
Muscle Biopsy: A Requirement for Precision Medicine in Adult-Onset Myopathy


Abstract
:1. Introduction
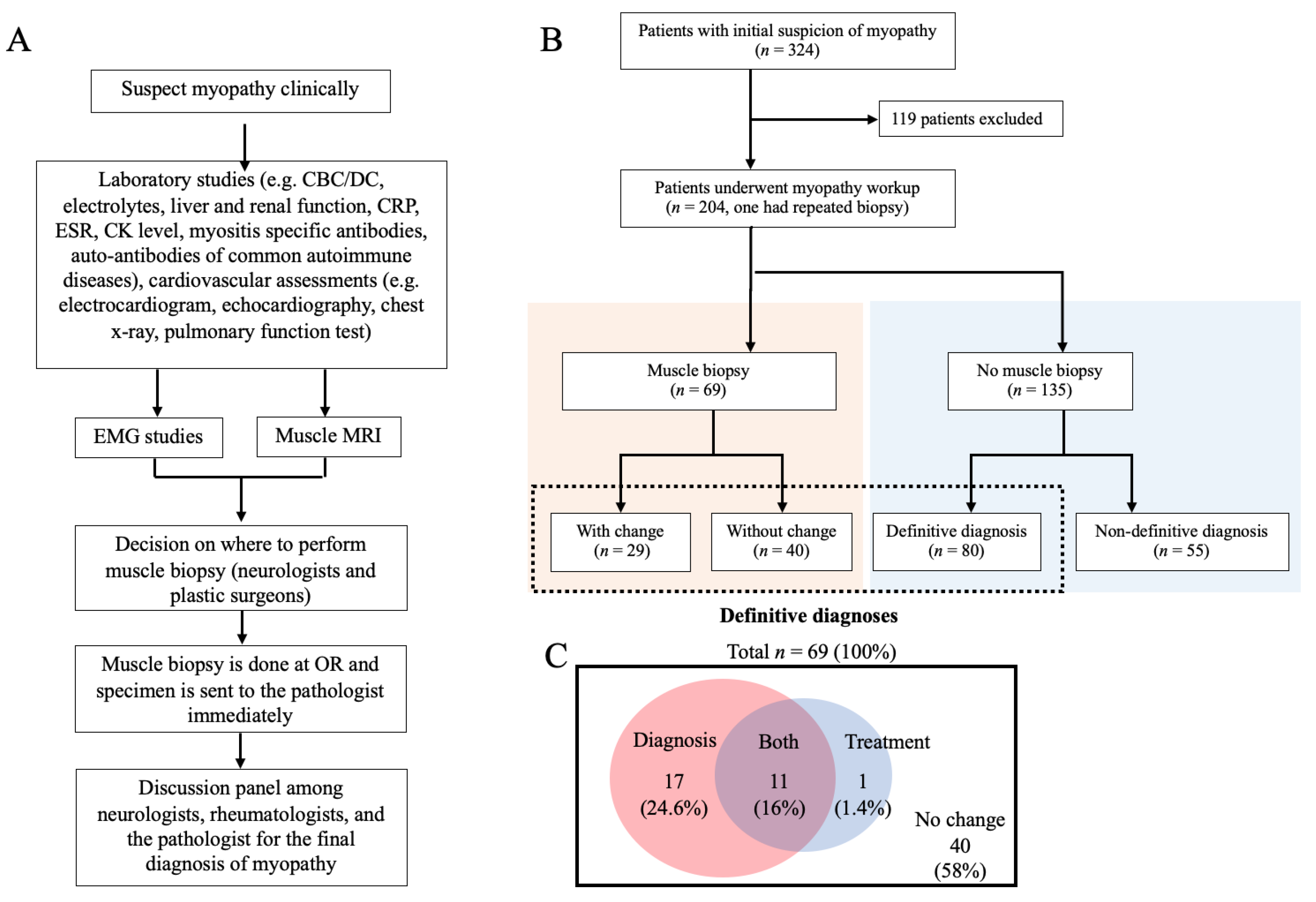
2. Method
2.1. Study Design
2.2. Patients
2.3. Muscle Biopsy Procedure and Interpretation
2.3.1. Muscle Biopsy Procedure
2.3.2. Histopathology Diagnosis
2.4. Clinical Information and Diagnostic Criteria
2.4.1. NCS and EMG
2.4.2. MRI
2.4.3. Diagnosis of IIMs
2.4.4. Genetic Tests
2.4.5. Other Criteria
2.5. Statistical Analysis
3. Results
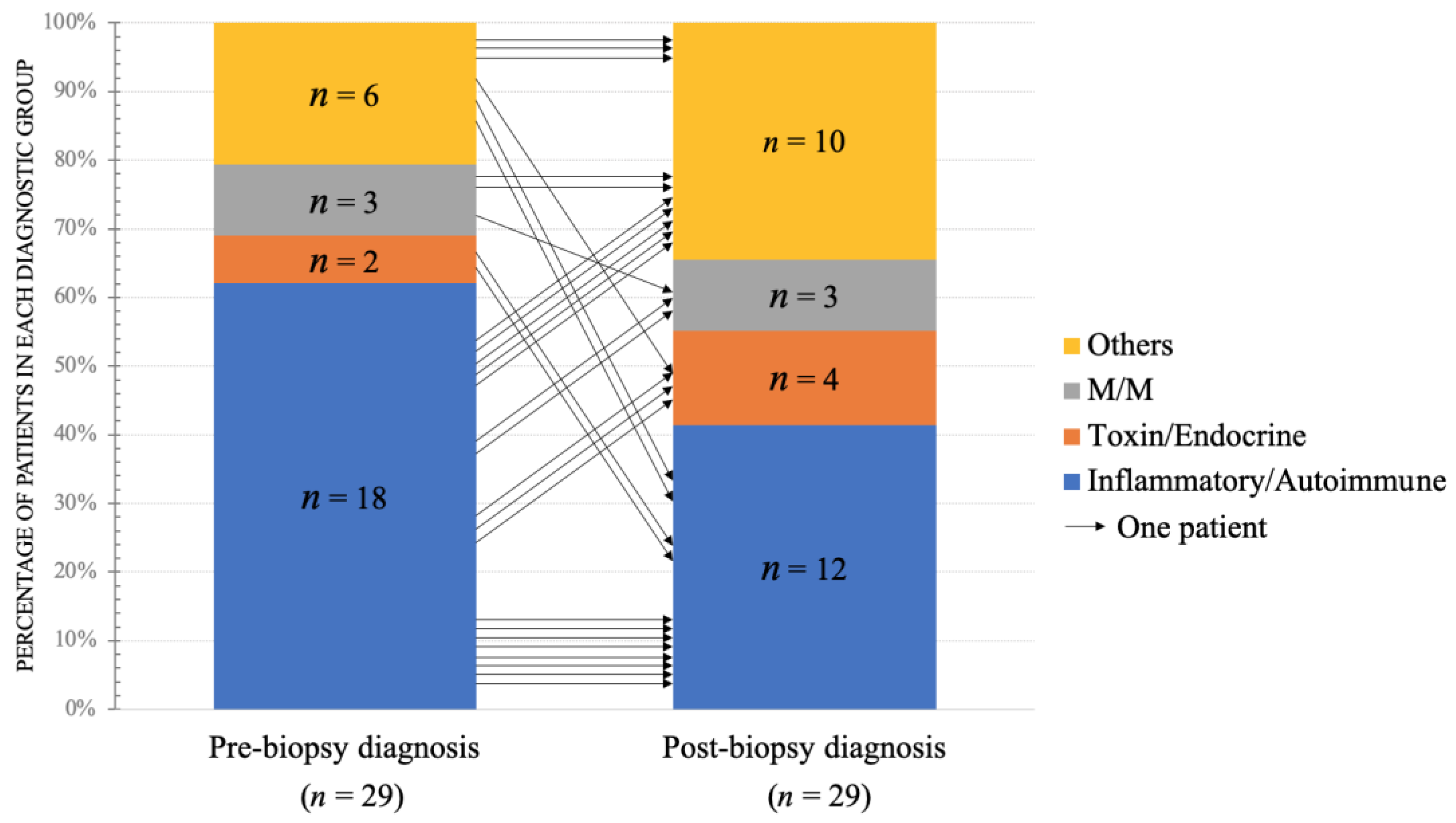
3.1. The Changes of Pre- and Post-Biopsy Diagnoses
3.2. The Changes of Pre- and Post-Biopsy Treatments
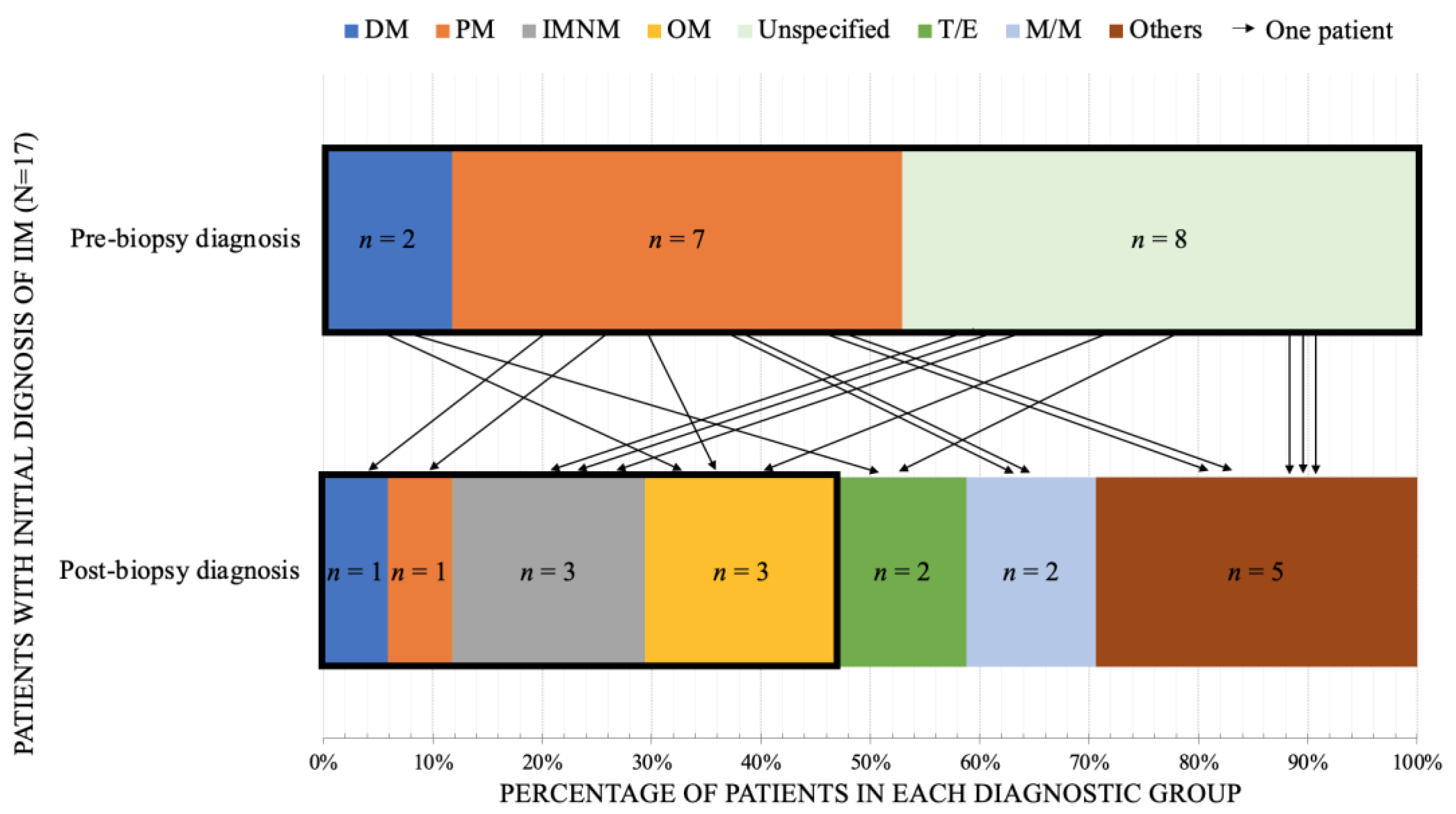
3.3. The Special Considerations in IIM
3.4. Myopathy Patients without Muscle Biopsy
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghirardello, A.; Borella, E.; Beggio, M.; Franceschini, F.; Fredi, M.; Doria, A. Myositis autoantibodies and clinical phenotypes. Autoimmun. Highlights 2014, 5, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimori, T.; Imura, Y.; Nakashima, R.; Yoshifuji, H. Autoantibodies in idiopathic inflammatory myopathy: An update on clinical and pathophysiological significance. Curr. Opin. Rheumatol. 2007, 19, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Tanaka, S.; Ceribelli, A.; Calise, S.J.; Chan, E.K.L. A Comprehensive Overview on Myositis-Specific Antibodies: New and Old Biomarkers in Idiopathic Inflammatory Myopathy. Clin. Rev. Allergy Immunol. 2017, 52, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceribelli, A.; De Santis, M.; Isailovic, N.; Gershwin, M.E.; Selmi, C. The Immune Response and the Pathogenesis of Idiopathic Inflammatory Myositis: A Critical Review. Clin. Rev. Allergy Immunol. 2017, 52, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Mariampillai, K.; Granger, B.; Amelin, D.; Guiguet, M.; Hachulla, E.; Maurier, F.; Meyer, A.; Tohmé, A.; Charuel, J.-L.; Musset, L.; et al. Development of a New Classification System for Idiopathic Inflammatory Myopathies Based on Clinical Manifestations and Myositis-Specific Autoantibodies. JAMA Neurol. 2018, 75, 1528–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platteel, A.C.; Wevers, B.A.; Lim, J.; Bakker, J.A.; Bontkes, H.J.; Curvers, J.; Damoiseaux, J.; Heron, M.; de Kort, G.; Limper, M.; et al. Frequencies and clinical associations of myositis-related antibodies in The Netherlands: A one-year survey of all Dutch patients. J. Transl. Autoimmun. 2019, 2, 100013. [Google Scholar] [CrossRef] [PubMed]
- Cassandrini, D.; Rodolico, C.; Trovato, R.; Rubegni, A.; Lenzi, S.; Fiorillo, C.; Baldacci, J.; Minetti, C.; Astrea, G.; Bruno, C.; et al. Congenital myopathies: Clinical phenotypes and new diagnostic tools. Ital. J. Pediatr. 2017, 43, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, J.; Gonçalves, A.; Taipa, R.; Melo-Pires, M.; Oliveira, E.M.; Costa, J.L.; Machado, J.C.; Medeiros, E.; Coelho, T.; Santos, M.; et al. New massive parallel sequencing approach improves the genetic characterization of congenital myopathies. J. Hum. Genet. 2016, 61, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Savarese, M. Next-generation sequencing approaches for the diagnosis of skeletal muscle disorders. Curr. Opin. Neurol. 2016, 29, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Bonnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Beroud, C.; Mathews, K.; et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. [Google Scholar] [CrossRef]
- Babic Bozovic, I.; Maver, A.; Leonardis, L.; Meznaric, M.; Osredkar, D.; Peterlin, B. Diagnostic yield of exome sequencing in myopathies: Experience of a Slovenian tertiary centre. PLoS ONE 2021, 16, e0252953. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Kang, P.B. Update on the genetics of limb girdle muscular dystrophy. Semin. Pediatr. Neurol. 2012, 19, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.P.; Cooper, S.T.; Evesson, F.J.; Seto, J.T.; Chiotis, M.; Tay, V.; Compton, A.G.; Cairns, A.G.; Corbett, A.; MacArthur, D.G.; et al. Limb-girdle muscular dystrophy: Diagnostic evaluation, frequency and clues to pathogenesis. Neuromuscul. Disord. 2008, 18, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Georganopoulou, D.G.; Moisiadis, V.G.; Malik, F.A.; Mohajer, A.; Dashevsky, T.M.; Wuu, S.T.; Hu, C.-K. A Journey with LGMD: From Protein Abnormalities to Patient Impact. Protein J. 2021, 40, 466–488. [Google Scholar] [CrossRef] [PubMed]
