
Sphingosine-1-Phosphate Levels Are Higher in Male Patients with Non-Classic Fabry Disease

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Patients
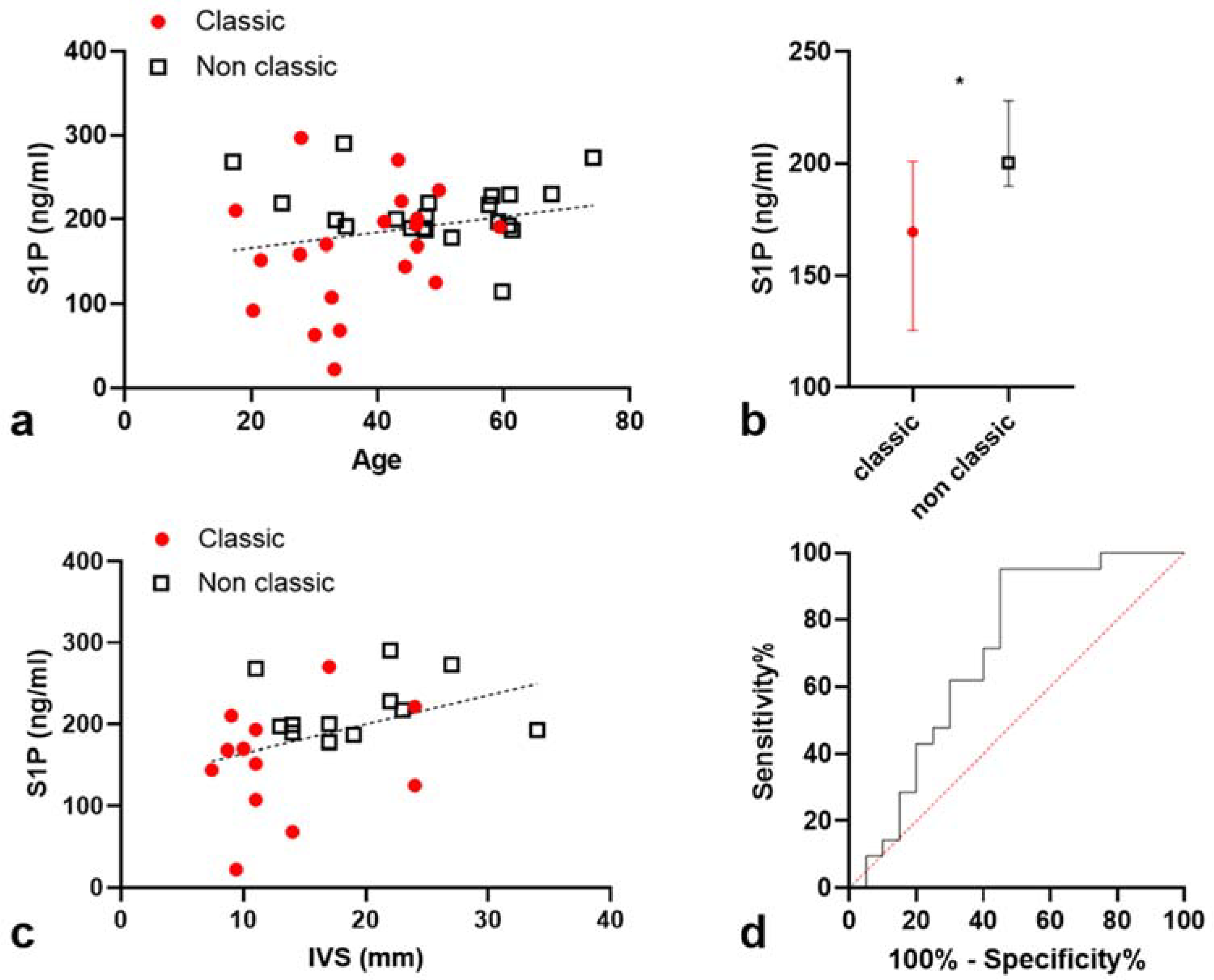
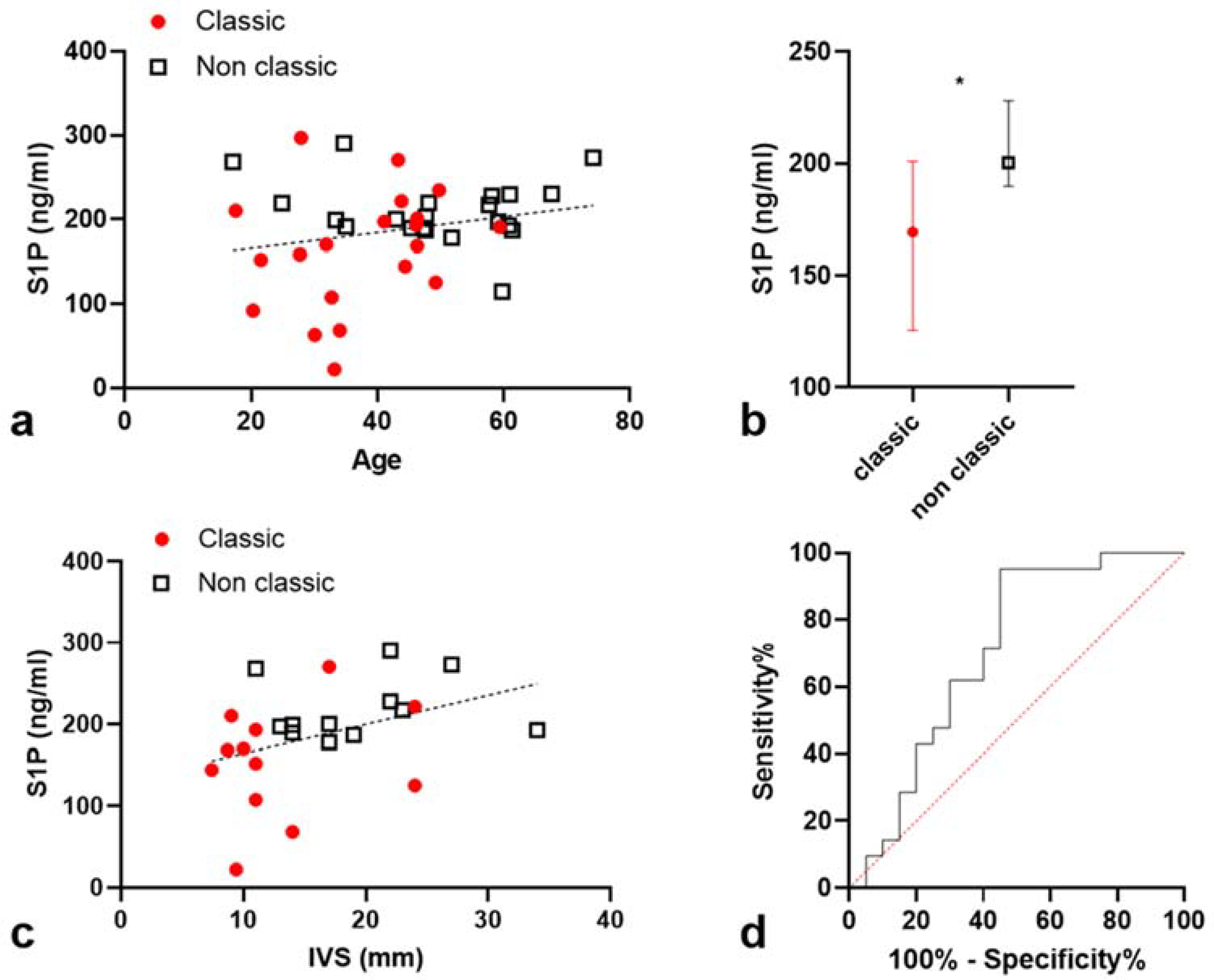
2.2. S1P Levels Are Higher in Non-Classic Patients
2.3. Correlation between S1P Levels and Interventricular Septum Thickness (IST)
2.3.1. S1P Levels Are Correlated with IST in a Univariate Model
2.3.2. Multivariate Model
2.4. S1P Levels Remain Associated with Non-Classic Phenotype in a Multivariate Logistic Regression Model
2.5. S1P Level Is Higher in Non-Classic Patients Compared to Classic Patients Sharing the Same Genotype
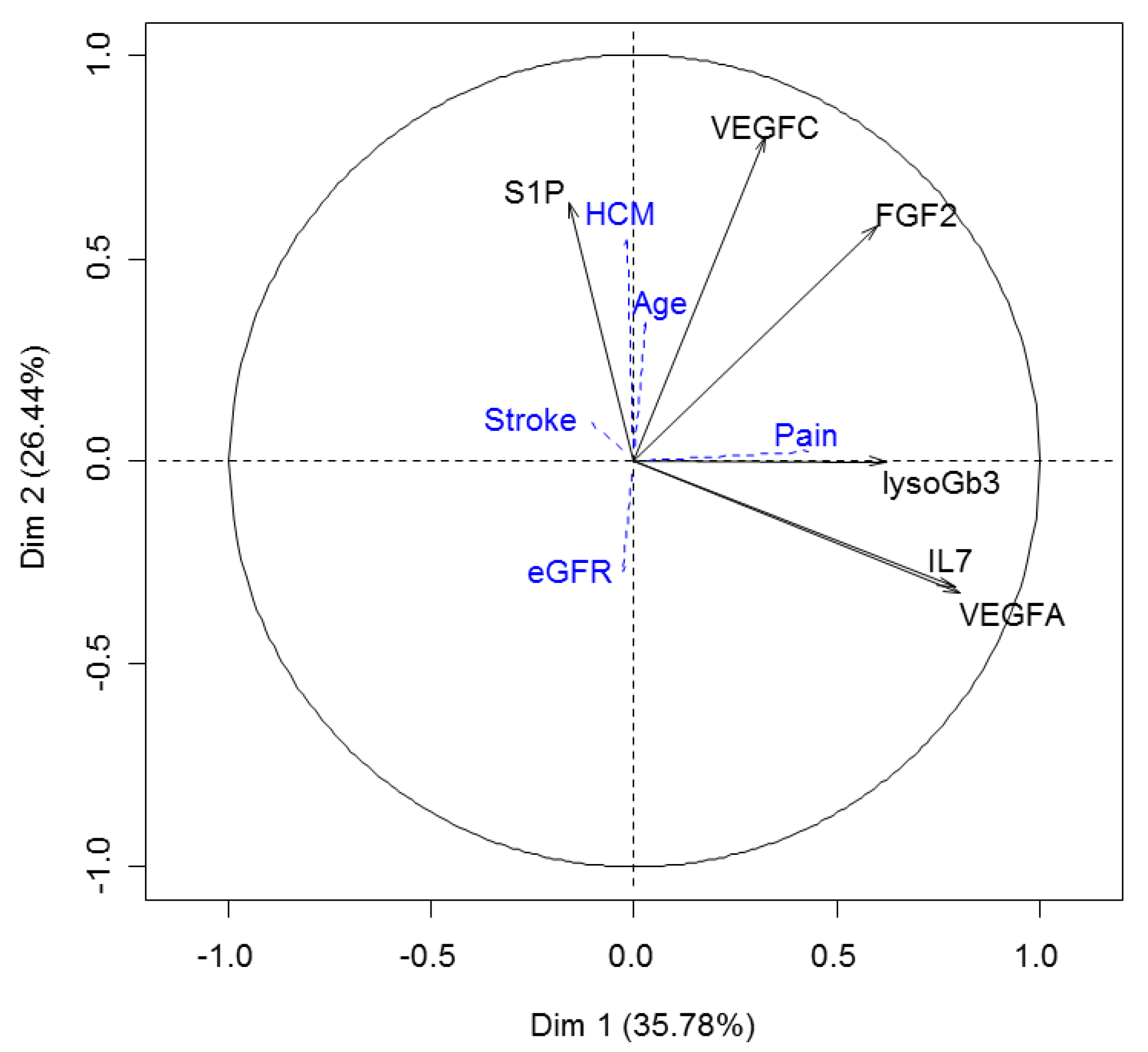
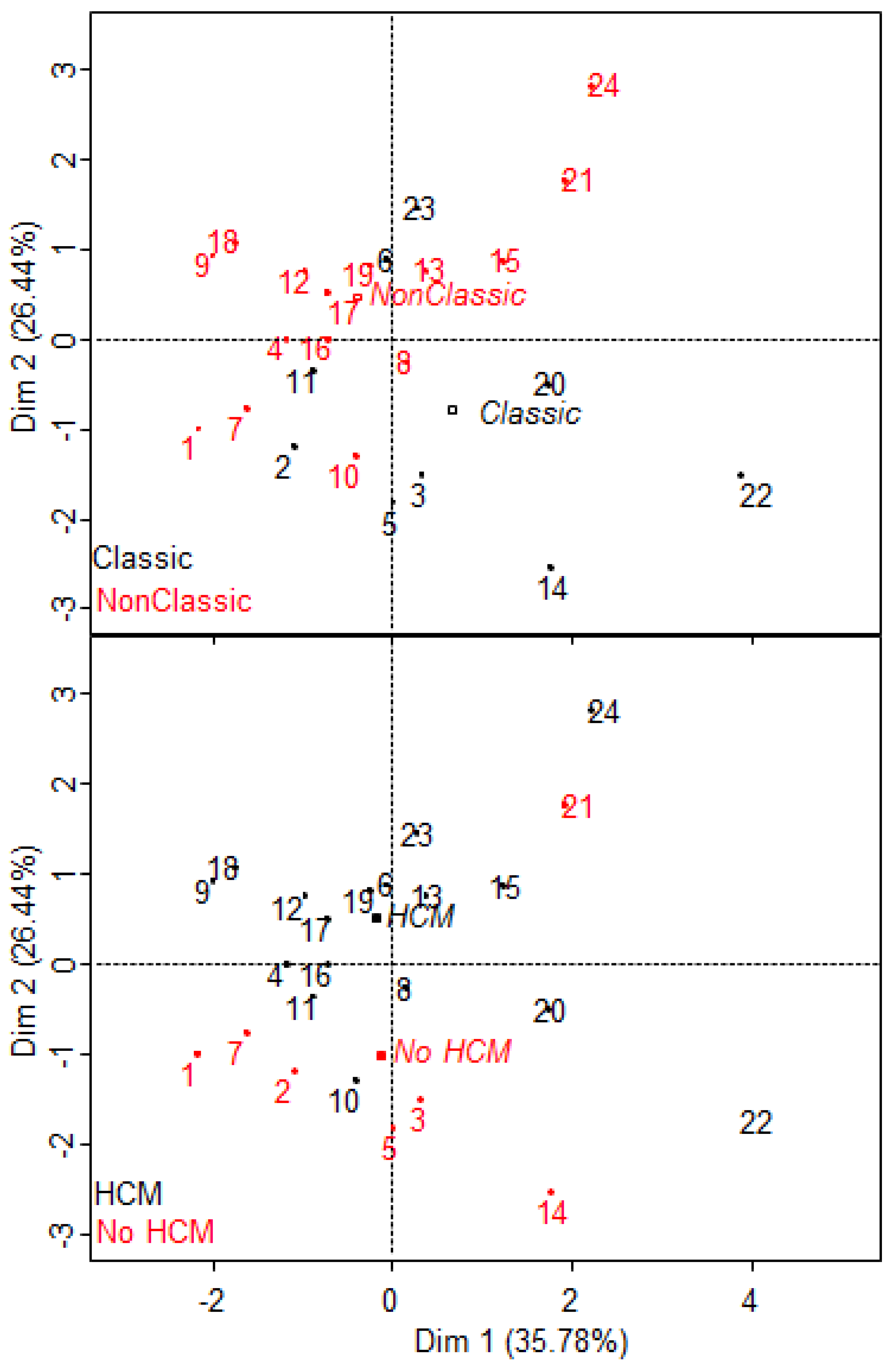
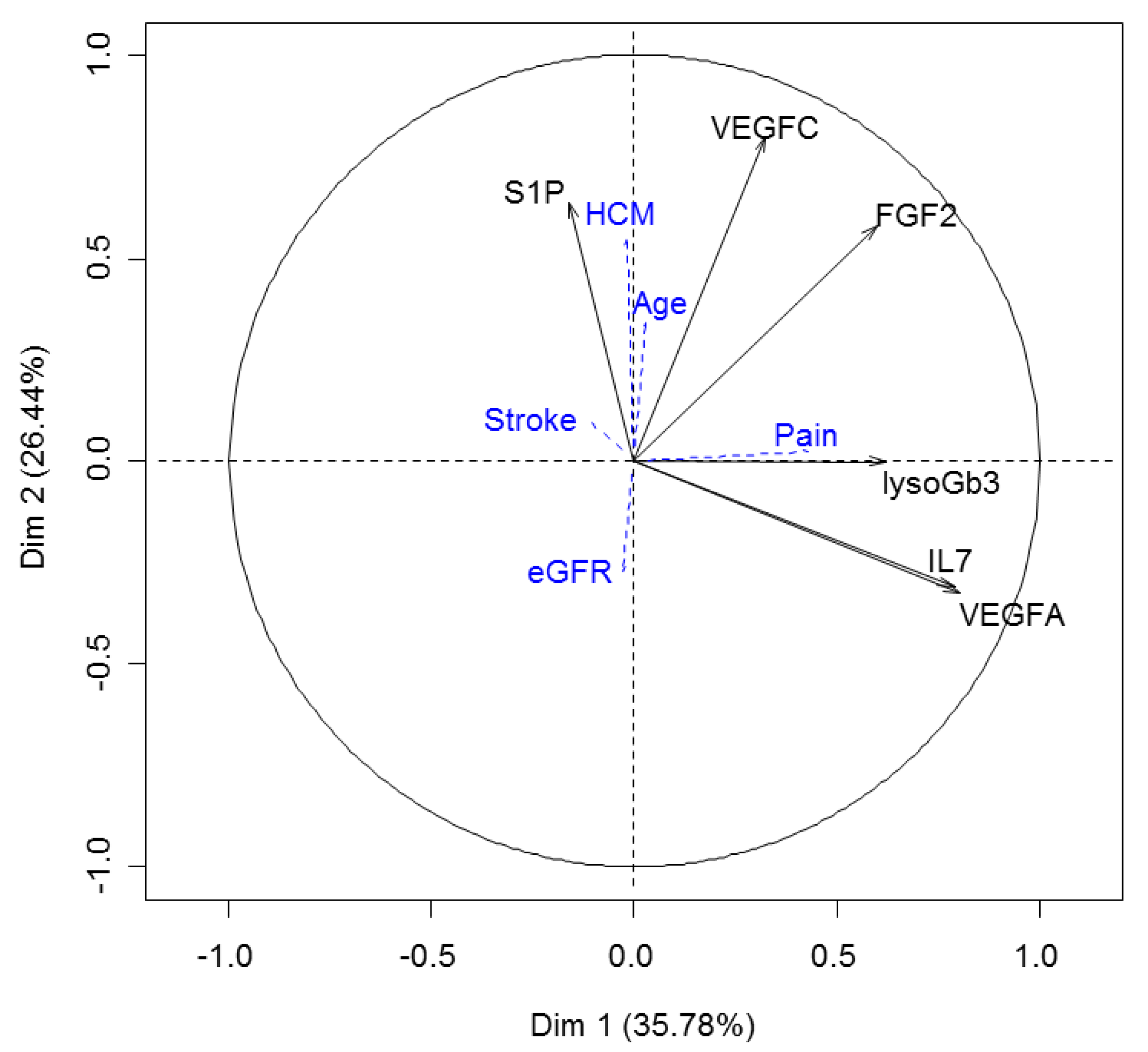
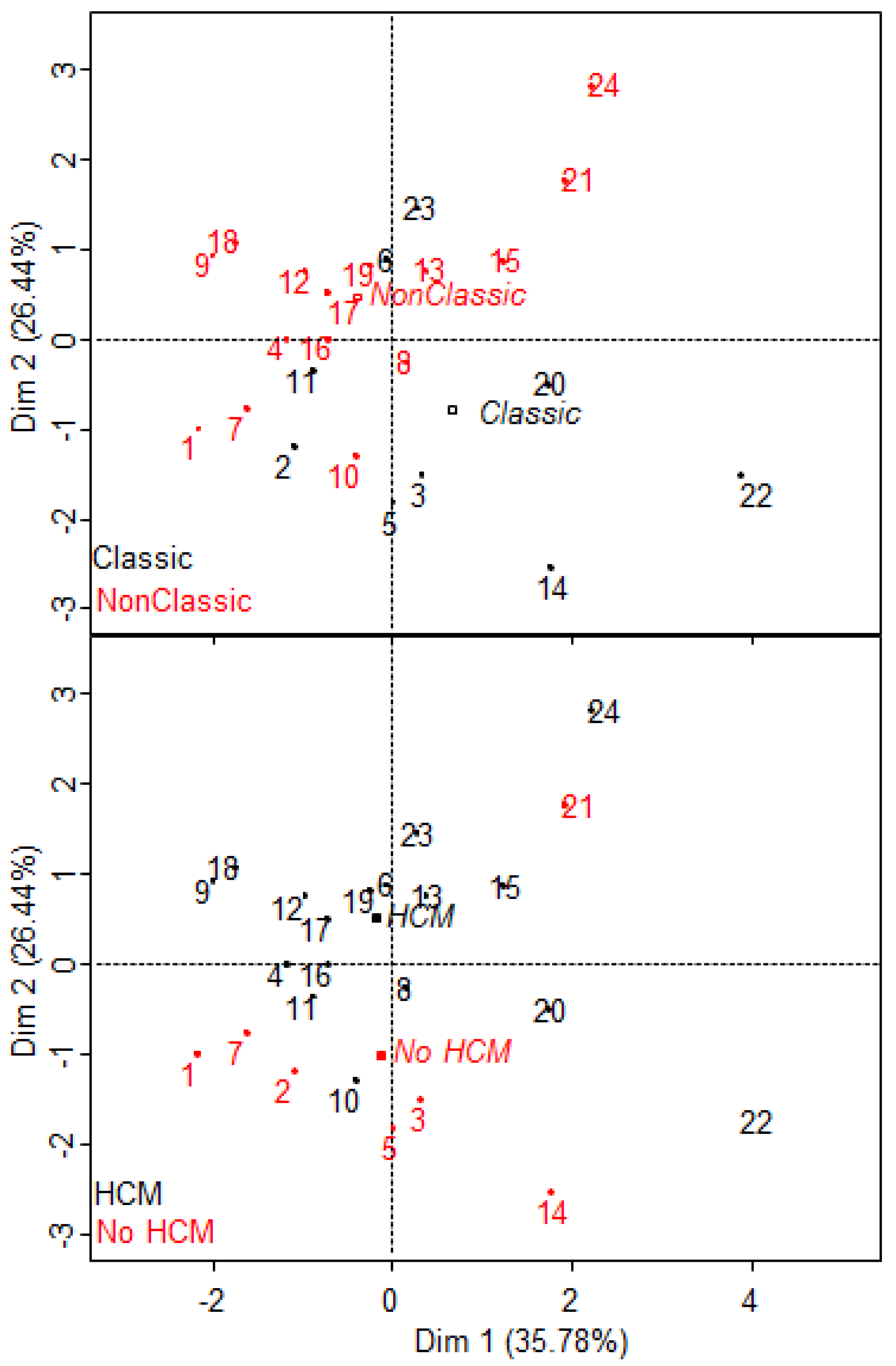
2.6. S1P Provides Additional Information to the Proteomic Signature of Fabry Disease
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brady, R.O.; Schiffmann, R. Clinical features of and recent advances in therapy for Fabry disease. JAMA 2000, 284, 2771–2775. [Google Scholar] [CrossRef] [PubMed]
- Rubino, M.; Monda, E.; Lioncino, M.; Caiazza, M.; Palmiero, G.; Dongiglio, F.; Fusco, A.; Cirillo, A.; Cesaro, A.; Capodicasa, L.; et al. Diagnosis and management of cardiovascular involvement in Fabry disease. Heart Fail. Clin. 2022, 18, 39–49. [Google Scholar] [CrossRef]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Levade, T.; Hachulla, E.; Knebelmann, B.; Lacombe, D.; Seguin, V.L.; Nguyen, K.; Noël, E.; Rabès, J.-P. Challenging the traditional approach for interpreting genetic variants: Lessons from Fabry disease. Clin. Genet. 