

Metronomic Chemotherapy in Pediatric Oncology: From Preclinical Evidence to Clinical Studies



Abstract
1. Introduction
2. Methodology
3. Preclinical Activity of Metronomic Chemotherapy in Pediatric Tumor Models
3.1. Preclinical Models of Neuroblastoma
3.2. Pediatric Brain Tumor Models
3.3. Soft Tissue and Bone Sarcoma Models
3.4. Pediatric Retinoblastoma Models
3.5. Acute Lymphoblastic Leukemia Models
3.6. Metronomic Combined Schedules in Multiple Pediatric Tumor Models

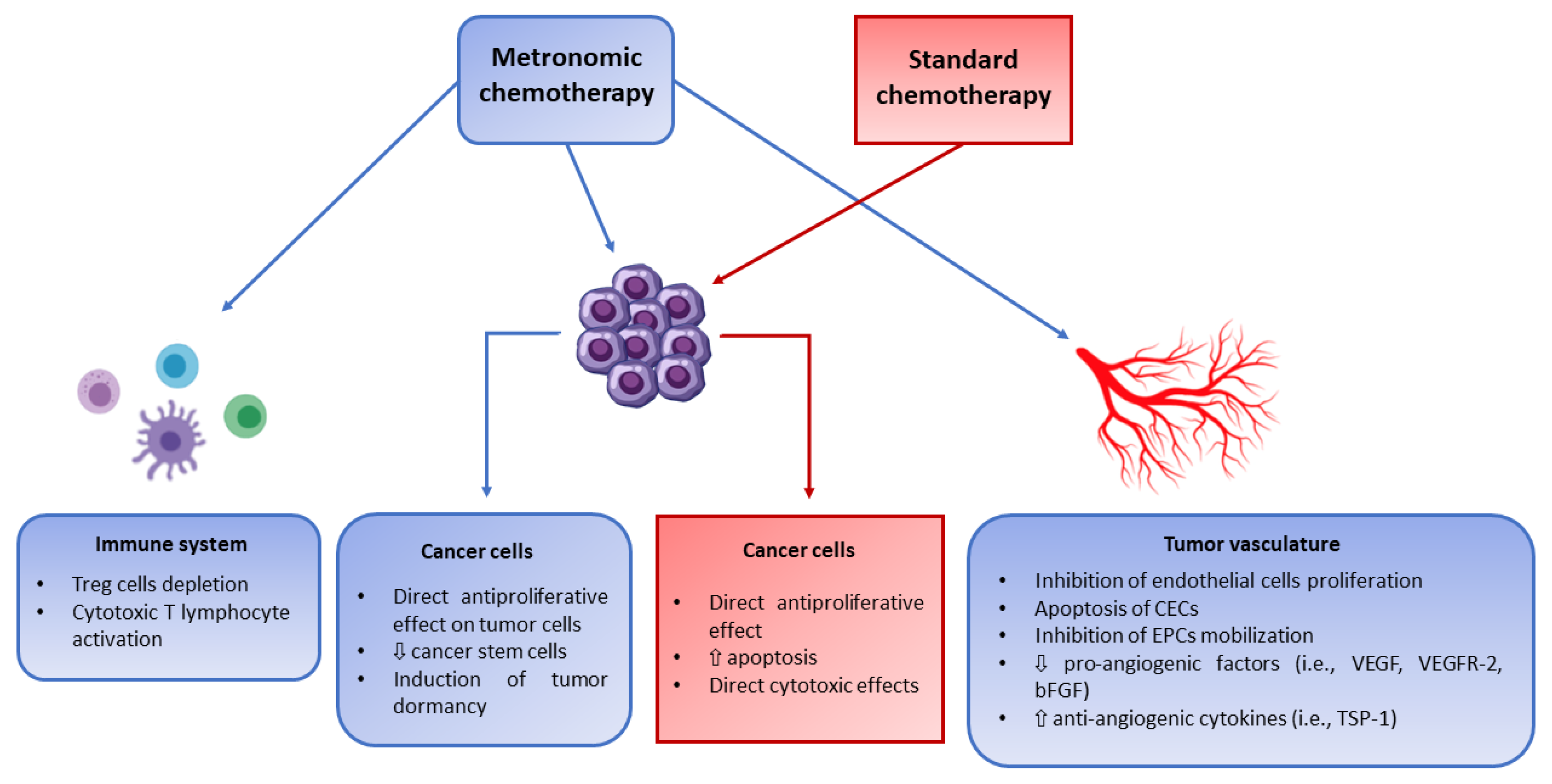{kind=link}
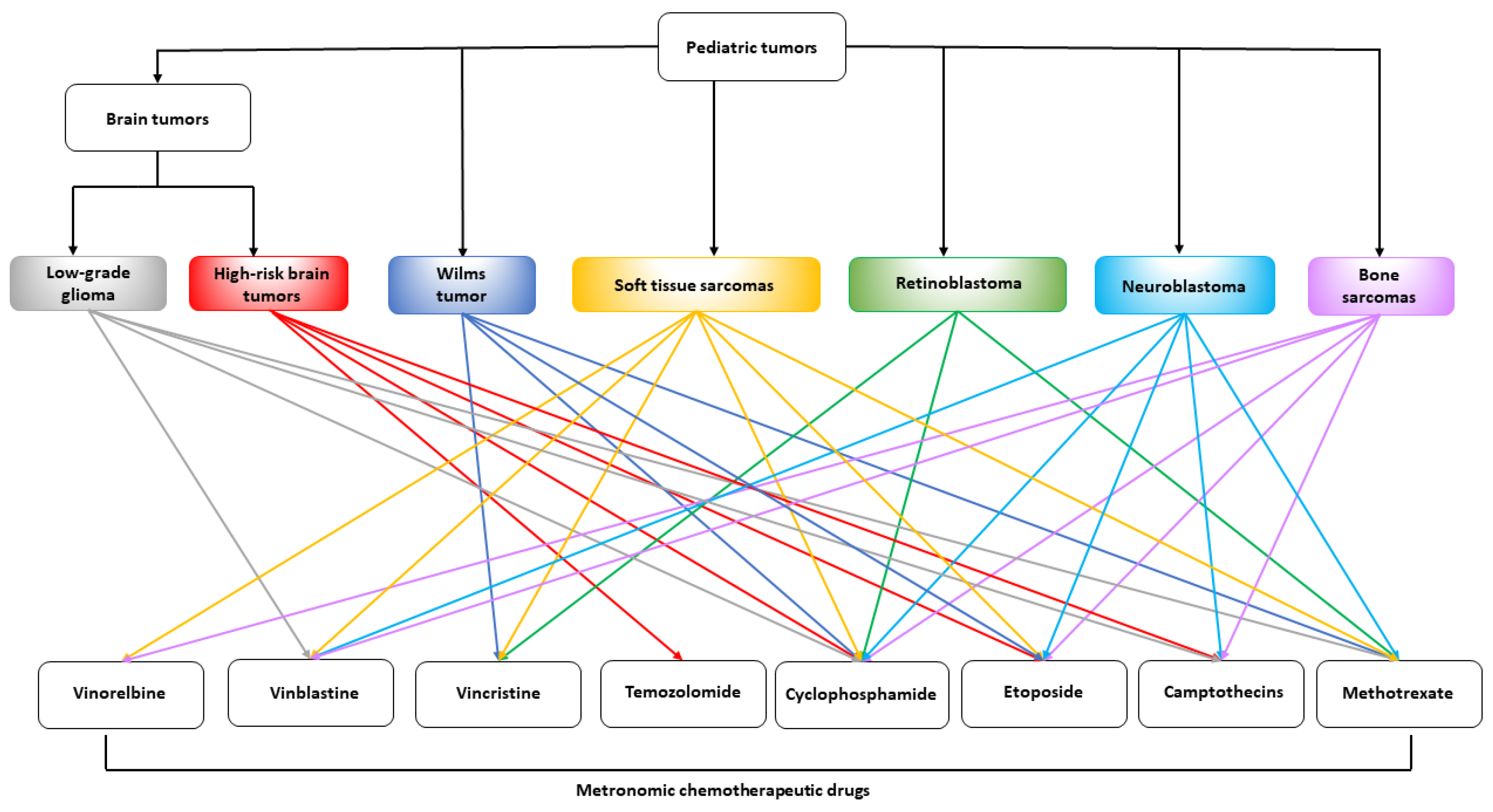{kind=link}
| Metronomic Regimen | Preclinical Tumor Model | Results | Reference |
|---|---|---|---|
| Neuroblastoma | |||
| Vinblastine (1.5 mg/m2 i.p. every 3 days), anti-VEGFR-2 antibody DC101 (800 μg/mouse, i.p. every 3 days), or combination, for >6 months | Human SK-N-MC neuroepithelioma and SK-N-AS neuroblastoma cell lines implanted s.c. in CB-17 SCID mice, aged 4 to 6 weeks | Combined treatment induced complete and sustained tumor regression, without marked toxicity or appearance of drug resistance. | Klement et al. [12] |
| Topotecan (0.36 mg/kg, i.p. 5x/week), anti-VEGF antibody A4.6.1 (100 μg, i.p. twice a week), or combination for a total of 5 weeks | Human NGP-GFP neuroblastoma xenograft in female NCR athymic mice, aged 4 to 6 weeks | LDM topotecan either with or without anti-VEGF antibody significantly suppresses NB xenograft growth. All treated mice showed reduced tumor vascularization. Only combined treatment significantly stopped the regrowth. LDM topotecan increased apoptosis of neuroblastoma cells. | Kim et al. [20] |
| Vinblastine (0.625–250 pM) and rapamycin (1.56–1000 pM), administered for 144 h, thrice a week | In vitro ECs (HUVEC and EA.hy926) and ECs preincubated with CM from the human NB cell line HTLA-230. In vivo CAM assay with HTLA-230-derived CM, HTLA-230-derived tumor xenografts, and human NB biopsy specimens | Significant antiproliferative effect in ECs preincubated with HTLA-230 CM after combination at low doses. Combination of 50 pM vinblastine and 0.5 nM rapamycin was synergistic in arresting the cell cycle and increased apoptosis of ECs. The combination markedly inhibited the angiogenic effects induced in vivo in the CAM assay. | Marimpietri et al. [19] |
| Topotecan (0.5 mg/kg daily i.p.), bevacizumab (5 mg/kg daily i.p.) or sunitinib (40 mg/kg daily p.o.), or combination, for 2 weeks | Unmodified CHLA-20 and NB-1691, and shHIF-1α-modified NB-1691 and SKNAS neuroblastoma cell lines injected into the retroperitoneal space of CB-17 SCID mice | Combined LDM treatment significantly reduced tumor growth and downregulated the expression of VEGF and GLUT3. | Hartwich et al. [22] |
| In vitro: topotecan (5 nM, 2×/week for 3 weeks) In vivo: topotecan (0.1 mg/kg/day i.p. for 6 or 15 weeks) | In vitro WT or MYCN-amplified neuroblastoma cell lines (i.e., STA-NB-10 and CLB-Ma). In vivo STA-NB-10 xenografts established in female CD1: Foxn1nu/nu mice, 6 to 10 weeks old | Induction of DNA-damage and a tumor-inhibiting favorable SASP selectively in MYCN-amplified cells in vitro. Tumor regression and prolonged survival, with no evident toxicity in vivo. Significant reduction of MYCN mRNA and protein expression both in vitro and in vivo Decreased VEGF-A expression and tumor vascularization in vivo. | Taschner-Mandl et al. [26] |
| Cyclophosphamide (mCTX, 40 mg/kg/day, p.o. through the drinking water), calorie restricted KD, optimized KD, or combination | Non-MYCN-amplified SH-SY5Y and MYCN-amplified SK-N-BE(2) NB cell lines injected s.c. in female CD1 nude mice, aged 5 to 6 weeks | mCTX induced greater tumor inhibition in MYCN-amplified xenografts, diminution of blood vessel density, and intratumoral bleeding, decreased Bcl-2 expression, and increased caspase-3 cleavage. Combining mCTX with calorie restricted KD resulted in tumor regression and complete growth arrest of both NB xenografts. Combining mCTX with optimized KD resulted in significant tumor growth suppression and prolonged survival, especially in SH-SY5Y xenografts, and inhibition of angiogenesis. | Morscher et al. [32] Aminzadeh-Gohari et al. [33] |
