
Heparanase Inhibition Prevents Liver Steatosis in E0 Mice


Abstract
:1. Background
2. Methods
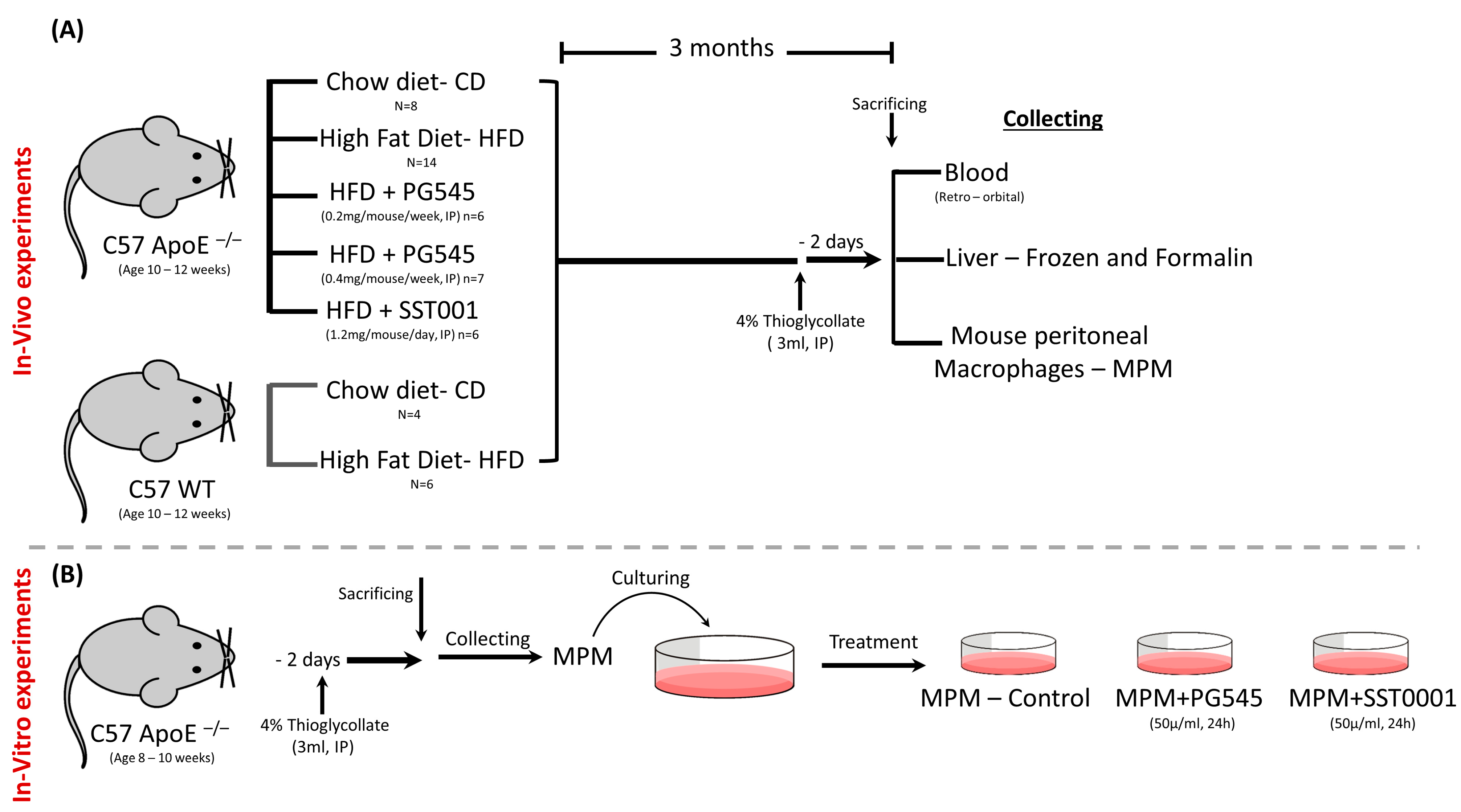
2.1. In Vivo Experiments: Animals
- (1)
- ApoE−/− fed with chow diet (CD), n = 8, (E0-CD);
- (2)
- ApoE−/− fed with high-fat diet (HFD, TD.88137, 42% of calories from fat) and treated with saline, n = 14, (E0-HFD);
- (3)
- ApoE−/− fed with HFD and treated with PG545 at low dose (0.2 mg/mouse/once weekly, 6.4 mg/kg), n = 6;
- (4)
- ApoE−/− fed with HFD and treated with PG545 at high dose (0.4 mg/mouse/once weekly, 13.3 mg/kg), n = 7;
- (5)
- ApoE−/− fed with HFD and treated with SST0001 (1.2 mg/mouse/daily), n = 6
- (6)
- WT fed with CD, n = 4;
- (7)
- WT fed with HFD, n = 6.