- Reddy, H.M.; Cho, K.-A.; Lek, M.; Estrella, E.; Valkanas, E.; Jones, M.D.; Mitsuhashi, S.; Darras, B.T.; Amato, A.A.; Lidov, H.G.; et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J. Hum. Genet. 2017, 62, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Ghaoui, R.; Cooper, S.T.; Lek, M.; Jones, K.; Corbett, A.; Reddel, S.W.; Needham, M.; Liang, C.; Waddell, L.B.; Nicholson, G.; et al. Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy: Outcomes and Lessons Learned. JAMA Neurol. 2015, 72, 1424–1432. [Google Scholar] [CrossRef]
- Ashton, C.; Paramalingam, S.; Stevenson, B.; Brusch, A.; Needham, M. Idiopathic inflammatory myopathies: A review. Intern. Med. J. 2021, 51, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Tanboon, J.; Nishino, I. Classification of idiopathic inflammatory myopathies: Pathology perspectives. Curr. Opin. Neurol. 2019, 32, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Melli, G.; Chang, Y.-J.; Skolasky, R.; Corse, A.M.; Wagner, K.R.; Cornblath, D.R. Open muscle biopsy in suspected myopathy: Diagnostic yield and clinical utility. Eur. J. Neurol. 2009, 17, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.E.; Tjärnlund, A.; Bottai, M.; Werth, V.P.; Pilkington, C.; De Visser, M.; Alfredsson, L.; Amato, A.A.; Barohn, R.J.; Liang, M.H.; et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann. Rheum. Dis. 2017, 76, 1955–1964. [Google Scholar] [CrossRef]
- Tenny, S.O.; Schmidt, K.P.; Follett, K.A. Association of Preoperative Diagnosis with Clinical Yield of Muscle Biopsy. Cureus 2018, 10, e3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubowitz, V.; Sewry, C.; Oldfors, A. Muscle Biopsy: A Practical Approach, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Cai, C.; Anthony, D.C.; Pytel, P. A pattern-based approach to the interpretation of skeletal muscle biopsies. Mod. Pathol. 2019, 32, 462–483. [Google Scholar] [CrossRef] [PubMed]
- Uruha, A.; Goebel, H.H.; Stenzel, W. Updates on the Immunopathology in Idiopathic Inflammatory Myopathies. Curr. Rheumatol. Rep. 2021, 23, 56. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Marozzo, R.; Pegoraro, V.; Sacconi, S. Diagnostic challenges in metabolic myopathies. Expert Rev. Neurother. 2020, 20, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Amato, A. Electrodiagnostic evaluation of myopathies. Phys. Med. Rehabilitation Clin. N. Am. 2013, 24, 193–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caetano, A.P.; Alves, P. Advanced MRI Patterns of Muscle Disease in Inherited and Acquired Myopathies: What the Radiologist Should Know. Semin. Musculoskelet. Radiol. 2019, 23, e82–e106. [Google Scholar] [CrossRef] [PubMed]
- Ten Dam, L.; Van Der Kooi, A.J.; Verhamme, C.; Wattjes, M.P.; De Visser, M. Muscle imaging in inherited and acquired muscle diseases. Eur. J. Neurol. 2016, 23, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Wattjes, M.P.; Kley, R.A.; Fischer, D. Neuromuscular imaging in inherited muscle diseases. Eur. Radiol. 2010, 20, 2447–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, Y.; Wadhwa, V.; Phillips, L.; Pezeshk, P.; Chhabra, A. MR imaging of skeletal muscle signal alterations: Systematic approach to evaluation. Eur. J. Radiol. 2016, 85, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, J.B.; Nico, M.A.; Omond, A.G.; Aivazoglou, L.U.; Jorge, R.B.; Zanoteli, E.; Fernandes, A.R.C. Diagnostic Imaging of Inflammatory Myopathies: New Concepts and a Radiological Approach. Curr. Rheumatol. Rep. 2019, 21, 8. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.G.; Gao, B.-X.; Chen, H.; Yang, M.-X.; Chen, X.-L.; Yan, R.; Lu, X.; Shi, K.-N.; Chan, Q.; Wang, G.-C. An efficacy analysis of whole-body magnetic resonance imaging in the diagnosis and follow-up of polymyositis and dermatomyositis. PLoS ONE 2017, 12, e0181069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, H.; Kirkhus, E.; Garen, T.; Walle-Hansen, R.; Merckoll, E.; Molberg, Ø. Comparative analyses of muscle MRI and muscular function in anti-synthetase syndrome patients and matched controls: A cross-sectional study. Arthritis Res. Ther. 2017, 19, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dion, E.; Cherin, P.; Payan, C.; Fournet, J.-C.; Papo, T.; Maisonobe, T.; Auberton, E.; Chosidow, O.; Godeau, P.; Piette, J.-C.; et al. Magnetic resonance imaging criteria for distinguishing between inclusion body myositis and polymyositis. J. Rheumatol. 2002, 29, 1897–1906. [Google Scholar] [PubMed]