| Topotecan (1.0 mg/kg/day p.o.), pazopanib (150 mg/kg/day p.o.), or combination | SK-N-BE(2) and SH-SY5Y s.c. (into inguinal fat pad) NB xenografts, BE(2)-c and NUB-72 metastatic (into lateral tail vein) NB xenografts in NOD/SCID mice | Combined treatment slowed tumor growth in SK-N-BE(2) and SH-SY5Y models, and limited micrometastasis in BE(2)-c and NUB-7 models. Significant reduction in viable CECs and CEPs and tumor microvessel density in SH-SY5Y xenografts. | Kumar et al. [29] |
| Topotecan (1.0 mg/kg/day p.o.), pazopanib (150 mg/kg/day p.o.), or combination for 28, 56, and 80 days | SK-N-BE(2) s.c. xenograft–bearing NOD/SCID mice, 4 to 8 weeks old | Only the animals receiving the combination survived up to 80 days. All three durations of combined treatment significantly reduced microvessel density. Higher proliferative and mitotic indices after 28 days of combined treatment. | Kumar et al. [30] |
| In vitro: crizotinib, LDM topotecan, or combination (ratio 20:1, 0,01–10 μM) continuously for 6 days In vivo: topotecan (1 mg/kg/day p.o.), crizotinib (50 mg/kg/day p.o.) for 9 days | In vitro ALKF1174L-mutated SH-SY5Y cell line and MYCN-amplified SK-N-BE(2), KELLY, and LAN-5 cell lines. In vivo ALKF1174L-mutated SH-SY5Y and KELLY xenografts in female NOD/SCID mice, 4 to 6 weeks old. | Combined treatment resulted in a synergistic antiproliferative effect only in SH-SY5Y cells in vitro and a significantly delayed tumor growth of KELLY xenografts, and complete regression of SH-SY5Y tumors in vivo. | Zhang et al. [31] |
| Brain tumor | |||
| Bolus at day 1 with 6 mg/kg carboplatin i.p. and 4 mg/kg etoposide i.p., followed by 2 mg/kg carboplatin + 2 mg/kg etoposide and 2 mg/kg of PEX i.p. for 2 days, every 3 days. Treatment duration ≥ 120 days | Intracranial human U87 glioblastoma xenografts in 5-week-old Swiss male nude mice | This combination was more effective than chemotherapy alone and induced better survival, substantial reduction in tumor volume, vascularization and proliferation index, increased apoptosis, and no complications. | Bello et al. [37] |
| DC101 (800 μg/mouse i.p. every 3 days), LDM cyclophosphamide (20 mg/kg/day p.o. via the drinking water), MTD cyclophosphamide (100 mg/kg i.p. on days 1, 3, and 5 of a 21-day cycle), and their combinations | Athymic nude mice bearing s.c. rat C6 glioma xenografts | The combination of LDM cyclophosphamide with potent inhibition of angiogenesis by DC101 caused a significant and selective elimination of TSLCs from the glioma. | Folkins et al. [5] |
| Low-dose topotecan (1 mg/kg daily, 10 total doses) | Female athymic nude (NCr/nu) mice bearing human U251-HRE glioblastoma xenografts | Sustained inhibition of xenograft tumor progression, with prominent diminution of HIF-1α protein amount, angiogenesis, and expression of genes targeted by HIF-1 in tumor mass | Rapisarda et al. [38] |
| Very low-dose topotecan (0.5 mg/kg daily, for 10 days) + bevacizumab | Female athymic nude (NCr/nu) mice bearing human U251-HRE glioblastoma xenografts | The combination significantly suppressed glioblastoma growth. Addition of topotecan clearly abrogated HIF-1 transcriptional activity in the tumor microenvironment, significantly inhibited proliferation, and induced apoptosis. | Rapisarda et al. [39] |
| In vitro: LDM TMZ (1–100 μM daily for 144 h) In vivo rat model: conventional TMZ (7 mg/kg p.o. for 5 days) vs. LDM TMZ (1 or 2 mg/kg p.o., every day for 16 days) In vivo mouse model: conventional TMZ (2.5 or 1.25 mg/kg p.o., for 5 days) vs. LDM TMZ (0.5 and 0.25 mg/kg p.o., daily for 25 days) | In vitro C6/LacZ rat glioma cells and U-87MG human glioblastoma cells. In vivo male Sprague-Dawley (SD) rats (200–250 g) bearing intracranial C6/LacZ glioma and male Balb/c-nu mice (6 weeks) bearing intracranial U-87MG glioblastoma | In vitro C6/LacZ rat glioma cells were more resistant to LDM TMZ than U-87MG human glioblastoma cells. In the orthotopic C6/LacZ glioma model, LDM TMZ significantly hampered tumor growth and angiogenesis and promoted apoptosis, compared to the conventional schedule. In the orthotopic U-87MG xenograft, TMZ even at a very low dose (0.25 mg/kg daily for 25 days) markedly reduced the microvessel density. | Kim et al. [40] |