2.2. In Vitro Experiments
2.2.1. MPM Isolation and Treatment
2.2.2. Serum Analysis
2.2.3. Real-Time Polymerase Chain Reaction (qPCR)
2.2.4. Cholesterol and Triglycerides Mass in MPM and Liver Tissue
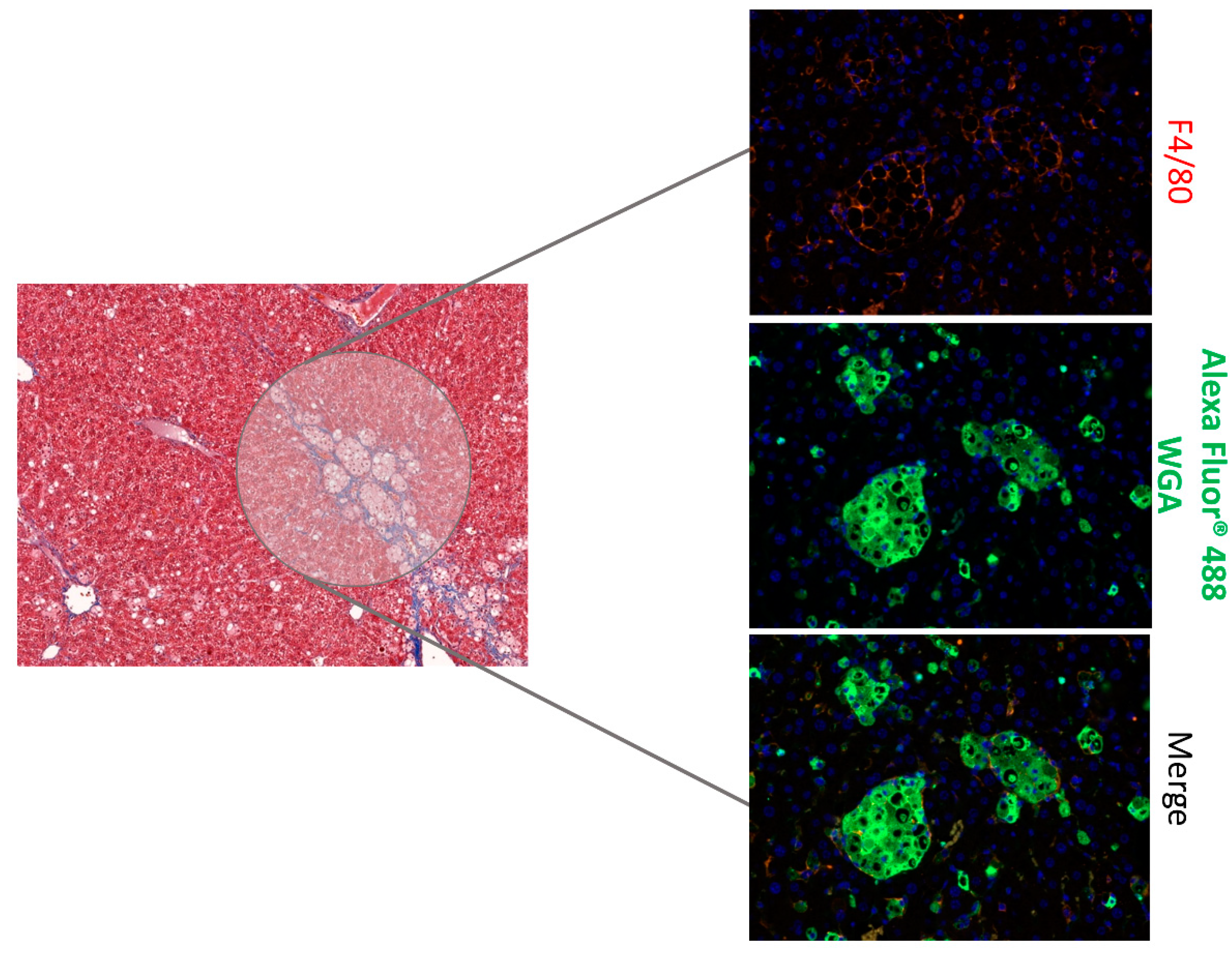
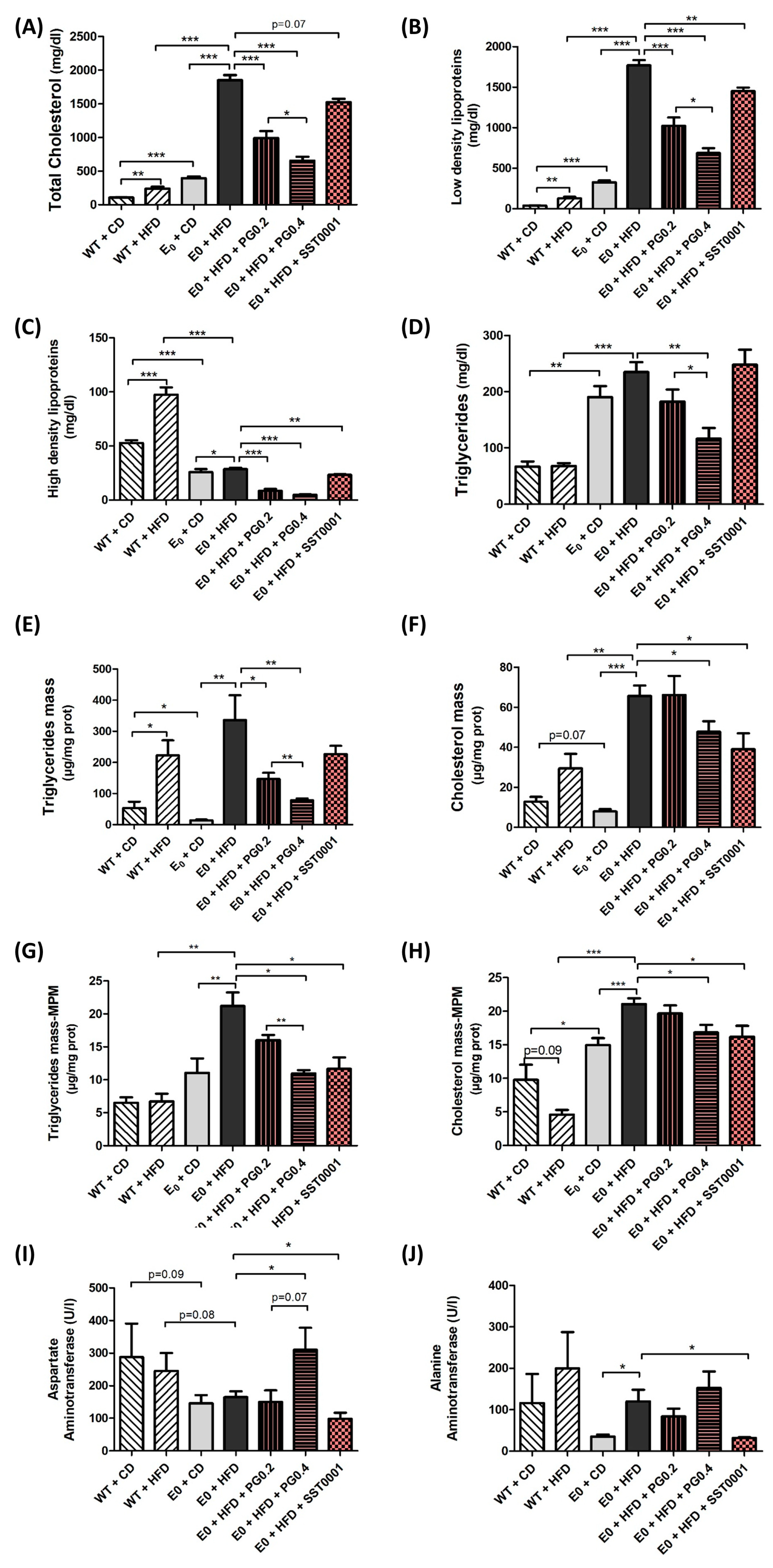
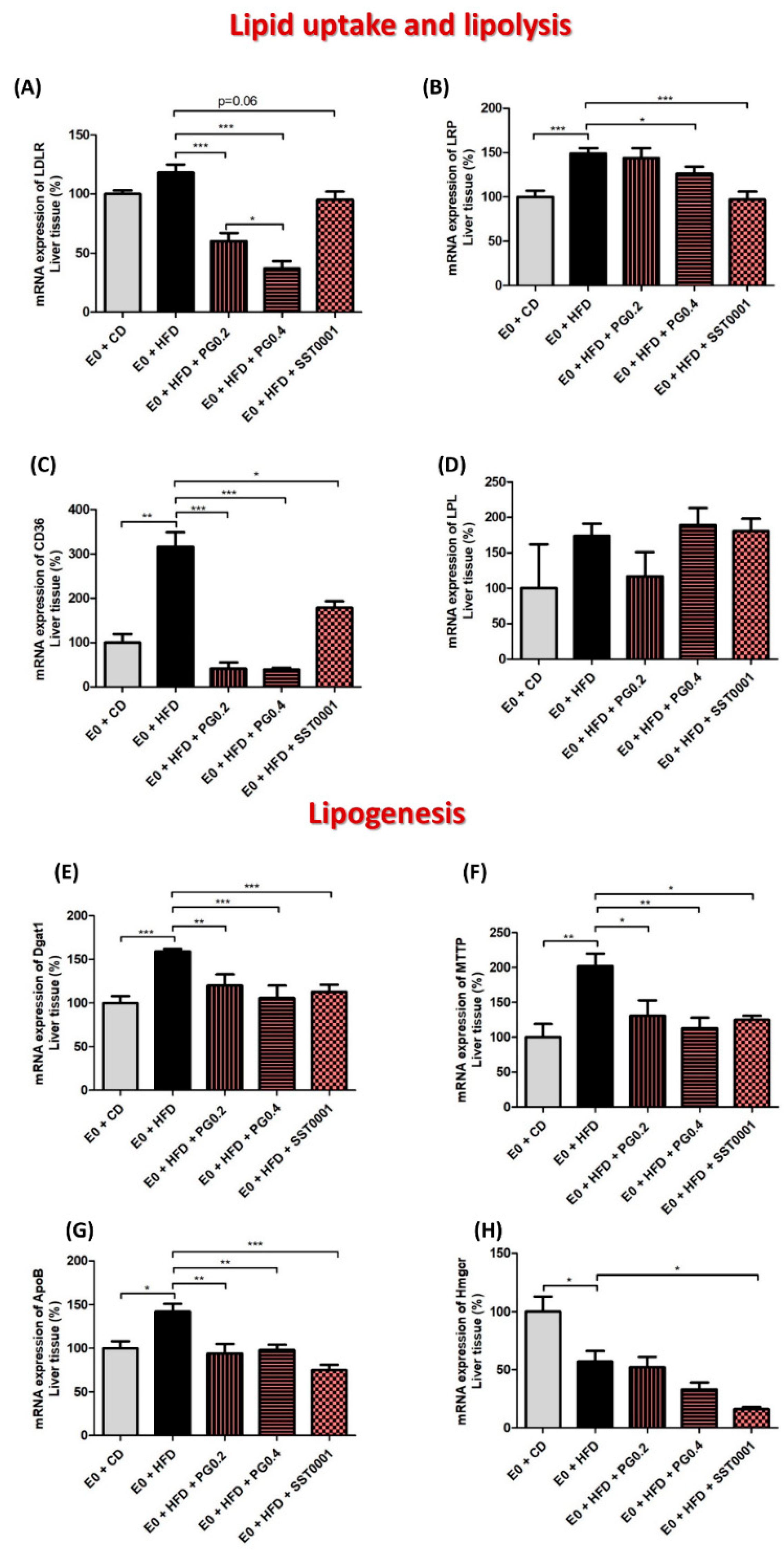
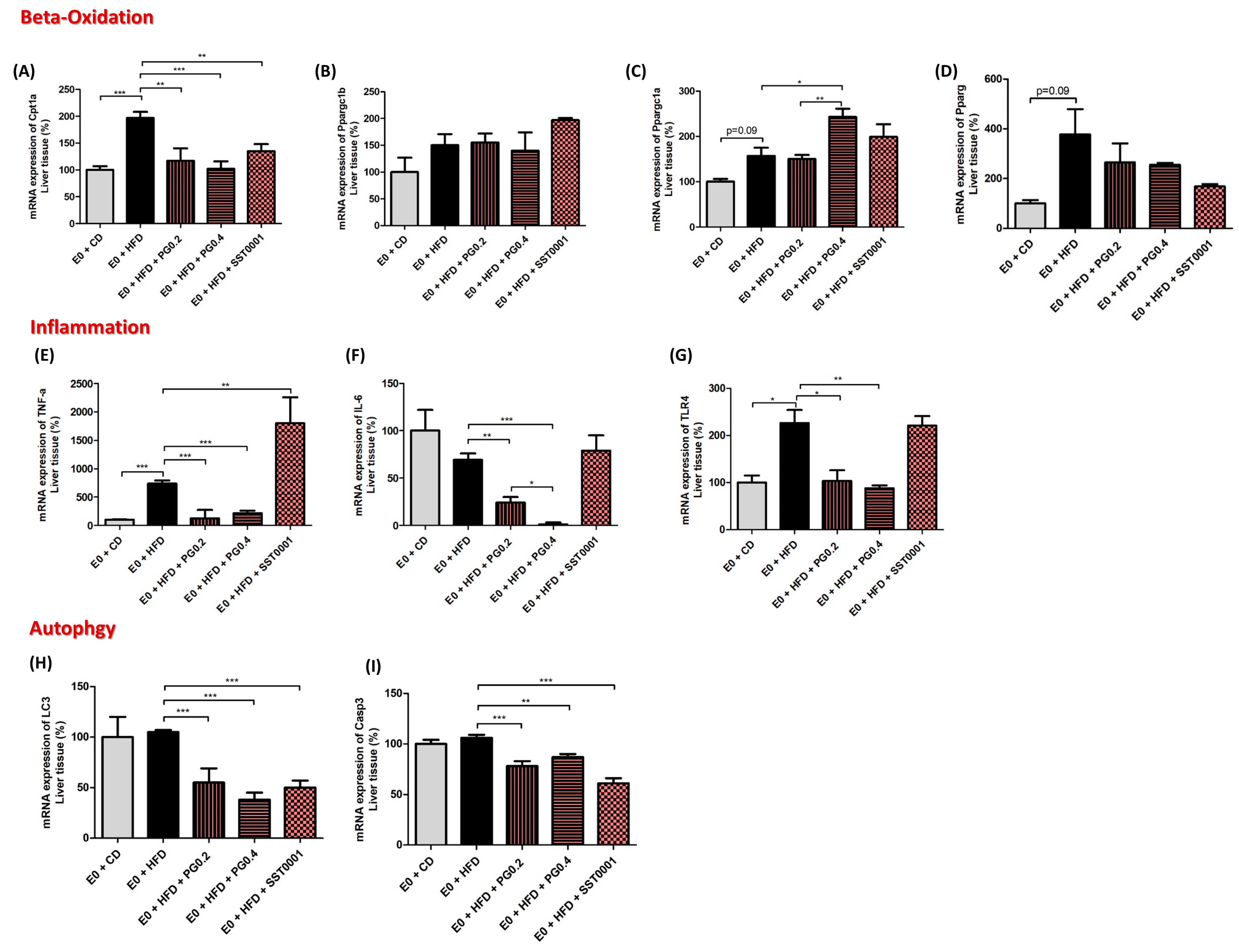
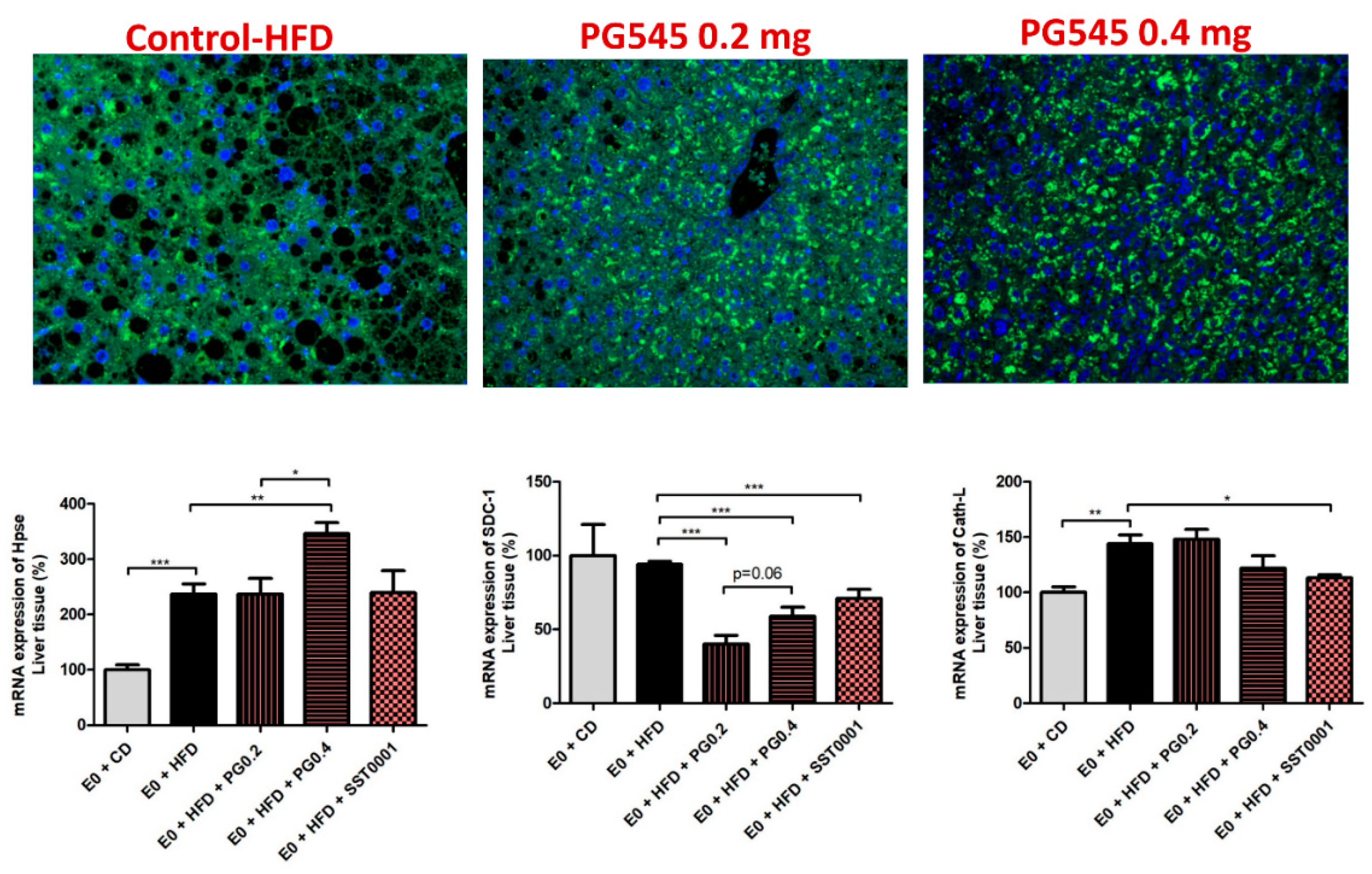
3. Results
4. Discussion
5. Conclusions
6. Limitations
- Mouse model used: To study the effect of PG545 on liver fibrosis, the mouse model of CCl4, induced liver fibrosis, should be utilized, as animals in this model rapidly develop severe liver fibrosis upon exposure to CCl4, whereas E0 mice are not a fibrosis model;
- Sample size: We used a small number of animals in each treatment group. Increasing the number of animals in the study groups would absolutely strengthen our results. However, it is noteworthy to emphasize the consistent effect of heparanase inhibition in all the animals in the same group.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated with Long-term Outcomes of Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e310. [Google Scholar] [CrossRef] [Green Version]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease- Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Stepanova, M.; Rafiq, N.; Makhlouf, H.; Younoszai, Z.; Agrawal, R.; Goodman, Z. Pathologic criteria for nonalcoholic steatohepatitis: Interprotocol agreement and ability to predict liver-related mortality. Hepatology 2011, 53, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Jojima, T.; Tomotsune, T.; Iijima, T.; Akimoto, K.; Suzuki, K.; Aso, Y. Empagliflozin (an SGLT2 inhibitor), alone or in combination with linagliptin (a DPP-4 inhibitor), prevents steatohepatitis in a novel mouse model of non-alcoholic steatohepatitis and diabetes. Diabetol. Metab. Syndr. 2016, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Singal, A.K. Trends in Liver Disease Etiology Among Adults Awaiting Liver Transplantation in the United States, 2014–2019. JAMA Netw. Open 2020, 3, e1920294. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G. Hepatic fatty acid trafficking: Multiple forks in the road. Adv. Nutr. 2013, 4, 697–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Hall, C.; Glaser, S.; Francis, H.; Meng, F.; Alpini, G. Pathogenesis of Kupffer Cells in Cholestatic Liver Injury. Am. J. Pathol. 2016, 186, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, J.D.; Guo, G.L. Regulation of Hepatic Stellate Cells and Fibrogenesis by Fibroblast Growth Factors. BioMed Res. Int. 2016, 2016, 8323747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Wang, F.; Jin, C.; Huang, X.; Miller, D.L.; Basilico, C.; McKeehan, W.L. Role of fibroblast growth factor type 1 and 2 in carbon tetrachloride-induced hepatic injury and fibrogenesis. Am. J. Pathol. 2003, 163, 1653–1662. [Google Scholar] [CrossRef] [Green Version]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic Steatohepatitis Is the Second Leading Etiology of Liver Disease Among Adults Awaiting Liver Transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef]
- Charlton, M.R.; Burns, J.M.; Pedersen, R.A.; Watt, K.D.; Heimbach, J.K.; Dierkhising, R.A. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 2011, 141, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Graubard, B.I.; Quraishi, S.; Zeuzem, S.; Davila, J.A.; El-Serag, H.B.; McGlynn, K.A. Population-attributable fractions of risk factors for hepatocellular carcinoma in the United States. Am. J. Gastroenterol. 2013, 108, 1314–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoen, L.F.; Guimaraes, E.L.; Dolle, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Huang, H.; Shi, M.; Yang, W.; Kuang, J.; Yan, J. Autophagy mediated by endoplasmic reticulum stress enhances the caffeine-induced apoptosis of hepatic stellate cells. Int. J. Mol. Med. 2017, 40, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymann, F.; Tacke, F. Immunology in the liver—From homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M. Lessons from animal models of NASH. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2005, 33, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N.; Lau, M.Y.; Kao, E.; Petrovic, L.M.; Lee, A.S. Liver-Specific Loss of Glucose-Regulated Protein 78 Perturbs the Unfolded Protein Response and Exacerbates a Spectrum of Liver Diseases in Mice. Hepatology 2011, 54, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vreys, V.; David, G. Mammalian heparanase: What is the message? J. Cell. Mol. Med. 2007, 11, 427–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secchi, M.F.; Masola, V.; Zaza, G.; Lupo, A.; Gambaro, G.; Onisto, M. Recent data concerning heparanase: Focus on fibrosis, inflammation and cancer. Biomol. Concepts 2015, 6, 415–421. [Google Scholar] [CrossRef]
- Levidiotis, V.; Freeman, C.; Punler, M.; Martinello, P.; Creese, B.; Ferro, V.; van der Vlag, J.; Berden, J.H.; Parish, C.R.; Power, D.A. A synthetic heparanase inhibitor reduces proteinuria in passive Heymann nephritis. J. Am. Soc. Nephrol. JASN 2004, 15, 2882–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabelink, T.J.; van den Berg, B.M.; Garsen, M.; Wang, G.; Elkin, M.; van der Vlag, J. Heparanase: Roles in cell survival, extracellular matrix remodelling and the development of kidney disease. Nat. Rev. Nephrol. 2017, 13, 201–212. [Google Scholar] [CrossRef]
- Nadir, Y.; Brenner, B. Heparanase multiple effects in cancer. Thromb. Res. 2014, 133, S90–S94. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2016, 29, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Zaza, G.; Secchi, M.F.; Gambaro, G.; Lupo, A.; Onisto, M. Heparanase is a key player in renal fibrosis by regulating TGF-beta expression and activity. Bba-Mol. Cell Res. 2014, 1843, 2122–2128. [Google Scholar] [CrossRef]
- Masola, V.; Gambaro, G.; Tibaldi, E.; Brunati, A.M.; Gastaldello, A.; D’Angelo, A.; Onisto, M.; Lupo, A. Heparanase and syndecan-1 interplay orchestrates fibroblast growth factor-2-induced epithelial-mesenchymal transition in renal tubular cells. J. Biol. Chem. 2012, 287, 1478–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, N.; Goldberg, R.; Neuman, T.; Garsen, M.; Zcharia, E.; Rubinstein, A.M.; van Kuppevelt, T.; Meirovitz, A.; Pisano, C.; Li, J.P.; et al. Heparanase Is Essential for the Development of Diabetic Nephropathy in Mice. Diabetes 2012, 61, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Chang, J.; Lee, P.H.; Lin, D.Y.; Wu, C.C.; Jeng, L.B.; Lin, Y.J.; Mok, K.T.; Lee, W.C.; Yeh, H.Z.; et al. Adjuvant heparanase inhibitor PI-88 therapy for hepatocellular carcinoma recurrence. World J. Gastroenterol. 