- Zheng, Y.; Liu, L.; Wang, L.; Xiao, J.; Wang, Z.; Lv, H.; Zhang, W.; Yuan, Y. Magnetic resonance imaging changes of thigh muscles in myopathy with antibodies to signal recognition particle. Rheumatology 2015, 54, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filli, L.; Maurer, B.; Manoliu, A.; Andreisek, G.; Guggenberger, R. Whole-body MRI in adult inflammatory myopathies: Do we need imaging of the trunk? Eur. Radiol. 2015, 25, 3499–3507. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.; Swigris, J.J.; Brown, K.K. Myositis-related interstitial lung disease and antisynthetase syndrome. J. Bras. Pneumol. 2011, 37, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Troyanov, Y.; Targoff, I.N.; Tremblay, J.-L.; Goulet, J.-R.; Raymond, Y.; Senécal, J.-L. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: Analysis of 100 French Canadian patients. Medicine 2005, 84, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Toxic and drug-induced myopathies. J. Neurol. Neurosurg. Psychiatry 2009, 80, 832–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.K.; Woo, J.; Assantachai, P.; Auyeung, T.-W.; Chou, M.-Y.; Iijima, K.; Jang, H.C.; Kang, L.; Kim, M.; Kim, S.; et al. Asian Working Group for Sarcopenia: 2019 Consensus Update on Sarcopenia Diagnosis and Treatment. J. Am. Med. Dir. Assoc. 2020, 21, 300–307.e2. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, R.J.; Hasni, S. Pathogenesis and Management of Sarcopenia. Clin. Geriatr. Med. 2017, 33, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantino, U.; Scimeca, M.; Piccirilli, E.; Tancredi, V.; Baldi, J.; Gasbarra, E.; Bonanno, E. Sarcopenia: A histological and immunohistochemical study on age-related muscle impairment. Aging Clin. Exp. Res. 2015, 27 (Suppl. S1), S51–S60. [Google Scholar] [CrossRef] [PubMed]
- Tubridy, N.; Fontaine, B.; Eymard, B. Congenital myopathies and congenital muscular dystrophies. Curr. Opin. Neurol. 2001, 14, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Shieh, P.B. Muscular dystrophies and other genetic myopathies. Neurol. Clin. 2013, 31, 1009–1029. [Google Scholar] [CrossRef] [PubMed]
- Mah, J.K.; Korngut, L.; Fiest, K.M.; Dykeman, J.; Day, L.J.; Pringsheim, T.; Jette, N. A Systematic Review and Meta-analysis on the Epidemiology of the Muscular Dystrophies. Can. J. Neurol. Sci. 2016, 43, 163–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, J. Current Classification and Management of Inflammatory Myopathies. J. Neuromuscul. Dis. 2018, 5, 109–129. [Google Scholar] [CrossRef] [PubMed]
- Shaibani, A.; Jabari, D.; Jabbour, M.; Arif, C.; Lee, M.; Rahbar, M.H. Diagnostic outcome of muscle biopsy. Muscle Nerve 2015, 51, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Malartre, S.; Bachasson, D.; Mercy, G.; Sarkis, E.; Anquetil, C.; Benveniste, O.; Allenbach, Y. MRI and muscle imaging for idiopathic inflammatory myopathies. Brain Pathol. 2021, 31, e12954. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Hwu, W.L.; Lee, N.C. Pompe disease: Early diagnosis and early treatment make a difference. Pediatr. Neonatol. 2013, 54, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peruzzo, P.; Pavan, E.; Dardis, A. Molecular genetics of Pompe disease: A comprehensive overview. Ann. Transl. Med. 2019, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Lin, W.-C.; Lin, P.-Y.; Weng, M.-Y.; Sun, Y.-T. The significance of myositis autoantibodies in idiopathic inflammatory myopathy concomitant with interstitial lung disease. Neurol. Sci. 2021, 42, 2855–2864. [Google Scholar] [CrossRef]
- Huang, H.L.; Lin, W.-C.; Yeh, C.-C.; Sun, Y.-T. Serological risk factors for concomitant interstitial lung disease in patients with idiopathic inflammatory myopathy. J. Clin. Neurosci. 2020, 74, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Qiang, J.K.; Kim, W.B.; Baibergenova, A.; Alhusayen, R. Risk of Malignancy in Dermatomyositis and Polymyositis. J. Cutan. Med. Surg. 2016, 21, 131–136. [Google Scholar] [CrossRef] [PubMed]
- DeWane, M.E.; Waldman, R.; Lu, J. Dermatomyositis: Clinical features and pathogenesis. J. Am. Acad. Dermatol. 2020, 82, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Sawal, N.; Mukhopadhyay, S.; Rayancha, S.; Moore, A.; Garcha, P.; Kumar, A.; Kaul, V. A narrative review of interstitial lung disease in anti-synthetase syndrome: A clinical approach. J. Thorac. Dis. 2021, 13, 5556–5571. [Google Scholar] [CrossRef] [PubMed]
- Korsten, P.; Rademacher, J.-G.; Riedel, L.; Schnitzler, E.-M.; Olgemöller, U.; Seitz, C.S.; Schmidt, J.; Larsen, J.; Vasko, R. Antisynthetase Syndrome-Associated Interstitial Lung Disease: Monitoring of Immunosuppressive Treatment Effects by Chest Computed Tomography. Front. Med. 2020, 7, 609595. [Google Scholar] [CrossRef] [PubMed]
