombinant rhinoviruses among circulating picornaviruses. Emerg. Infect. Dis. 2009, 15, 719. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.K.; Khuri-Bulos, N.; Williams, J.V.; Shehabi, A.A.; Faouri, S.; Al Jundi, I.; Chen, Q.; Heil, L.; Mohamed, Y.; Morin, L.-L. Human rhinovirus C associated with wheezing in hospitalised children in the Middle East. J. Clin. Virol. 2009, 46, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, C.L.; Knowles, N.J.; Simmonds, P. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J. Gen. Virol. 2013, 94, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Henquell, C.; Mirand, A.; Deusebis, A.-L.; Regagnon, C.; Archimbaud, C.; Chambon, M.; Bailly, J.-L.; Gourdon, F.; Hermet, E.; Dauphin, J.-B. Prospective genotyping of human rhinoviruses in children and adults during the winter of 2009–2010. J. Clin. Virol. 2012, 53, 280–284. [Google Scholar] [CrossRef]
- Arakawa, M.; Okamoto-Nakagawa, R.; Toda, S.; Tsukagoshi, H.; Kobayashi, M.; Ryo, A.; Mizuta, K.; Hasegawa, S.; Hirano, R.; Wakiguchi, H. Molecular epidemiological study of human rhinovirus species A, B and C from patients with acute respiratory illnesses in Japan. J. Med. Microbiol. 2012, 61, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, P.; McIntyre, C.; Savolainen-Kopra, C.; Tapparel, C.; Mackay, I.M.; Hovi, T. Proposals for the classification of human rhinovirus species C into genotypically assigned types. J. Gen. Virol. 2010, 91, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.G.; Bates, P.J.; Bardin, P.G.; Papi, A.; Leir, S.H.; Fraenkel, D.J.; Meyer, J.; Lackie, P.M.; Sanderson, G.; Holgate, S.; et al. Rhinoviruses infect the lower airways. J. Infect. Dis. 2000, 181, 1875–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jartti, T.; Jartti, L.; Ruuskanen, O.; Söderlund-Venermo, M. New respiratory viral infections. Curr. Opin. Pulm. Med. 2012, 18, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, P.G.; Britto, M.C.; Correia, J.B.; Duarte Mdo, C.; Fonceca, A.M.; Rose, K.; Hopkins, M.J.; Cuevas, L.E.; McNamara, P.S. Viral and atypical bacterial detection in acute respiratory infection in children under five years. PLoS ONE 2011, 6, e18928. [Google Scholar] [CrossRef] [Green Version]
- Cheuk, D.K.; Tang, I.W.; Chan, K.H.; Woo, P.C.; Peiris, M.J.; Chiu, S.S. Rhinovirus infection in hospitalized children in Hong Kong: A prospective study. Pediatr. Infect. Dis. J. 2007, 26, 995–1000. [Google Scholar] [CrossRef]
- Teeratakulpisarn, J.; Pientong, C.; Ekalaksananan, T.; Ruangsiripiyakul, H.; Uppala, R. Rhinovirus infection in children hospitalized with acute bronchiolitis and its impact on subsequent wheezing or asthma: A comparison of etiologies. Asian Pac. J. Allergy Immunol. 2014, 32, 226–234. [Google Scholar]
- Nour, A.; Alsayed, A.R.; Basheti, I. Prevalence of Asthma amongst Schoolchildren in Jordan and Staff Readiness to Help. Healthcare 2023, 11, 183. [Google Scholar] [CrossRef]
- Alsayed, A.R. Illustrating How to Use the Validated Alsayed_v1 Tools to Improve Medical Care: A Particular Reference to the Global Initiative for Asthma 2022 Recommendations. Patient Prefer. Adherence 2023, 17, 1161–1179. [Google Scholar] [CrossRef]