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Ouyang, Y.; Wang, Z.; Ren, H.; Shen, P.; Wang, W.; Xu, Y.; Ni, L.; Yu, X.; Chen, X.; et al. Genotype: A crucial but not unique factor affecting the clinical phenotypes in Fabry disease. PLoS ONE 2016, 11, e0161330. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. International collaborative Fabry disease study group safety and efficacy of recombinant human alpha-galactosidase a replacement therapy in Fabry’s disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A.; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in Fabry disease: A randomized controlled trial. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef]
- Lenders, M.; Brand, E. Mechanisms of neutralizing anti-drug antibody formation and clinical relevance on therapeutic efficacy of enzyme replacement therapies in Fabry disease. Drugs 2021, 81, 1969–1981. [Google Scholar] [CrossRef]
- Germain, D.P.; Elliott, P.M.; Falissard, B.; Fomin, V.V.; Hilz, M.J.; Jovanovic, A.; Kantola, I.; Linhart, A.; Mignani, R.; Namdar, M.; et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol. Genet. Metab. Rep. 2019, 19, 100454. [Google Scholar] [CrossRef]
- Song, H.-Y.; Yang, Y.-P.; Chien, Y.; Lai, W.-Y.; Lin, Y.-Y.; Chou, S.-J.; Wang, M.-L.; Wang, C.-Y.; Leu, H.-B.; Yu, W.-C.; et al. Reversal of the inflammatory responses in Fabry patient IPSC-derived cardiovascular endothelial cells by CRISPR/Cas9-corrected mutation. Int. J. Mol. Sci. 2021, 22, 2381. [Google Scholar] [CrossRef]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Cammisa, M.; Correra, A.; Andreotti, G.; Cubellis, M.V. Fabry_CEP: A tool to identify