- Long, K.; Danoff, S.K. Interstitial Lung Disease in Polymyositis and Dermatomyositis. Clin. Chest Med. 2019, 40, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, J.; Baborie, A. Muscle biopsy: What and why and when? Pract. Neurol. 2020, 20, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Gibreel, W.O.; Selcen, D.; Zeidan, M.M.; Ishitani, M.B.; Moir, C.R.; Zarroug, A.E. Safety and yield of muscle biopsy in pediatric patients in the modern era. J. Pediatr. Surg. 2014, 49, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Driessen, J.; Willems, S.; Dercksen, S.; Giele, J.; Van Der Staak, F.; Smeitink, J. Anesthesia-related morbidity and mortality after surgery for muscle biopsy in children with mitochondrial defects. Pediatr. Anesthesia 2007, 17, 16–21. [Google Scholar] [CrossRef]
- Neves, M., Jr.; Barreto, G.; Boobis, L.; Harris, R.; Roschel, H.; Tricoli, V.; Ugrinowitsch, C.; Negrão, C.; Gualano, B. Incidence of adverse events associated with percutaneous muscular biopsy among healthy and diseased subjects. Scand. J. Med. Sci. Sports 2012, 22, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, B. The muscle biopsy technique. Historical and methodological considerations. Scand. J. Med. Sci. Sports 2016, 27, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Olivier, P.A.; De Paepe, B.; Aronica, E.; Berfelo, F.; Colman, R.; Amato, A.; Dimitri, D.; Gallardo, E.; Gherardi, R.; Goebel, H.-H.; et al. Idiopathic inflammatory myopathy: Interrater variability in muscle biopsy reading. Neurology 2019, 93, e889–e894. [Google Scholar] [CrossRef] [PubMed]
- Meola, G.; Bugiardini, E.; Cardani, R. Muscle biopsy. J. Neurol. 2012, 259, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Preusse, C.; Goebel, H.H.; Held, J.; Wengert, O.; Scheibe, F.; Irlbacher, K.; Koch, A.; Heppner, F.L.; Stenzel, W. Immune-mediated necrotizing myopathy is characterized by a specific Th1-M1 polarized immune profile. Am. J. Pathol. 2012, 181, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Hayat, G.; Kalia, J.S.; Guzman, M.A. Idiopathic Inflammatory Myopathies: Clinical Approach and Management. Front. Neurol. 2016, 7, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 69) | Without Change (n = 40) | With Change (n = 29) | p-Value | |
|---|---|---|---|---|
| Age (year, mean ± SD) | 54 ± 13.7 | 55 ± 13.8 | 53 ± 13.9 | 0.258 |
| Gender (male, n (%)) | 27 (39.1) | 18 (45) | 9 (31) | 0.126 |
| CK level (U/L, mean ± SD) | 3031 ± 7409 | 2468 ± 4239 | 3703 ± 10,282 | 0.495 |
| EMG findings (n (%)) | 0.517 | |||
| Myopathic change | 43 (62.3) | 25 (62.5) | 18 (62.1) | |
| Neuropathic change | 6 (8.7) | 2 (5) | 4 (13.8) | |
| Mixed | 9 (13) | 6 (15) | 3 (10.3) | |
| Normal | 2 (3) | 2 (5) | 0 (0) | |
| Not available | 9 (13) | 5 (12.5) | 4 (13.8) | |
| Muscle MRI (n (%)) | 0.909 | |||
| Abnormal findings | 46 (66.7) | 25 (62.5) | 21 (72.4) | |
| Unremarkable | 14 (20.3) | 11 (27.5) | 3 (10.3) | |
| Not available | 9 (13) | 4 (10) | 5 (17.3) | |
| Pre-biopsy primary diagnosis (n (%)) | 1.000 | |||
| Inflammatory/Autoimmune | 44 (63.8) | 26 (65) | 18 (62) | |
| Toxin/Endocrine | 2 (2.9) | 0 (0) | 2 (7) | |
| Metabolic/Mitochondrial diseases | 8 (11.6) | 5 (12.5) | 3 (10) | |
| Others | 15 (21.7) | 9 (22.5) | 6 (21) | |
| Pre-biopsy treatment (n (%)) | 0.990 | |||
| Steroid/immune-modulating therapy | 38 (55.1) | 24 (60) | 14 (48.3) | |
| Discontinue myotoxic agent | 3 (4.3) | 0 (0) | 3 (10.3) | |
| Other types of medication | 10 (14.5) | 5 (12.5) | 5 (17.3) | |
| No treatment | 18 (26.1) | 11 (27.5) | 7 (24.1) |
| Case | Post-Biopsy Diagnosis | Clinical Information | |
|---|---|---|---|
| 1 | Metabolic myopathy (MADD) | Clinical presentation | A 37-year-old female diagnosed with polymyositis for years without muscle biopsy had yearly deterioration. She had a poor response to immune therapy, even with rituximab. The steroid was discontinued for an extended period. |
| NE | Axial weakness and proximal limb weakness with MRC scale 3 over 4 limbs. | ||
| Lab | CK level (505 U/L); positive anti-PM-Scl 75 antibody; negative acetylcholine receptor antibody The MS/MS for various lengths of fatty acid was done after a muscle biopsy, which showed elevations of long- to mid-chain fatty acid. | ||
| NCS/EMG | Essentially normal. | ||
| MRI | Diffuse muscular swelling and enhancement at bilateral thighs, especially at soleus muscles. | ||
| Muscle pathology | Myopathic changes with intracellular lipid accumulation (Sudan III and oil-red-O are both positive). Type 1 muscle atrophy with focal type 2 muscle grouping without active inflammatory myopathy. The result suggested metabolic myopathy. | ||
| Diagnosis and outcome | The genetic test confirmed the compound heterozygous mutation of the ETFDH gene. The patient completely recovered after the carnitine supplement. | ||