- AL-awaisheh, R.a.I.; Alsayed, A.R.; Basheti, I.A. Assessing the Pharmacist’s Role in Counseling Asthmatic Adults Using the Correct Inhaler Technique and Its Effect on Asthma Control, Adherence, and Quality of Life. Patient Prefer. Adherence 2023, 17, 961–972. [Google Scholar] [CrossRef]
- Stein, R.T.; Sherrill, D.; Morgan, W.J.; Holberg, C.J.; Halonen, M.; Taussig, L.M.; Wright, A.L.; Martinez, F.D. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet 1999, 354, 541–545. [Google Scholar] [CrossRef]
- Sigurs, N.; Aljassim, F.; Kjellman, B.; Robinson, P.D.; Sigurbergsson, F.; Bjarnason, R.; Gustafsson, P.M. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 2010, 65, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Camargo, C.A., Jr.; Raita, Y.; Freishtat, R.J.; Fujiogi, M.; Hahn, A.; Mansbach, J.M.; Spergel, J.M.; Pérez-Losada, M.; Hasegawa, K. Nasopharyngeal airway dual-transcriptome of infants with severe bronchiolitis and risk of childhood asthma: A multicenter prospective study. J. Allergy Clin. Immunol. 2022, 150, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Raita, Y.; Pérez-Losada, M.; Freishtat, R.J.; Hahn, A.; Castro-Nallar, E.; Ramos-Tapia, I.; Stearrett, N.; Bochkov, Y.A.; Gern, J.E.; Mansbach, J.M.; et al. Nasopharyngeal metatranscriptome profiles of infants with bronchiolitis and risk of childhood asthma: A multicentre prospective study. Eur. Respir. J. 2021, 60, 2102293. [Google Scholar] [CrossRef]
- Ooka, T.; Raita, Y.; Fujiogi, M.; Freishtat, R.J.; Gerszten, R.E.; Mansbach, J.M.; Zhu, Z.; Camargo, C.A., Jr.; Hasegawa, K. Proteomics endotyping of infants with severe bronchiolitis and risk of childhood asthma. Allergy 2022, 77, 3350–3361. [Google Scholar] [CrossRef] [PubMed]
- Raita, Y.; Pérez-Losada, M.; Freishtat, R.J.; Harmon, B.; Mansbach, J.M.; Piedra, P.A.; Zhu, Z.; Camargo, C.A.; Hasegawa, K. Integrated omics endotyping of infants with respiratory syncytial virus bronchiolitis and risk of childhood asthma. Nat. Commun. 2021, 12, 3601. [Google Scholar] [CrossRef]
- Zhu, Z.; Camargo, C.A., Jr.; Raita, Y.; Fujiogi, M.; Liang, L.; Rhee, E.P.; Woodruff, P.G.; Hasegawa, K. Metabolome subtyping of severe bronchiolitis in infancy and risk of childhood asthma. J. Allergy Clin. Immunol. 2022, 149, 102–112. [Google Scholar] [CrossRef]
- Palmenberg, A.C.; Spiro, D.; Kuzmickas, R.; Wang, S.; Djikeng, A.; Rathe, J.A.; Fraser-Liggett, C.M.; Liggett, S.B. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 2009, 324, 55–59. [Google Scholar] [CrossRef] [Green Version]
- Lewis-Rogers, N.; Bendall, M.L.; Crandall, K.A. Phylogenetic relationships and molecular adaptation dynamics of human rhinoviruses. Mol. Biol. Evol. 2009, 26, 969–981. [Google Scholar] [CrossRef]
- Nix, W.A.; Oberste, M.S.; Pallansch, M.A. Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J. Clin. Microbiol. 2006, 44, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Leitch, E.M.; Harvala, H.; Robertson, I.; Ubillos, I.; Templeton, K.; Simmonds, P. Direct identification of human enterovirus serotypes in cerebrospinal fluid by amplification and sequencing of the VP1 region. J. Clin. Virol. 2009, 44, 119–124. [Google Scholar] [CrossRef]