2014, 20, 11384–11393. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Kleeff, J.; Shi, X.; Buchler, M.W.; Friess, H. Heparanase expression in hepatocellular carcinoma and the cirrhotic liver. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2003, 26, 192–198. [Google Scholar] [CrossRef]
- El-Assal, O.N.; Yamanoi, A.; Ono, T.; Kohno, H.; Nagasue, N. The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 1299–1305. [Google Scholar]
- Ikeguchi, M.; Hirooka, Y.; Kaibara, N. Heparanase gene expression and its correlation with spontaneous apoptosis in hepatocytes of cirrhotic liver and carcinoma. Eur. J. Cancer 2003, 39, 86–90. [Google Scholar] [CrossRef]
- Hamoud, S.; Muhammad, R.S.; Abu-Saleh, N.; Hassan, A.; Zohar, Y.; Hayek, T. Heparanase Inhibition Reduces Glucose Levels, Blood Pressure, and Oxidative Stress in Apolipoprotein E Knockout Mice. BioMed. Res. Int. 2017, 2017, 7357495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shekh Muhammad, R.; Niroz, A.S.; Kinaneh, S.; Agbaria, M.; Sabo, E.; Grajeda-Iglesias, C.; Volkova, N.; Hamoud, S. Heparanase inhibition attenuates atherosclerosis progression and liver steatosis in E0 mice. Atherosclerosis 2018, in press. [Google Scholar]
- Ritchie, J.P.; Ramani, V.C.; Ren, Y.; Naggi, A.; Torri, G.; Casu, B.; Penco, S.; Pisano, C.; Carminati, P.; Tortoreto, M.; et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 1382–1393. [Google Scholar] [CrossRef] [Green Version]
- Shafat, I.; Ben-Arush, M.W.; Issakov, J.; Meller, I.; Naroditsky, I.; Tortoreto, M.; Cassinelli, G.; Lanzi, C.; Pisano, C.; Ilan, N.; et al. Pre-clinical and clinical significance of heparanase in Ewing’s sarcoma. J. Cell Mol. Med. 2011, 15, 1857–1864. [Google Scholar] [CrossRef]