| LDM TMZ (0.5 or 2 mg/kg/die p.o., 5 days per week for 21 days) vs. standard TMZ (30 mg/kg/die p.o. for 5 days, or 10 mg/kg/die, 5 days per week for 21 days) | TMZ-resistant RG2 glioma cells implanted s.c. in Fischer rats | TMZ significantly decreased the Treg/CD4+ T cell proportion when given at very low doses but not at high standard doses. Treg depletion alone, detected in LDM TMZ-treated rats, was not sufficient to significantly inhibit tumor progression, compared to control. | Banissi et al. [6] |
| LDM TMZ (1.77 and 0.9 mg/kg/day, for a total of 5 weeks) + weekly morphine | Orthotopic human U87MG-luc2 glioblastoma xenograft, implanted intracranially in Foxn1 nude mice | Addition of morphine enhanced the antitumor efficacy and the long-term response to the TMZ metronomic regimens. | Iorio et al. [43] |
| MEDIC (medium-dose intermittent chemotherapy) Cyclophosphamide (140 mg/kg i.p., every 6 day) | SCID mice bearing s.c. xenografts of rat 9L gliosarcoma, mouse GL261 glioma, and human U251 glioblastoma | The MEDIC schedule activated robust and persistent immune responses, as well as induced a striking and extended tumor regression. More effective than the corresponding daily low-dose metronomic regimen. Induction of a strong CD8+ T cell response leading to tumor disappearance and gain of immune memory in the GL261 glioma xenograft. | Chen et al. [44] Doloff et al. [45] Wu et al. [46,47] |
| Every 6-day metronomic cyclophosphamide (140 mg/kg) or temozolomide (140, 200, and 240 mg/kg) | Orthotopic GL261 glioma model in immunocompetent C57BL/6 mice | Longer survival rate in mice treated with both regimes than in control animals. In total, 140 mg/kg temozolomide was the best dosing in reduction of tumor volume and improving survival. No tumor eradication was achieved. | Ferrer-font et al. [48] |
| Metronomic TMZ (25 mg/kg × 10 days) vs. standard TMZ (50 mg/kg × 5 days) + immunotherapy | GL261 and KR158 intracranial glioma models in C57BL/6 mice | Addition of metronomic TMZ to PD-1 immune checkpoint inhibition preserved the survival benefit gained with anti-PD-1 therapy alone in the GL261 model, while it was abolished by standard temozolomide. | Karachi et al. [50] |
| Soft tissue sarcoma | |||
| MTD doxorubicin (6 mg/kg once every 2 weeks), LDM doxorubicin (1.2 mg/kg, twice weekly for 4 weeks), DC101 (400 μg/dose every 3 days for a total of seven times), or combination | Female SCID mice (weight 18–22 g) harboring human RD rhabdomyosarcoma and SKLMS-1 leiomyosarcoma s.c. tumors | LDM doxorubicin alone was less effective than MTD doxorubicin. The combination of LDM doxorubicin and DC101 markedly reduced the volume of both SKLMS-1 and RD tumors, and the microvessel count, without overt toxicity. | Zhang et al. [53] |
| Bone sarcoma | |||
| MTD methotrexate (1.35 g/kg i.v., 6 h infusion, once weekly, at week 0, 1, 5, and 6), adriamycin and cisplatin (10 mg/kg and 20 mg/kg, respectively; once weekly, at week 2 and 7), LDM methotrexate (1.2 mg/kg i.v., bolus, twice a week), combination of MTD and LDM schedules, for a total of 8 weeks | 5-week-old SD rats bearing s.c. UMR 106 tumors | After the first 6 weeks of treatment, only the combination showed a protracted inhibition of tumor growth. Both MTD and combination schedules showed a much lower VEGF-A expression. | Zhu et al. [54] |
| Retinoblastoma | |||