- She, R.C.; Hymas, W.C.; Taggart, E.W.; Petti, C.A.; Hillyard, D.R. Performance of enterovirus genotyping targeting the VP1 and VP2 regions on non-typeable isolates and patient specimens. J. Virol. Methods 2010, 165, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Kiang, D.; Newbower, E.C.; Yeh, E.; Wold, L.; Chen, L.; Schnurr, D.P. An algorithm for the typing of enteroviruses and correlation to serotyping by viral neutralization. J. Clin. Virol. 2009, 45, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Haff, L.A. Improved quantitative PCR using nested primers. Genome Res. 1994, 3, 332–337. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.L. Epidemiology, Classification and Evolution of Human Rhinoviruses. Ph.D. Thesis, University of Edinburgh, Edinburgh, UK, 2013. [Google Scholar]
- Silva, P.A.; Diedrich, S.; de Paula Cardoso, D.d.D.; Schreier, E. Identification of enterovirus serotypes by pyrosequencing using multiple sequencing primers. J. Virol. Methods 2008, 148, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Vlasak, M.; Blomqvist, S.; Hovi, T.; Hewat, E.; Blaas, D. Sequence and structure of human rhinoviruses reveal the basis of receptor discrimination. J. Virol. 2003, 77, 6923–6930. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-M.; Monroe, S.; Rueckert, R. Role of maturation cleavage in infectivity of picornaviruses: Activation of an infectosome. J. Virol. 1993, 67, 2110–2122. [Google Scholar] [CrossRef] [Green Version]
- Hughes, P.J.; North, C.; Jellis, C.H.; Minor, P.D.; Stanway, G. The nucleotide sequence of human rhinovirus 1B: Molecular relationships within the rhinovirus genus. J. Gen. Virol. 1988, 69, 49–58. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1st-Round PCR Assay | OS Primer | OAS Primer | 2nd-Round PCR Assay | IS Primer | IAS Primer | PCR Product Length (bp) |
|---|---|---|---|---|---|---|
| Nested PCR | ||||||
| B | 92,580 = F187 | R 92383 | E | F VP1F | R VP1R | 665 |
| W | W:F | W:R | X | X:F | X:R | 929 |
| Y | Y:F | Y:R | Z | Z:F | Z:R | 563 |
| Semi-nested PCR | ||||||
| A | F 92378 | R 92379 a | D | F92580 = F187 | R 92379 a | 355 |
| C | F 92380 b | R 92383 | E | F VP1F b | R VP1R | 455 |
| Aetiology | Total n (%) | Age, n (%) | ||
|---|---|---|---|---|
| 1–6 Months n = 24 | >6–12 Months n = 35 | >12–24 Months n = 32 | ||
| Overall viral detection | 79 (86.8%) | 17 | 32 | 30 |
| ADV | 3 (3.3%) | 1 | 1 | 1 |
| BOV | 1 (1.1%) | 0 | 0 | 1 |
| COV | 1 (1.1%) | 0 | 0 | 1 |
| FLU | 13 (14.3%) | 2 | 4 | 7 |
| HRV | 15 (16.5%) | 3 | 4 | 8 |
| MPV | 3 (3.3%) | 1 | 2 | 0 |
| PF | 1 (1.1%) | 0 | 0 | 1 |
| RSV | 42 (46.2%) | 10 | 21 | 11 |
| Overall dual detection | 16 (17.6%) | 4 | 3 | 9 |
| RSV + HRV | 7 (7.7%) | 1 | 1 | 5 |
| RSV + FLU | 4 (4.4%) | 1 | 1 | 2 |
| HRV + FLU | 5 (5.5%) | 2 | 1 | 2 |
| Not found | 12 (13.2%) | 7 | 3 | 2 |
| Characteristics | Total n = 79 | RSV n = 42 | HRV n = 15 | FLU n = 13 |
|---|---|---|---|---|
| Male, n (%) | 45 (62.9) | 23 (59.1) | 8 (51.6) | 9 (70.0) |
| Body weight (kg), Mean ± SD | 8.30 ± 1.82 | 8.09 ± 1.82 | 8.52 ± 1.84 | 8.82 ± 1.86 |
| Breastfeeding, n (%) | 76 (94.1) | 42 (92.7) | 14 (96.8) | 10 (93.3) |
| Duration of breastfeeding (months) | 4 | 4 | 3 | 4 |
| First relative atopy, n (%) | 25 (31.6) | 18 (20.9) | 5 (19.4) | 4 (26.7) |
| Characteristics | Total n = 79 | RSV n = 42 | HRV n = 15 | FLU n = 13 |