fabry mutations responsive to pharmacological chaperones. Orphanet. J. Rare Dis. 2013, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenders, M.; Stappers, F.; Niemietz, C.; Schmitz, B.; Boutin, M.; Ballmaier, P.J.; Zibert, A.; Schmidt, H.; Brand, S.-M.; Auray-Blais, C.; et al. Mutation-specific Fabry disease patient-derived cell model to evaluate the amenability to chaperone therapy. J. Med. Genet. 2019, 56, 548–556. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [Green Version]
- Bichet, D.G.; Aerts, J.M.; Auray-Blais, C.; Maruyama, H.; Mehta, A.B.; Skuban, N.; Krusinska, E.; Schiffmann, R. Assessment of plasma Lyso-Gb3 for clinical monitoring of treatment response in migalastat-treated patients with fabry disease. Genet. Med. 2021, 23, 192–201. [Google Scholar] [CrossRef]
- Nowak, A.; Beuschlein, F.; Sivasubramaniam, V.; Kasper, D.; Warnock, D.G. Lyso-Gb3 associates with adverse long-term outcome in patients with Fabry disease. J. Med. Genet. 2021. [Google Scholar] [CrossRef]
- Simonetta, I.; Tuttolomondo, A.; Daidone, M.; Pinto, A. Biomarkers in Anderson-Fabry disease. Int. J. Mol. Sci. 2020, 21, 8080. [Google Scholar] [CrossRef]
- Tebani, A.; Mauhin, W.; Abily-Donval, L.; Lesueur, C.; Berger, M.G.; Nadjar, Y.; Berger, J.; Benveniste, O.; Lamari, F.; Laforêt, P.; et al. A proteomics-based analysis reveals predictive biological patterns in Fabry disease. J. Clin. Med. 2020, 9, 1325. [Google Scholar] [CrossRef]