| In vitro: melphalan or topotecan in a conventional (0.001–1000 μM or 0.001–10.000 nM, respectively) single 72 h exposure vs. metronomic (0.0001–100 μM or 0.0001–1000 nM, respectively) 7-day continuous exposure In vivo: topotecan LDM topotecan (0.6 mg/kg i.p., 5 days a week) for 2 weeks) vs. MTD (3 mg/kg i.p., once a week) for 2 weeks | In vitro: commercial Y79 and WERI-RB1 and patient-derived HSJD-RBT-7 and HSJD-RBT-8 retinoblastoma cell lines. HUVEC and EPC human vascular endothelial cells In vivo: Y79 retinoblastoma s.c. xenografts in female athymic nude mice, weighing 18–22 g | Continuous administration increased the sensitivity to chemotherapy of retinoblastoma and endothelial cells. The heavily pretreated HSJD-RBT-8 cells had enhanced chemosensitivity to the metronomic schedule compared to the conventional one. Both treatment regimens led to cell death through apoptosis and/or necrosis in all cell lines. Metronomic schedules significantly inhibited in vitro tube formation in endothelial cells. LDM topotecan significantly lowered tumor volumes compared to the MTD protocol and control group, with a similar toxicity profile and no weight loss. | Winter et al. [57] |
| Acute lymphoblastic leukemia | |||
| Dexamethasone (0.1, 1, and 7.6 μM) alone or in combination with low-dose arsenic trioxide (0.25 μM) for 72 h | GC-resistant ALL cell lines (CEM-C1-15, Jurkat, and MOLT-4), T-ALL, and precursor B-ALL cells from pediatric patients with poor response to prednisone | Low-dose arsenic trioxide significantly increased in vitro dexamethasone sensitivity. This combination reduced Akt phosphorylation, which is associated with an increase in Bad and decrease in XIAP protein. | Bornhauser et al. [60] |
| The 2-DG (0.2 to 10 mM) alone and in combination with dexamethasone (1 µM) for 24 h and 48 h | T-ALL cell lines: Molt-4 (GC resistance), Jurkat (GC resistance); CEM-C1-15 (GC resistance), CEM-C7-14(GC sensitive) B-ALL cell lines: Nalm-6 (GC sensitive), RS4:11 (GC sensitive) Burkitt lymphoma cell line: Raji (B-lineage, GC resistance) | Low-dose of 2-DG (1 mM) for 48 h induced apoptosis and cell-cycle arrest. Its combination with dexamethasone recovered the sensitivity of GC and produced a strong synergistic cytotoxic effect in GC-resistant Molt-4 and Raji cells. | Gu et al. [62] |
| L-ASNase 0.01 U/mL vs. 1 U/mL for 24 h | Cell lines: Jurkat and Reh (ALL), Jeko (mantle cell lymphoma), NK-YS (nasal-type NK-cell lymphoma) | Low-dose L-ASNase (0.01 U/mL) effectively killed Asn-dependent NK-YS cells, whereas clinically achievable intermediate doses (1 U/mL) induced robust apoptosis in Gln-dependent Jurkat and Jeko cell lines. | Sugimoto et al. [66] |
| Miscellaneous | |||
| In vitro: increasing concentrations of evofosfamide with or without the presence of low-dose topotecan (20 nmol/L) In vivo: evofosfamide, (50 mg/kg daily i.p., 5 days/week), LDM topotecan (1 mg/kg daily by oral gavage, 5 days/week), and their combination | In total, 5 different neuroblastoma cell lines (CHLA-15, CHLA-20, CHLA-90, SK-N-BE(2), and SH-SY5Y) and 3 rhabdomyosarcoma cell lines (RH4, RH30, and RD) in vitro. Aggressive s.c. xenografts using two neuroblastoma cell lines [CHLA-20 and SK-N-BE(2)] and two rhabdomyosarcoma cell lines (RH4 and RD); metastatic (intravenous) SK-N-BE(2) neuroblastoma model in NOD/SCID mice | Low-dose topotecan enhanced the cytotoxic effect of evofosfamide in all cell lines in vitro. Combined treatment resulted in a better antitumor effect than both monotherapies and induced complete tumor regression after 2 weeks of treatment in each s.c. xenograft. It also improved survival in the SK-N-BE(2) metastatic model. | Zhang et al. [67] |