|---|---|---|---|---|
| History of fever, n (%) | 72 (91.1) | 38 (90.5) | 11 (73.4) | 12 (92.3) |
| Temperature, °C Mean ± SD | 38.8 ± 0.9 | 38.2 ± 0.9 | 37.9 ± 0.9 | 38.2 ± 0.8 |
| 95% CI | 37.8–38.3 | 37.8–38.4 | 37.7–38.2 | 38.2–38.8 |
| Duration of fever, days Mean ± SD | 4.5 ± 3.0 | 4.4 ± 2.8 | 2.9 ± 2.9 | 4.5 ± 3.3 |
| 95% CI | 3.9–4.7 | 3.8–4.8 | 1.8–3.4 | 3.2–5.7 |
| SpO2 < 95%, n (%) | 105 (61.8) | 68 (61.8) | 18 (58.1) | 20 (66.7) |
| SpO2, % Min–Max | 69–99 | 69–99 | 71–99 | 87–99 |
| Mean ± SD | 93.6 ± 4.7 | 93.7 ± 4.9 | 94.1 ± 4.8 | 94.0 ± 3.2 |
| 95% CI | 93.1–94.4 | 92.8–94.6 | 92.3–95.9 | 92.8–95.2 |
| Specimen ID | Ct Value | Genotype- BLASTN | E-Value | Genotype- BLASTX | E-Value | Genotype- Phylogeny | Bootstrap | Distance |
|---|---|---|---|---|---|---|---|---|
| V14002321 | 18.06 | HRV-A_21 | 0 | HRV-A_21 | 2.1 × 10−117 | HRV-A_21 | 100 | 0.059 |
| V14001831 | 18.10 | HRV-C_32 | 0 | HRV-C | 1.8 × 10−93 | HRV-C_32 | 100 | 0.047 |
| V14001545 | 19.08 | HRV-B | 0 | HRV-B | 2.2 × 10−94 | HRV-B_79 | 55 | 0.150 |
| V14000746 | 19.12 | HRV-A_40 | 0 | HRV-A | 2.3 × 10−96 | HRV-A_40 | 96 | 0.055 |
| V17004616 | 33.00 | HRV-A | 0 | HRV-A | 4.7 × 10−31 | HRV-A_98 | <50 | 0.173 |
| V17004470 | 31.10 | HRV-A | 6.6 × 10−52 | HRV-A | 9.7 × 10−19 | HRV-A_13 | <50 | 0.236 |
| V17005728 | 26.69 | HRV-C | 0 | HRV-C | 2.1 × 10−86 | HRV-C_45 | 73 | 0.289 |
| V17006129 | 31.90 | HRV-C_7 | 0 | HRV-C | 1.4 × 10−137 | HRV-C_7 | 99 | 0.098 |
| V17006131 | 29.91 | HRV-C | 0 | HRV-C | 1.5 × 10−96 | HRV-C_45 | <50 | 0.296 |
| V17006286 | 33.07 | HRV-A | 4.2 × 10−69 | HRV-A | 5.6 × 10−21 | HRV-A_50 | 50 | 0.264 |
| V17004189 | 28.88 | HRV-A | 2 × 10−25 | HRV-A | 5.2 × 10−7 | NA | NA | NA |
| V17003665 | 30.09 | HRV-C | 1.7 × 10−46 | HRV-C | 2 × 10−17 | HRV-C_24 | 69 | 0.268 |
| V17004870 | 21.09 | HRV-C | 3 × 10−20 | NA | NA | HRV-C_32 | <50 | 0.927 |
| V17005031 | 31.54 | HRV-A | 1.4 × 10−121 | HRV-A | 1.4 × 10−53 | HRV-A_47 | 52 | 0.087 |
| V17004381 | 28.75 | HRV C | 9 × 10−66 | HRV C | 7 × 10−30 | HRV-C_36 | 100 | 0.066 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsayed, A.R.; Abed, A.; Abu-Samak, M.; Alshammari, F.; Alshammari, B. Etiologies of Acute Bronchiolitis in Children at Risk for Asthma, with Emphasis on the Human Rhinovirus Genotyping Protocol. J. Clin. Med. 2023, 12, 3909. https://doi.org/10.3390/jcm12123909
Alsayed AR, Abed A, Abu-Samak M, Alshammari F, Alshammari B. Etiologies of Acute Bronchiolitis in Children at Risk for Asthma, with Emphasis on the Human Rhinovirus Genotyping Protocol. Journal of Clinical Medicine. 2023; 12(12):3909. https://doi.org/10.3390/jcm12123909
Chicago/Turabian StyleAlsayed, Ahmad R., Anas Abed, Mahmoud Abu-Samak, Farhan Alshammari, and Bushra Alshammari. 2023. "Etiologies of Acute Bronchiolitis in Children at Risk for Asthma, with Emphasis on the Human Rhinovirus Genotyping Protocol" Journal of Clinical Medicine 12, no. 12: 3909. https://doi.org/10.3390/jcm12123909
APA StyleAlsayed, A. R., Abed, A., Abu-Samak, M., Alshammari, F., & Alshammari, B. (2023). Etiologies of Acute Bronchiolitis in Children at Risk for Asthma, with Emphasis on the Human Rhinovirus Genotyping Protocol. Journal of Clinical Medicine, 12(12), 3909. https://doi.org/10.3390/jcm12123909