- Cassinelli, G.; Lanzi, C.; Tortoreto, M.; Cominetti, D.; Petrangolini, G.; Favini, E.; Zaffaroni, N.; Pisano, C.; Penco, S.; Vlodavsky, I.; et al. Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochem. Pharmacol. 2013, 85, 1424–1432. [Google Scholar] [CrossRef]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzym. 1978, 52, 302–310. [Google Scholar]
- Aviram, M.; Vaya, J. Markers for low-density lipoprotein oxidation. Methods Enzym. 2001, 335, 244–256. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Planer, D.; Metzger, S.; Zcharia, E.; Wexler, I.D.; Vlodavsky, I.; Chajek-Shaul, T. Role of Heparanase on Hepatic Uptake of Intestinal Derived Lipoprotein and Fatty Streak Formation in Mice. PLoS ONE 2011, 6, e18370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacArthur, J.M.; Bishop, J.R.; Stanford, K.I.; Wang, L.; Bensadoun, A.; Witztum, J.L.; Esko, J.D. Liver heparan sulfate proteoglycans mediate clearance of triglyceride-rich lipoproteins independently of LDL receptor family members. J. Clin. Investig. 2007, 117, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Chen, K. Recent insights into factors affecting remnant lipoprotein uptake. Curr. Opin. Lipidol. 2010, 21, 396. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Fuki, I.V. Cell-surface heparan sulfate proteoglycans: Dynamic molecules mediating ligand catabolism. Curr. Opin. Lipidol. 1997, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Ho, C.T.; Liong, E.C.; Nanji, A.A.; Leung, T.M.; Lau, T.Y.; Fung, M.L.; Tipoe, G.L. Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1, and NF-kappa B pathways. Eur. J. Nutr. 2014, 53, 187–199. [Google Scholar] [CrossRef]
- Hu, J.; Wang, H.; Hu, Y.F.; Xu, X.F.; Chen, Y.H.; Xia, M.Z.; Zhang, C.; Xu, D.X. Cadmium induces inflammatory cytokines through activating Akt signaling in mouse placenta and human trophoblast cells. Placenta 2018, 65, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qin, W.; Zhang, J.P.; Hu, Z.Y.; Tong, J.W.; Ding, C.B.; Peng, Z.G.; Zhao, L.X.; Song, D.Q.; Jiang, J.D. HCV IRES-mediated core expression in zebrafish. PLoS ONE 2013, 8, e56985. [Google Scholar] [CrossRef] [PubMed]