| In vitro: 96 h exposure of temozolomide (0.3–1000 μmol/L) with or without talazoparib (10 nmol/L) In vivo: temozolomide (30 mg/kg/day × 5 days) and talazoparib (0.25 mg/kg twice daily × 5 days) alone or in two different combinations: high-dose temozolomide (30 mg/kg/day × 5) + talazoparib (0.1 mg/kg twice daily × 5) or low-dose temozolomide (12 mg/kg/day × 5) + talazoparib (0.25 mg/kg twice daily × 5) | Pediatric Preclinical Testing Program (PPTP) cell line panel including a total of 23 cell lines of rhabdomyosarcoma, rhabdoid, Ewing sarcoma, neuroblastoma, glioblastoma, ALL, AML, ALCL, and NHL origin, for cytotoxic in vitro assays. In vivo pediatric xenografts: C.B-17 scid−/− female mice for s.c. implantation of Wilms tumor, rhabdoid tumor, Ewing sarcoma, osteosarcoma, rhabdomyosarcoma, neuroblastoma, and non-glioblastoma brain tumors; BALB/c nu/nu mice for glioma models; female NOD.CB17-Prkdcscid/J mice for intravenous inoculation of human leukemia cells | In vitro, the combination resulted in a marked potentiation of temozolomide toxicity in Ewing sarcoma (50-fold) and ALL (30-fold) cell lines. In vivo, toxicity was comparable for both combinations. Both exhibited significant antitumor effects and induced total tumor regression in 5 of 10 Ewing xenografts, within 6 weeks of treatment. It was successful against xenografts with low MGMT expression (i.e., GBM2 glioblastoma and Rh28 rhabdomyosarcoma) and those with defective homologous recombination (i.e., KT-10 Wilms tumor). | Smith et al. [71] |
| In vitro: Topotecan for a total of 6 days, on three different ways of administration: continuous, for 8 h daily, or for 8 h every other day. In vivo: low-dose topotecan 0.6 mg/kg i.v. daily ×5 and 1.0 mg/kg every other day; increased dose 2 mg/kg daily ×5 and 3.3 mg/kg every other day (for 2 weeks, repeated every 21 days for 3 cycles) | In vitro: Daoy pediatric medulloblastoma and Rh30 pediatric rhabdomyosarcoma cells In vivo: Daoy and Rh30 s.c. xenografts in 4-week-old CBA/Caj tyhmectomized female mice | Topotecan IC50 values of ~2 nM for Daoy cells, while they increased with extending drug-free time in Rh30 cells. Rh30 xenografts regressed completely when treated with topotecan 0.6 mg/kg i.v. daily ×5. Increased doses of 2 mg/kg daily and 3.3 mg/kg every other day to obtain complete tumor regression of the less sensitive in vivo Daoy models. | Pawlik et al. [72] |
4. Pharmacokinetic Studies on Metronomic Chemotherapy in Preclinical, Pediatric Tumor Models
5. Clinical Studies on Metronomic Chemotherapy in Pediatric Patients
5.1. Retrospective Studies
5.2. Case Series
5.3. Phase I and Pilot Studies
5.4. Phase II Studies
5.5. Phase III Studies
6. Biomarkers of Metronomic Chemotherapy in Pediatric Patients
7. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bocci, G.; Kerbel, R.S. Pharmacokinetics of metronomic chemotherapy: A neglected but crucial aspect. Nat. Rev. Clin. Oncol. 2016, 13, 659–673. [Google Scholar] [CrossRef]
- Bocci, G.; Francia, G.; Man, S.; Lawler, J.; Kerbel, R.S. Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc. Natl. Acad. Sci. USA 2003, 100, 12917–12922. [Google Scholar] [CrossRef]
- Bocci, G.; Nicolaou, K.C.; Kerbel, R.S. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res. 2002, 62, 6938–6943. [Google Scholar]
- Natale, G.; Bocci, G. Does metronomic chemotherapy induce tumor angiogenic dormancy? A review of available preclinical and clinical data. Cancer Lett. 2018, 432, 28–37. [Google Scholar] [CrossRef]
- Folkins, C.; Man, S.; Xu, P.; Shaked, Y.; Hicklin, D.J.; Kerbel, R.S. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007, 67, 3560–3564. [Google Scholar] [CrossRef] [PubMed]
- Banissi, C.; Ghiringhelli, F.; Chen, L.; Carpentier, A.F. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol. Immunother. 2009, 58, 1627–1634. [Google Scholar] [CrossRef]
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A. International incidence of childhood cancer, 2001-10: A population-based registry study. Lancet. Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef]
- World Health Organization. Framework: WHO Global Initiative for Childhood Cancer; WHO: Geneva, Switzerland, 2020; ISBN 9789240025271.
- Pramanik, R.; Bakhshi, S. Metronomic therapy in pediatric oncology: A snapshot. Pediatr. Blood Cancer 2019, 66, e27811. [Google Scholar] [CrossRef] [PubMed]
- André, N.; Banavali, S.; Snihur, Y.; Pasquier, E. Has the time come for metronomics in low-income and middle-income countries? Lancet. Oncol. 2013, 14, e239–e248. [Google Scholar] [CrossRef]