- Brakch, N.; Dormond, O.; Bekri, S.; Golshayan, D.; Correvon, M.; Mazzolai, L.; Steinmann, B.; Barbey, F. Evidence for a role of sphingosine-1 phosphate in cardiovascular remodelling in Fabry disease. Eur. Heart J. 2010, 31, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Mirzaian, M.; Wisse, P.; Ferraz, M.J.; Marques, A.R.A.; Gabriel, T.L.; van Roomen, C.P.A.A.; Ottenhoff, R.; van Eijk, M.; Codée, J.D.C.; van der Marel, G.A.; et al. Accurate quantification of sphingosine-1-phosphate in normal and Fabry disease plasma, cells and tissues by LC-MS/MS with (13)C-encoded natural S1P as internal standard. Clin. Chim. Acta Int. J. Clin. Chem. 2016, 459, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Jozefczuk, E.; Guzik, T.J.; Siedlinski, M. Significance of sphingosine-1-phosphate in cardiovascular physiology and pathology. Pharmacol. Res. 2020, 156, 104793. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.; Colon-Moran, W.; Bhattarai, N. Characterization of the effects of immunomodulatory drug fingolimod (FTY720) on human T cell receptor signaling pathways. Sci. Rep. 2018, 8, 10910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, M.D.; Jones, D.E.; Goldman, M.D. Overview and safety of fingolimod hydrochloride use in patients with multiple sclerosis. Expert Opin. Drug Saf. 2014, 13, 989–998. [Google Scholar] [CrossRef]
- Józefczuk, E.; Nosalski, R.; Saju, B.; Crespo, E.; Szczepaniak, P.; Guzik, T.J.; Siedlinski, M. Cardiovascular effects of pharmacological targeting of sphingosine kinase 1. Hypertension 2020, 75, 383–392. [Google Scholar] [CrossRef]
- Mauhin, W.; Lidove, O.; Masat, E.; Mingozzi, F.; Mariampillai, K.; Ziza, J.