- Secchi, M.F.; Crescenzi, M.; Masola, V.; Russo, F.P.; Floreani, A.; Onisto, M. Heparanase and macrophage interplay in the onset of liver fibrosis. Sci. Rep. 2017, 7, 14956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, I.; Kang, H.S.; Yang, Y.; Pyun, K.H. IL-6 induces hepatic inflammation and collagen synthesis in vivo. Clin. Exp. Immunol. 1994, 95, 530–535. [Google Scholar] [CrossRef]
- Li, S.; Hong, M.; Tan, H.Y.; Wang, N.; Feng, Y.B. Insights into the Role and Interdependence of Oxidative Stress and Inflammation in Liver Diseases. Oxid Med. Cell Longev. 2016, 2016, 4234061. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.X.; Wong, C.W.; Feng, Y.B. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | Amplicon Length | ||
|---|---|---|---|---|
| 1 | LDLR | TCGACTCACGGGTTCAGATG | ACCAGTTCACCCCTCTAGGC | 104 |
| 2 | LRP1 | CTCCCACCGCTATGTGATCC | TCGCTGCCCACATACTTGTT | 132 |
| 3 | CD36 | CATTTGCAGGTCTATCTACG | CAATGTCTAGCACACCATAAG | 182 |
| 4 | LPL | GGATGGACGGTAACGGGAAT | ATAATGTTGCTGGGCCCGAT | 123 |
| 5 | DGAT1 | TCATACTCCATCATGTTCCTC | GAAGTAATAGAGATCTCGGTAGG | 174 |
| 6 | MTTP | TCCTGGACTTTTTGGATTTC | TTGAACTTACTAAGGAGGGC | 124 |
| 7 | ApoB | CTCCTACAAGAATAAGTATGGG | GAAGCGACTGTTGATCTTAG | 85 |
| 8 | HMGCoA reductase | GATAGCTGATCCTTCTCCTC | ATGCTGATCATCTTGGAGAG | 135 |
| 9 | IL-6 | AAGAAATGATGGATGCTACC | GAGTTTCTGTATCTCTCTGAAG | 164 |
| 10 | TLR4 | TCCCTGCATAGAGGTAGTTCC | TCCAGCCACTGAAGTTCTGA | 171 |
| 11 | TNF-a | CTATGTCTCAGCCTCTTCTC | CATTTGGGAACTTCTCATCC | 109 |
| 12 | nCEH | CTAGTGCAAAGATCAGCTAC | ATTTGCTCCGGAAAATAGAC | 118 |
| 13 | SIRT1 | AAACAGTGAGAAAATGCTGG | GGTATTGATTACCCTCAAGC | 75 |
| 14 | HPSE | CCAAGTGCTCGGGGTTAGAC | AGAAACTGTTGGGCTCATTGC | 159 |
| 15 | Cath L | CCCTATGAAGCGAAGGACGG | CTGGAGAGACGGATGGCTTG | 165 |
| 16 | Cpt1a | GGGAGGAATACATCTACCTG | GAAGACGAATAGGTTTGAGTTC | 183 |
| 17 | pPARgc1b | AAGAACTTCAGACGTGAGAG | TCAAAGCGCTTCTTTAGTTC | 161 |
| 18 | pPARgc1a | TCCTCTTCAAGATCCTGTTAC | CACATACAAGGGAGAATTGC | 78 |
| 19 | pPARg | AAAGACAACGGACAAATCAC | GGGATATTTTTGGCATACTCTG | 195 |
| 20 | LC3 | GAACCGCAGACGCATCTCT | TGATCACCGGGATCTTACTGG | 171 |
| 21 | Casp3 | GAGTCCACTGACTTGCTCCC | AGCTTGGAACGGTACGCTAA | 117 |
| 22 | Rpl13a | AAGCAGGTACTTCTGGGCCG | GGGGTTGGTATTCATCCGCT | 126 |
| Control (n = 5) | PG545 (mg/Mouse) | p-Value | ||||
|---|---|---|---|---|---|---|