- Newman, E.A.; Abdessalam, S.; Aldrink, J.H.; Austin, M.; Heaton, T.E.; Bruny, J.; Ehrlich, P.; Dasgupta, R.; Baertschiger, R.M.; Lautz, T.B.; et al. Update on neuroblastoma. J. Pediatr. Surg. 2019, 54, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Klement, G.; Baruchel, S.; Rak, J.; Man, S.; Clark, K.; Hicklin, D.J.; Bohlen, P.; Kerbel, R.S. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J. Clin. Investig. 2000, 105, R15–R24. [Google Scholar] [CrossRef] [PubMed]
- Nör, J.E.; Christensen, J.; Mooney, D.J.; Polverini, P.J. Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am. J. Pathol. 1999, 154, 375–384. [Google Scholar] [CrossRef]
- Tran, J.; Rak, J.; Sheehan, C.; Saibil, S.D.; LaCasse, E.; Korneluk, R.G.; Kerbel, R.S. Marked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochem. Biophys. Res. Commun. 1999, 264, 781–788. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.S.; Schechner, J.S.; Adida, C.; Mesri, M.; Rothermel, A.L.; Li, F.; Nath, A.K.; Pober, J.S.; Altieri, D.C. Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am. J. Pathol. 2000, 156, 393–398. [Google Scholar] [CrossRef]
- Vacca, A.; Iurlaro, M.; Ribatti, D.; Minischetti, M.; Nico, B.; Ria, R.; Pellegrino, A.; Dammacco, F. Antiangiogenesis is produced by nontoxic doses of vinblastine. Blood 1999, 94, 4143–4155. [Google Scholar] [CrossRef] [PubMed]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef]
- Misawa, A.; Hosoi, H.; Tsuchiya, K.; Sugimoto, T. Rapamycin inhibits proliferation of human neuroblastoma cells without suppression of MycN. Int. J. cancer 2003, 104, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Marimpietri, D.; Nico, B.; Vacca, A.; Mangieri, D.; Catarsi, P.; Ponzoni, M.; Ribatti, D. Synergistic inhibition of human neuroblastoma-related angiogenesis by vinblastine and rapamycin. Oncogene 2005, 24, 6785–6795. [Google Scholar] [CrossRef]
- Kim, E.S.; Soffer, S.Z.; Huang, J.; McCrudden, K.W.; Yokoi, A.; Manley, C.A.; Middlesworth, W.; Kandel, J.J.; Yamashiro, D.J. Distinct response of experimental neuroblastoma to combination antiangiogenic strategies. J. Pediatr. Surg. 2002, 37, 518–522. [Google Scholar] [CrossRef]
- Puppo, M.; Battaglia, F.; Ottaviano, C.; Delfino, S.; Ribatti, D.; Varesio, L.; Bosco, M.C. Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia-inducible factor-1alpha and -2alpha. Mol. Cancer Ther. 2008, 7, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Hartwich, J.; Orr, W.S.; Ng, C.Y.; Spence, Y.; Morton, C.; Davidoff, A.M. HIF-1α activation mediates resistance to anti-angiogenic therapy in neuroblastoma xenografts. J. Pediatr. Surg. 2013, 48, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Meitar, D.; Crawford, S.E.; Rademaker, A.W.; Cohn, S.L. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1996, 14, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Sköldenberg, E.G.; Larsson, A.; Jakobson, A.; Hedborg, F.; Kogner, P.; Christofferson, R.H.; Azarbayjani, F. The angiogenic growth factors HGF and VEGF in serum and plasma from neuroblastoma patients. Anticancer Res. 2009, 29, 3311–3319. [Google Scholar]
- Taschner-Mandl, S.; Schwarz, M.; Blaha, J.; Kauer, M.; Kromp, F.; Frank, N.; Rifatbegovic, F.; Weiss, T.; Ladenstein, R.; Hohenegger, M.; et al. Metronomic topotecan impedes tumor growth of MYCNamplified neuroblastoma cells in vitro and in vivo by therapy induced senescence. Oncotarget 2016, 7, 3571–3586. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Natarajan, V.; Demidenko, Z.N.; Burdelya, L.G.; Gudkov, A.V.; Blagosklonny, M.V. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 13314–13318. [Google Scholar] [CrossRef]
- Rapisarda, A.; Uranchimeg, B.; Sordet, O.; Pommier, Y.; Shoemaker, R.H.; Melillo, G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: Mechanism and therapeutic implications. Cancer Res. 2004, 64, 1475–1482. [Google Scholar] [CrossRef]