-M.; Benveniste, O. Innate and adaptive immune response in Fabry disease. JIMD Rep. 2015, 22, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of classical and nonclassical Fabry disease: A multicenter study. J. Am. Soc. Nephrol. JASN 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Snider, A.J.; Orr Gandy, K.A.; Obeid, L.M. Sphingosine kinase: Role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie 2010, 92, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signalling. Development 2014, 141, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Mahajan-Thakur, S.; Böhm, A.; Jedlitschky, G.; Schrör, K.; Rauch, B.H. Sphingosine-1-phosphate and its receptors: A mutual link between blood coagulation and inflammation. Mediat. Inflamm. 2015, 2015, 831059. [Google Scholar] [CrossRef] [Green Version]
- Lépine, S.; Allegood, J.C.; Park, M.; Dent, P.; Milstien, S.; Spiegel, S. Sphingosine-1-Phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 2011, 18, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef] [PubMed]
- Augusto, J.B.; Johner, N.; Shah, D.; Nordin, S.; Knott, K.D.; Rosmini, S.; Lau, C.; Alfarih, M.; Hughes, R.; Seraphim, A.; et al. The myocardial phenotype of Fabry disease pre-hypertrophy and pre-detectable storage. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.J.; Marques, A.R.A.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef] [Green Version]
- Mauhin, W.; Benveniste, O.; Amelin, D.; Montagner, C.; Lamari, F.; Caillaud, C.; Douillard, C.; Dussol, B.; Leguy-Seguin, V.; D’Halluin, P.; et al. Cornea verticillata and acroparesthesia efficiently discriminate clusters of severity in Fabry disease. PLoS ONE 2020, 15, e0233460. [Google Scholar] [CrossRef] [PubMed]