| 2 | Muscular dystrophy | Clinical presentation | A 45-year-old female with insidious onset, progressive bilateral lower limbs weakness for 3 years |
| NE | Proximal weakness with MRC scale 4 over bilateral lower limbs and positive Gowers’ sign. | ||
| Lab | CK level (7354 U/L); elevated liver enzymes. | ||
| NCS/ENG | Diffuse myopathic change with some neuropathic change. | ||
| MRI | Diffuse muscular atrophy with fat replacement of bilateral thighs. | ||
| Muscle pathology | Marked fiber degeneration and regeneration, along with occasional fiber necrosis and endomysial fibrosis. Despite the degenerative change, clumps or chain of nuclei were rarely seen. About 10% of fibers show internal nuclei. There was mild and focal endomysial infiltration of mononuclear cells, consist of mainly T-lymphocytes. No excessive storage of glycogen or intracellular lipid. The result suggested a muscular dystrophy. | ||
| Diagnosis and outcome | Molecular diagnosis was not done, because the patient was lost to follow-up after being discharged. | ||
| 3 | Metabolic or mitochondrial myopathy | Clinical presentation | A 51-year-old male had polymyositis under daily prednisolone 30 mg. Subacute onset bilateral lower limbs weakness for 2 years. |
| NE | Proximal weakness over 4r limbs; positive Gowers’ sign. | ||
| Lab | Elevated CK level (1197 U/L); absence of myositis autoantibodies. | ||
| NCS/ENG | Myopathic changes without irritability. | ||
| MRI | Non-specific, minimal edema with asymmetric distribution at the muscles of both thighs. | ||
| Muscle pathology | Minimal myopathic changes with presence of intracellular lipid deposition. Gomori trichrome showed increased mitochondria, COX staining was intact, but SDH staining was lost. The result suggested either metabolic or mitochondrial myopathy. | ||
| Diagnosis and outcome | Molecular diagnosis was not done because the patient was lost to follow-up after being discharged. | ||
| 4 | Sarcopenia | Clinical presentation | A 72-year-old male ILD was in the treatment for pulmonary tuberculosis had insidious onset progressive exertional dyspnea and general weakness for four months. |
| NE | Atrophy over the bilateral shoulder and pelvic girdles, proximal weakness with MRC scale 4, and positive Gowers’ sign. | ||
| Lab | Normal CK level; presence of anti-Ku and anti-PL-12 antibodies. | ||
| NCS/ENG | Neuropathic changes. | ||
| MRI | Non-specific, minimal edema with symmetric distribution of both gluteal and thigh areas. | ||
| Muscle pathology | Myopathic changes with predominant type 2 muscle atrophy, worst in type 2B muscles. Electronic microscopic findings showed degenerative changes and sarcolemmal fold in some atrophic fibers. Sarcopenia secondary to malnutrition was considered. | ||
| Diagnosis and outcome | He showed much improvement after nutritional support and regained body weight 8 months later. | ||
| 5 | Mycobacterium-related granulomatous myopathy | Clinical presentation | A 61-year-old female had completed the modified radical mastectomy and combined chemo-radiotherapy for her breast cancer. She had subacute onset left hand and bilateral lower limbs progressive swelling and weakness for one month. |
| NE | MRC scale 4 of bilateral upper limbs, MRC scale 5 of bilateral lower limbs, and preserved deep tendon reflexes. | ||
| Lab | CK level (3364 U/L); elevated liver enzymes; positive ANA-cytoplasm; elevated rheumatoid factor. | ||
| NCS/ENG | Sensorimotor polyneuropathy and irritable myopathy. | ||
| MRI | Infiltration and edema at subcutaneous regions and muscles suggesting dermatomyositis. | ||
| Muscle pathology | Granulomatous inflammation with necrosis and positive immunostain with CD 68. The acid fast stain was negative. Skin biopsy: palisading necrotizing granulomatous dermatitis. The causative pathogen was identified as Mycobacterium haemophilium. | ||
| Diagnosis and outcome | After completing clarithromycin and ciprofloxacin treatments, she was recovered entirely many months later. | ||
| 6 | Fibromyalgia | Clinical presentation | A 67-year-old female had insidious onset neck tightness, neck weakness, and fatigue for years. |
| NE | Full muscle strength. | ||
| Lab | Normal CK level; weakly positivity for anti-SRP and anti- SAE1. | ||
| NCS/ENG | Myopathic change. | ||
| MRI | Cervical spine MRI showed herniated intervertebral discs at multiple levels. | ||
| Muscle pathology | Mild myopathic change without specific pattern. | ||
| Diagnosis and outcome | After excluding myopathy, duloxetine was tried, and the patient got extraordinary improvements. | ||