- Kumar, S.; Mokhtari, R.B.; Sheikh, R.; Wu, B.; Zhang, L.; Xu, P.; Man, S.; Oliveira, I.D.; Yeger, H.; Kerbel, R.S.; et al. Metronomic oral topotecan with pazopanib is an active antiangiogenic regimen in mouse models of aggressive pediatric solid tumor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5656–5667. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mokhtari, R.B.; Oliveira, I.D.; Islam, S.; Toledo, S.R.C.; Yeger, H.; Baruchel, S. Tumor dynamics in response to antiangiogenic therapy with oral metronomic topotecan and pazopanib in neuroblastoma xenografts. Transl. Oncol. 2013, 6, 493–503. [Google Scholar] [CrossRef][Green Version]
- Zhang, L.; Wu, B.; Baruchel, S. Oral Metronomic Topotecan Sensitizes Crizotinib Antitumor Activity in ALKF1174L Drug-Resistant Neuroblastoma Preclinical Models. Transl. Oncol. 2017, 10, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Morscher, R.J.; Aminzadeh-Gohari, S.; Hauser-Kronberger, C.; Feichtinger, R.G.; Sperl, W.; Kofler, B. Combination of metronomic cyclophosphamide and dietary intervention inhibits neuroblastoma growth in a CD1-nu mouse model. Oncotarget 2016, 7, 17060–17073. [Google Scholar] [CrossRef]
- Aminzadeh-Gohari, S.; Feichtinger, R.G.; Vidali, S.; Locker, F.; Rutherford, T.; O’Donnel, M.; Stöger-Kleiber, A.; Mayr, J.A.; Sperl, W.; Kofler, B. A ketogenic diet supplemented with medium-chain triglycerides enhances the anti-tumor and anti-angiogenic efficacy of chemotherapy on neuroblastoma xenografts in a CD1-nu mouse model. Oncotarget 2017, 8, 64728–64744. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet. Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Udaka, Y.T.; Packer, R.J. Pediatric Brain Tumors. Neurol. Clin. 2018, 36, 533–556. [Google Scholar] [CrossRef]
- Bello, L.; Lucini, V.; Carrabba, G.; Giussani, C.; Machluf, M.; Pluderi, M.; Nikas, D.; Zhang, J.; Tomei, G.; Villani, R.M.; et al. Simultaneous inhibition of glioma angiogenesis, cell proliferation, and invasion by a naturally occurring fragment of human metalloproteinase-2. Cancer Res. 2001, 61, 8730–8736. [Google Scholar]
- Bello, L.; Carrabba, G.; Giussani, C.; Lucini, V.; Cerutti, F.; Scaglione, F.; Landré, J.; Pluderi, M.; Tomei, G.; Villani, R.; et al. Low-dose chemotherapy combined with an antiangiogenic drug reduces human glioma growth in vivo. Cancer Res. 2001, 61, 7501–7506. [Google Scholar]
- Rapisarda, A.; Zalek, J.; Hollingshead, M.; Braunschweig, T.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Hewitt, S.M.; Shoemaker, R.H.; et al. Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res. 2004, 64, 6845–6848. [Google Scholar] [CrossRef]
- Rapisarda, A.; Hollingshead, M.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Gehrs, B.; Raffeld, M.; Kinders, R.J.; Parchment, R.; et al. Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol. Cancer Ther. 2009, 8, 1867–1877. [Google Scholar] [CrossRef]
- Kim, J.T.; Kim, J.-S.; Ko, K.W.; Kong, D.-S.; Kang, C.-M.; Kim, M.H.; Son, M.J.; Song, H.S.; Shin, H.-J.; Lee, D.-S.; et al. Metronomic treatment of temozolomide inhibits tumor cell growth through reduction of angiogenesis and augmentation of apoptosis in orthotopic models of gliomas. Oncol. Rep. 2006, 16, 33–39. [Google Scholar] [CrossRef][Green Version]
- Carli, M.; Donnini, S.; Pellegrini, C.; Coppi, E.; Bocci, G. Opioid receptors beyond pain control: The role in cancer pathology and the debated importance of their pharmacological modulation. Pharmacol. Res. 2020, 159, 104938. [Google Scholar] [CrossRef]
- Kang, S.M.; Rosales, J.L.; Meier-Stephenson, V.; Kim, S.; Lee, K.Y.; Narendran, A. Genome-wide loss-of-function genetic screening identifies opioid receptor μ1 as a key regulator of L-asparaginase resistance in pediatric acute lymphoblastic leukemia. Oncogene 2017, 36, 5910–5913. [Google Scholar] [CrossRef] [PubMed]