- Whybra, C.; Kampmann, C.; Krummenauer, F.; Ries, M.; Mengel, E.; Miebach, E.; Baehner, F.; Kim, K.; Bajbouj, M.; Schwarting, A.; et al. The Mainz severity score index: A new instrument for quantifying the anderson-fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin. Genet. 2004, 65, 299–307. [Google Scholar] [CrossRef]
- Chen, F.; Du, M.; Blumberg, J.B.; Ho Chui, K.K.; Ruan, M.; Rogers, G.; Shan, Z.; Zeng, L.; Zhang, F.F. Association among dietary supplement use, nutrient intake, and mortality among U.S. adults: A cohort study. Ann. Intern. Med. 2019, 170, 604–613. [Google Scholar] [CrossRef]
- Mauhin, W.; Lidove, O.; Amelin, D.; Lamari, F.; Caillaud, C.; Mingozzi, F.; Dzangué-Tchoupou, G.; Arouche-Delaperche, L.; Douillard, C.; Dussol, B.; et al. Deep characterization of the anti-drug antibodies developed in fabry disease patients, a prospective analysis from the French multicenter cohort FFABRY. Orphanet. J. Rare Dis. 2018, 13, 127. [Google Scholar] [CrossRef] [Green Version]
- Kanda, Y. Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Classic | Non-Classic | All | |
|---|---|---|---|
| N | 20 | 21 | 41 |
| Age at diagnosis (years) | 23.7 [16.8–35.8] | 45.4 [32.2–55.3] | 32.7 [20.0–49.2] |
| Age at sampling | 37.6 [29.5–46.2] | 48.1 [43.0–59.8] | 45.3 [33.2–51.8] |
| Patients treated (n) | 18 | 16 | 34 |
| Cumulative treatment exposure (years) | 11.4 [5.6–13.0] | 4.4 [1.3–7.5] | 6.6 [3.8–12.8] |
| eGFR in mL/min/1,73 m2 | 104.9 [61.8–118.2] | 99.5 [65.9–114–4] | 102.7 [61.8–118.2] |
| Kidney transplant (n) | 4 | 0 | 4 |
| Hypertrophic cardiomyopathy (n) | 7 | 18 | 25 |
| Arrhythmia (n) | 7 | 12 | 19 |
| Interventricular Septum Thickness (mm) | 11 [9–15] | 18 [14–22] | 14 [11–22] |
| recent IST assessment available in n | 12 | 12 | 24 |
| Ischemic stroke | 1 | 1 | 2 |