| 0.2 (n = 5) | 0.4 (n = 5) | Control vs. 0.2 | Control vs. 0.4 | 0.2 vs. 0.4 | ||
| TC (mg/dL) | 652 ± 21 | 397 ± 29 | 274 ± 13 | 0.0001 | p < 0.0001 | 0.0055 |
| TG (mg/dL) | 230 ± 23 | 189 ± 24 | 136 ± 20 | 0.2677 | 0.0169 | 0.1326 |
| LDL (mg/dL) | 542 ± 19 | 339 ± 22 | 237 ± 10 | 0.0003 | p < 0.0001 | 0.0035 |
| HDL (mg/dL) | 53 ± 0.8 | 19 ± 3.6 | 10 ± 0.8 | p < 0.0001 | p < 0.0001 | 0.0337 |
| PD (nmol/mL) | 985 ± 15 | 870 ± 36 | 746 ± 21 | 0.0214 | p < 0.0001 | 0.0196 |
| TBARS (nmol MDA/mL) | 27.3 ± 1.0 | 19.5 ± 2.0 | 12.3 ± 0.6 | 0.0101 | p < 0.0001 | 0.0103 |
| PON1 activity (U/mL) | 532 ± 15 | 326 ± 28 | 226 ± 16 | 0.0002 | p < 0.0001 | 0.0156 |
| Glucose | 166 ± 10 | 178 ± 13 | 159 ± 11 | NS † | NS † | NS † |
| Creatinine | 0.41 ± 0.01 | 0.41 ± 0.01 | 0.40± 0.01 | NS † | NS † | NS † |
| Blood Urea Nitrogen (mg/dL) | 26.0 ± 0.7 | 24.2 ± 1.4 | 30.0 ± 2.2 | NS † | NS † | NS † |
| ALT (U/L) | 161 ± 13 | 66 ± 7 | 108 ± 24 | 0.0003 | 0.0900 | 0.1379 |
| AST (U/L) | 149 ± 7 | 116 ± 12 | 223 ± 31 | 0.0540 | 0.0525 | 0.0135 |
| ALP (U/L) | 54 ± 4 | 57 ± 7 | 66 ± 4 | 0.7595 | 0.0832 | 0.3106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinaneh, S.; Hijaze, W.; Mansour-Wattad, L.; Hammoud, R.; Zaidani, H.; Kabala, A.; Hamoud, S. Heparanase Inhibition Prevents Liver Steatosis in E0 Mice. J. Clin. Med. 2022, 11, 1672. https://doi.org/10.3390/jcm11061672
Kinaneh S, Hijaze W, Mansour-Wattad L, Hammoud R, Zaidani H, Kabala A, Hamoud S. Heparanase Inhibition Prevents Liver Steatosis in E0 Mice. Journal of Clinical Medicine. 2022; 11(6):1672. https://doi.org/10.3390/jcm11061672
Chicago/Turabian StyleKinaneh, Safa, Walaa Hijaze, Lana Mansour-Wattad, Rawan Hammoud, Hisam Zaidani, Aviva Kabala, and Shadi Hamoud. 2022. "Heparanase Inhibition Prevents Liver Steatosis in E0 Mice" Journal of Clinical Medicine 11, no. 6: 1672. https://doi.org/10.3390/jcm11061672
APA StyleKinaneh, S., Hijaze, W., Mansour-Wattad, L., Hammoud, R., Zaidani, H., Kabala, A., & Hamoud, S. (2022). Heparanase Inhibition Prevents Liver Steatosis in E0 Mice. Journal of Clinical Medicine, 11(6), 1672. https://doi.org/10.3390/jcm11061672