| 7 | Diabetic amyotrophy | Clinical presentation | A 68-year-old T2DM male with rosuvastatin use had acute onset lower limbs weakness and lower backache during hospitalization for infection of unknown origin. |
| NE | MRC scale 2 and 4 on bilateral proximal and distal lower limbs, respectively. | ||
| Lab | Normal CK level and absence of myositis autoantibodies. | ||
| NCS/ENG | Bilateral upper lumbar radiculopathy. | ||
| MRI | Spinal MRI showed posterior herniation of L4-5 disc with mild compression of thecal sac and nerve roots, and central herniations of C5-6 disc with compression of the thecal sac. | ||
| Muscle pathology | Biopsy was performed due to suspicion of superimposed statin-induced myopathy. Diffuse atrophic fibers with minimal perivascular infiltration of mononuclear cells; almost absence of endomysial or perimysial infiltration of mononuclear cells. | ||
| Diagnosis and outcome | The patient was diagnosed with diabetic amyotrophy. His muscle strength was improved 6 months later after strict glycemic control. | ||
| 8 | Steroid-related myopathy | Clinical presentation | A 66-year-old female with ILD, taking prednisolone, was admitted for pneumonia. |
| NE | Muscle powers were full in 4 limbs, negative Gowers’ sign, presence of mechanic’s hand. | ||
| Lab | Normal CK level; elevated anti-SSA antibody (184 U/mL); strong positive anti-Jo-1 and anti-Ro-52 antibodies. | ||
| NCS/ENG | Sensorimotor polyneuropathy; myopathic changes in bilateral vastus medialis without irritability. | ||
| MRI | Edematous change and rim enhancement at bilateral sartorius, gracilis and rectus femoris muscles. | ||
| Muscle pathology | Chronic myopathic changes with type 2 fiber atrophy. No fiber necrosis or phagocytosis with minimal infiltration of mononuclear cells. | ||
| Diagnosis and outcome | Azathioprine and pirfenidone were added for ILD. The steroid was not suspended because the benefit outweighs the adverse effect in her deteriorating clinical course. | ||
| 9 | Steroid-related myopathy | Clinical presentation | A 59-year-old female with hypothyroidism and skin disease treated with methylprednisolone. She presented with subacute onset progressive four limbs weakness for 3 months. |
| NE | Drowsy consciousness, dysarthria, quadriparesis with MRC scale 2 on all limbs, areflexia except normal deep tendon reflexes on bilateral brachioradialis. | ||
| Lab | Normal CK level with strong positivity for anti-PL-12 and anti-Ro-52 antibodies. | ||
| NCS/ENG | NCS showed sensorimotor polyneuropathy. EMG was performed only at resting state because she could not cooperate for minimal and maximal effort due to decreased conscious level. Increased resting activities in right gastrocnemius, abductor pollicis brevis and semimembranosus muscles. | ||
| MRI | Edema at the subcutaneous regions and muscles of bilateral gluteal regions and thighs, suggesting dermatomyositis. Septic arthritis of both hip joints, and minimal effusions of both knee joints. | ||
| Muscle pathology | Severe type 2 fiber atrophy. No fiber necrosis, phagocytosis or presence of internal nuclei. Minimal endomysial infiltration of mononuclear cells and negative immunostain with MHC class I. The NADH-TR stain showed atrophic, degenerative fibers. COX, SDH and AMPDA were all intact. No excessive storage of glycogen or intracellular lipid. Steroid myopathy or hypothyroidism-related changes were suggested. | ||
| Case | Pre-Biopsy Main Diagnosis | Pre-Biopsy Differential Diagnosis | Post-Biopsy Diagnosis | Clinical Decision |
|---|---|---|---|---|
| 1 | SLE-related myopathy | CIDP or mononeuritis multiplex | Steroid-related myopathy | Steroid-sparing plaquenil as therapy |
| 2 | IIM (PM) with CTD and ILD | Pompe disease due to presence of paraspinal myotonia | Malnutrition sarcopenia | Nutritional support |
| 3 | IIM | Metabolic, endocrine, or drug-related myopathy | IIM (OM) | Began steroid therapy |
| 4 | Undetermined myopathy | Fibromyalgia | No evidence of myopathy | Began duloxetine therapy |
| 5 | Sjögren syndrome with IIM (DM) | IIM (OM) | Higher dose of steroid for therapy | |
| 6 | IIM | Granulomatous myopathy due to Mycobacterium infection | Began treatment for Mycobacterium infection | |
| 7 | Myasthenia gravis | Undetermined myopathy | Steroid-related myopathy | Steroid-sparing azathioprine therapy |
| 8 | IIM (PM) | Metabolic myopathy | Metabolic myopathy | Began carnitine-L therapy |
| 9 | Steroid-related myopathy | Worsening pre-existing IIM (PM) | Residual activity of IIM (PM) | Re-started steroid therapy |