| White matter lesions n/n with brain MRI | 4/12 | 4/9 | 8/21 |
| FFABRY Heart score | 1 [0] | 2 [1–3] | 1 [1–3] |
| FFABRY Kidney score | 1 [0–4] | 0 [0–2] | 1 [0–2] |
| FFABRY Neurological score | 1 [0.1] | 0 [0.1] | 1 [0.1] |
| FFABRY Total score | 3 [1–6] | 4 [2–6] | 3.5 [1–6] |
| MSSI cardiovascular | 2 [0–6.25] | 9 [2–13] | 3 [0–12] |
| MSSI renal | 2 [0–8] | 0 [0–8] | 0 [0–8] |
| MSSI neurological | 6 [2–9.5] | 2 [0–5] | 5 [1–8] |
| MSSI general | 4.5 [3.5–7.5] | 2 [1–4] | 4 [2–6] |
| MSSI total | 19.5 [13.5–29.5] | 20 [12–24] | 20 [12–26] |
| LysoGb3 (ng/mL) | 18.9 [10.6–48.8] | 7.1 [2.6–22.1] | 13.8 [6.5–31.8] |
| Treated patients (n) | 18.7 [10.5–43.0] (17) | 7.1 [2.6–22.1] (15) | |
| Untreated patients (n) | 109 (1) | 5.6 [2.6–24.6] (4) |
| Risk of Hypertrophic Cardiomyopathy | Odds Ratio | Lower 95%CI | Upper 95%CI |
|---|---|---|---|
| multivariate analysis (age, lysoGb3, S1P, Phenotype) | |||
| (Intercept) | 0.00000038 | 1.93 × 10−12 | 0.075 |
| age | 1.33000000 | 1.05 | 1.690 |
| lysoGb3 | 1.05000000 | 0.979 | 1.130 |
| S1P | 1.01000000 | 0.986 | 1.030 |
| Phenotype (Non-Classic) | 16.20000000 | 0.409 | 643.000 |
| univariate analysis | |||
| age | 1.250000 | 1.090000000 | 1.4300 |
| lysoGb3 | 0.997 | 0.971 | 1.02 |
| S1P | 1.010 | 0.9990 | 1.02 |
| Phenotype (Non-Classic) | 10.300 | 2.21 | 47.80 |
| Risk of classic phenotype | odds ratio | Lower 95%CI | Upper 95%CI |
| multivariate analysis (age, lysoGb3, S1P and treatment exposure) | |||
| (Intercept) | 111.000 | 0.401 | 30,700.000 |
| age | 0.961 | 0.890 | 1.040 |
| lysoGb3 | 1.040 | 0.992 | 1.080 |
| S1P | 0.974 | 0.951 | 0.998 |
| cumulative treatment exposure | 1.160 | 0.980 | 1.380 |
| multivariate analysis (age, lysoGb3, S1P and treatment exposure) with stepwise selection based on p-value | |||
| (Intercept) | 96.50 | 1.040 | 8940.000 |
| lysoGb3 | 1.05 | 1.010 | 1.100 |
| S1P | 0.97 | 0.946 | 0.994 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauhin, W.; Tebani, A.; Amelin, D.; Abily-Donval, L.; Lamari, F.; London, J.; Douillard, C.; Dussol, B.; Leguy-Seguin, V.; Noel, E.; et al. Sphingosine-1-Phosphate Levels Are Higher in Male Patients with Non-Classic Fabry Disease. J. Clin. Med. 2022, 11, 1233. https://doi.org/10.3390/jcm11051233
Mauhin W, Tebani A, Amelin D, Abily-Donval L, Lamari F, London J, Douillard C, Dussol B, Leguy-Seguin V, Noel E, et al. Sphingosine-1-Phosphate Levels Are Higher in Male Patients with Non-Classic Fabry Disease. Journal of Clinical Medicine. 2022; 11(5):1233. https://doi.org/10.3390/jcm11051233
Chicago/Turabian StyleMauhin, Wladimir, Abdellah Tebani, Damien Amelin, Lenaig Abily-Donval, Foudil Lamari, Jonathan London, Claire Douillard, Bertrand Dussol, Vanessa Leguy-Seguin, Esther Noel, and et al. 2022. "Sphingosine-1-Phosphate Levels Are Higher in Male Patients with Non-Classic Fabry Disease" Journal of Clinical Medicine 11, no. 5: 1233. https://doi.org/10.3390/jcm11051233
APA StyleMauhin, W., Tebani, A., Amelin, D., Abily-Donval, L., Lamari, F., London, J., Douillard, C., Dussol, B., Leguy-Seguin, V., Noel, E., Masseau, A., Lacombe, D., Maillard, H., Bekri, S., Lidove, O., & Benveniste, O. (2022). Sphingosine-1-Phosphate Levels Are Higher in Male Patients with Non-Classic Fabry Disease. Journal of Clinical Medicine, 11(5), 1233. https://doi.org/10.3390/jcm11051233