| 10 | IIM (PM) | IIM (OM) | Higher dose of steroid | |
| 11 | Metabolic myopathy | Congenital myopathy | Myopathy with positive Ro-52 myositis antibody | Began steroid and azathioprine therapy |
| Diagnosis | Post-Biopsy Non-IIMs | Post-Biopsy IIMs | Total |
|---|---|---|---|
| Pre-biopsy IIMs | 10 | 8 | 18 |
| Pre-biopsy non-IIMs | 7 | 4 | 11 |
| Total | 17 | 12 | 29 |
| Case | Pre-Biopsy Diagnosis | Post-Biopsy Diagnosis | Clinical Information | |
|---|---|---|---|---|
| EMG: Normal Findings | ||||
| 1 | Inflammatory myopathy | Metabolic myopathy (MADD) | Clinical presentation | A 37-year-old female diagnosed with polymyositis had yearly deterioration for years without a muscle biopsy. She had a poor response to immune therapy, even with rituximab. The steroid was discontinued for an extended period. |
| NE | Proximal weakness with MRC scale 3 over upper and lower limbs and truncal weakness. | |||
| Lab | CK level 505 U/L, positive anti-PM-Scl 75 antibody, negative acetylcholine receptor antibody. The MS/MS for various lengths of fatty acid was done after a muscle biopsy, which showed elevations of long- and middle-chain fatty acid. | |||
| MRI | Diffuse muscular swelling, infiltration, and enhancement at bilateral thighs, especially at soleus muscles. | |||
| Muscle pathology | Myopathic changes with intracellular lipid accumulation (Sudan III and oil-red-O were positive). Type 1 muscle atrophy with focal type 2 muscle grouping without active inflammatory myopathy. The result suggested metabolic myopathy. | |||
| Diagnosis and outcome | The genetic test confirmed the compound heterozygous mutation of the ETFDH gene. The patient completely recovered after the carnitine supplement. | |||
| 2 | Hereditary congenital myopathy | FSHD | Clinical presentation | A 61-year-old female had insidious onset, progressive proximal lower limbs and axial weakness for more than 5 years. |
| NE | Diffuse muscle atrophy, MRC scale 4 over bilateral upper and lower limbs, negative percussion myotonia, diffuse hyperreflexia. | |||
| Lab | Normal CK level. | |||
| MRI | Muscle atrophy of left semitendinosus, semimembranosus, gastrocnemius muscles, bilateral vastus lateralis, intermedius, medialis muscles, and gluteus maximus muscles. | |||
| Muscle pathology | Chronic myopathic changes with fiber necrosis, primarily type 1 fiber atrophy. No central core or excessive storage of glycogen by PAS stain. No rimmed vacuoles. No endomysial infiltration of mononuclear cells. No fatty spilling from the replacing adipose tissue. | |||
| Diagnosis and outcome | The genetic test confirmed FSHD (D4Z4 deletion 27Kb). The patient needed a cane one year later. | |||
| EMG: Neuropathic findings | ||||
| 3 | Pompe disease | Pseudodeficiency of GAA | Clinical presentation | A 58-year-old male treated with a statin for his hyperlipidemia had insidious onset lower limbs predominant myalgia. |
| NE | Preserved muscle strength and deep tendon reflex. | |||
| Lab | Elevated CK level (730 U/L), reduced GAA activity, no abnormal organic acids found in the urine. | |||
| MRI | Unremarkable. | |||
| Muscle pathology | Normal appearing muscle; no rimmed vacuoles or ragged red fibers; no structural myopathy on NADH-TR stain. The COX, SDH and AMPDA are all intact. No excessive storage of glycogen or intracellular lipid. | |||
| Diagnosis and outcome | Confirm psedodeficiency of GAA caused by c.1726G>A (p.Gly576Ser) homozygous and c.2065G>A(p.Glu689Lys) homozygous mutation. Statin was discontinued. | |||
| 4 | Congenital NMJ disorder or motor neuron disease | Motor neuron disease | Clinical presentation | A 25-year-old female had insidious onset, progressive dysphagia and general weakness for 3 months. |
| NE | Atrophy over tongue, biceps, triceps, and deltoid; MRC scale 3 over right proximal upper limb and scale 4 on distal part, while the rest of muscle power was full; generalized hyperreflexia | |||
| Lab | Normal CK level, more than 10% decremental change on RNST. | |||
| MRI | No signal change at the muscles of both thighs. | |||
| Muscle pathology | Minimal histological change with mild, variable fiber size and small angulated fibers. No endomysial fibrosis, fiber necrosis, phagocytosis, regeneration, internal nuclei, inclusions or infiltration of inflammatory cells. Ultrastructure images showed intact myofibers, regular Z lines, except for some small foci of mild loss of myofilaments. | |||
| Diagnosis and outcome | WES showed FUS mutation. The patient underwent tracheotomy due to respiratory failure and expired after ventilator withdrawal in hospice care. | |||
| 5 | Non-specific myopathy | Non-specific myopathy | Clinical presentation | A 41-year-old female had insidious onset, progressive weakness over bilateral lower limbs and jaw, face muscle while chewing for one year. |
| NE | Proximal weakness of MRC scale 4 over all limbs, and positive Gowers’ sign. | |||
| Lab | Normal CK level, negative autoimmune serology tests, normal GAA activity; absence of abnormal organic acid in urine. | |||
| MRI | No image evidence of muscular atrophy or edematous change. | |||
| Muscle pathology | Scattered small, angulated fibers without grouping. No inflammatory cells indented. Unremarkable findings by Gomori trichome and NADH-TR stain. COX, SDH, and AMPDA were intact. No excessive storage of glycogen or intracellular lipid. | |||
| Diagnosis and outcome | WES showed a negative result. | |||
| 6 | Inflammatory myopathy | Sarcopenia | Clinical presentation | A 72-year-old male had interstitial lung disease. He was in the treatment for pulmonary tuberculosis and had insidious onset progressive exertional dyspnea and general weakness for four months. |
| NE | Atrophy over the bilateral shoulder and pelvic girdles, proximal weakness with MRC scale 4, and positive Gowers’ sign. | |||
| Lab | Normal CK level, presence of anti-Ku and anti-PL-12 antibodies. | |||
| MRI | Non-specific, minimal edema with symmetric distribution of both gluteal and thigh areas. | |||
| Muscle pathology | Myopathic changes with predominant type 2 muscle atrophy, worst in type 2B muscles. Electronic microscope showed degenerative changes and sarcolemmal fold in some atrophic fibers. Sarcopenia secondary to malnutrition was considered. | |||
| Diagnosis and outcome | Nutritional support was the mainstay of treatment, and he showed much improvement after he regained body weight 8 months later. | |||
| 7 | Statin related myositis | Diabetic amyotrophy | Clinical presentation | A 68-year-old T2DM male using rosuvastatin had acute onset lower limbs weakness and lower backache during hospitalization for infection of unknown origin. |
| NE | MRC scale 2 and 4 on bilateral proximal and distal lower limbs, respectively. | |||
| Lab | Normal CK level and absence of myositis autoantibodies. | |||
| MRI | Spinal MRI showed posterior herniation of L4-5 disc with mild compression of thecal sac and nerve roots, and central herniations of C5-6 disc with compression of the thecal sac. | |||
| Muscle pathology | Chronic neuropathic changes, diffuse atrophic fibers with minimal perivascular infiltration of mononuclear cells. Almost absent of endomysial or perimysial infiltration mononuclear cells. | |||
| Diagnosis and outcome | The patient was diagnosed with diabetic amyotrophy. His regained muscle strength 6 months later after strict glycemic control. | |||
| 8 | Inflammatory myopathy | IMNM | Clinical presentation | A 43-year-old male with T2DM and statin use had acute onset bilateral thighs pain for one day; relapse and remitting a few times. |
| NE | Positive Gowers’ sign, diffuse hyporeflexia | |||
| Lab | Elevated CK level (4284 U/L), weak positivity for anti-MDA5 antibody. | |||
| MRI | Not performed. | |||
| Muscle pathology | Active myopathic damage with necrotic fibers and minimal inflammatory infiltration. Samples were stained with ATPase 9.4, 4.3 and 4.6 and showed type 2 fiber predominance (particular 2B fiber). No inclusions, rimmed vacuoles, split fibers or lobulated fibers. No excessive storage of glycogen or intracellular lipid. The Gomori trichrome was unremarkable. The result suggested IMNM or myotoxic myopathy. | |||
| Diagnosis and outcome | Urine organic acid and MS/MS were normal. Statin was discontinued. | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.-J.; Liao, W.-A.; Lin, P.-Y.; Sun, Y.-T. Muscle Biopsy: A Requirement for Precision Medicine in Adult-Onset Myopathy. J. Clin. Med. 2022, 11, 1580. https://doi.org/10.3390/jcm11061580
Wu M-J, Liao W-A, Lin P-Y, Sun Y-T. Muscle Biopsy: A Requirement for Precision Medicine in Adult-Onset Myopathy. Journal of Clinical Medicine. 2022; 11(6):1580. https://doi.org/10.3390/jcm11061580
Chicago/Turabian StyleWu, Meng-Ju, Wei-An Liao, Po-Yu Lin, and Yuan-Ting Sun. 2022. "Muscle Biopsy: A Requirement for Precision Medicine in Adult-Onset Myopathy" Journal of Clinical Medicine 11, no. 6: 1580. https://doi.org/10.3390/jcm11061580
APA StyleWu, M.-J., Liao, W.-A., Lin, P.-Y., & Sun, Y.-T. (2022). Muscle Biopsy: A Requirement for Precision Medicine in Adult-Onset Myopathy. Journal of Clinical Medicine, 11(6), 1580. https://doi.org/10.3390/jcm11061580