- Iorio, A.L.; da Ros, M.; Genitori, L.; Lucchesi, M.; Colelli, F.; Signorino, G.; Cardile, F.; Laffi, G.; de Martino, M.; Pisano, C.; et al. Tumor response of temozolomide in combination with morphine in a xenograft model of human glioblastoma. Oncotarget 2017, 8, 89595–89606. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Doloff, J.C.; Waxman, D.J. Intermittent metronomic drug schedule is essential for activating antitumor innate immunity and tumor xenograft regression. Neoplasia 2014, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Doloff, J.C.; Waxman, D.J. VEGF receptor inhibitors block the ability of metronomically dosed cyclophosphamide to activate innate immunity-induced tumor regression. Cancer Res. 2012, 72, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide schedule-dependence of innate immune cell recruitment and tumor regression in an implanted glioma model. Cancer Lett. 2014, 353, 272–280. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8(+) T-cell responses and immune memory. Oncoimmunology 2015, 4, e1005521. [Google Scholar] [CrossRef]
- Ferrer-Font, L.; Arias-Ramos, N.; Lope-Piedrafita, S.; Julià-Sapé, M.; Pumarola, M.; Arús, C.; Candiota, A.P. Metronomic treatment in immunocompetent preclinical GL261 glioblastoma: Effects of cyclophosphamide and temozolomide. NMR Biomed. 2017, 30, e3748. [Google Scholar] [CrossRef]
- Delgado-Goñi, T.; Julià-Sapé, M.; Candiota, A.P.; Pumarola, M.; Arús, C. Molecular imaging coupled to pattern recognition distinguishes response to temozolomide in preclinical glioblastoma. NMR Biomed. 2014, 27, 1333–1345. [Google Scholar] [CrossRef]
- Karachi, A.; Yang, C.; Dastmalchi, F.; Sayour, E.J.; Huang, J.; Azari, H.; Long, Y.; Flores, C.; Mitchell, D.A.; Rahman, M. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro. Oncol. 2019, 21, 730–741. [Google Scholar] [CrossRef]
- Loeb, D.M.; Thornton, K.; Shokek, O. Pediatric soft tissue sarcomas. Surg. Clin. N. Am. 2008, 88, 615–627. [Google Scholar] [CrossRef]
- Grohar, P.J.; Janeway, K.A.; Mase, L.D.; Schiffman, J.D. Advances in the Treatment of Pediatric Bone Sarcomas. Am. Soc. Clin. Oncol. Educ. book. Am. Soc. Clin. Oncol. Annu. Meet. 2017, 37, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, D.; Hicklin, D.J.; Hannay, J.A.F.; Ellis, L.M.; Pollock, R.E. Combined anti-fetal liver kinase 1 monoclonal antibody and continuous low-dose doxorubicin inhibits angiogenesis and growth of human soft tissue sarcoma xenografts by induction of endothelial cell apoptosis. Cancer Res. 2002, 62, 2034–2042. [Google Scholar] [PubMed]
- Zhu, X.; Yin, H.; Mei, J. Inhibition of tumors cell growth in osteosarcoma-bearing SD rats through a combination of conventional and metronomic scheduling of neoadjuvant chemotherapy. Acta Pharmacol. Sin. 2010, 31, 970–976. [Google Scholar] [CrossRef][Green Version]
- Dimaras, H.; Kimani, K.; Dimba, E.A.O.; Gronsdahl, P.; White, A.; Chan, H.S.L.; Gallie, B.L. Retinoblastoma. Lancet 2012, 379, 1436–1446. [Google Scholar] [CrossRef]
- Abramson, D.H.; Shields, C.L.; Munier, F.L.; Chantada, G.L. Treatment of Retinoblastoma in 2015: Agreement and Disagreement. JAMA Ophthalmol. 2015, 133, 1341–1347. [Google Scholar] [CrossRef]
- Winter, U.; Mena, H.A.; Negrotto, S.; Arana, E.; Pascual-Pasto, G.; Laurent, V.; Suñol, M.; Chantada, G.L.; Carcaboso, A.M.; Schaiquevich, P. Schedule-Dependent Antiangiogenic and Cytotoxic Effects of Chemotherapy on Vascular Endothelial and Retinoblastoma Cells. PLoS ONE 2016, 11, e0160094. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Inaba, H.; Annesley, C.; Beck, J.; Colace, S.; Dallas, M.; DeSantes, K.; Kelly, K.; Kitko, C.; Lacayo, N.; et al. Pediatric Acute Lymphoblastic Leukemia, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2020, 18, 81–112. [Google Scholar] [CrossRef] [PubMed]
- Tissing, W.J.E.; Meijerink, J.P.P.; den Boer, M.L.; Pieters, R. Molecular determinants of glucocorticoid sensitivity and resistance in acute lymphoblastic leukemia. Leukemia 2003, 17, 17–25. [Google Scholar] [CrossRef]
- Bornhauser, B.C.; Bonapace, L.; Lindholm, D.; Martinez, R.; Cario, G.; Schrappe, M.; Niggli, F.K.; Schäfer, B.W.; Bourquin, J.P. Low-dose arsenic trioxide sensitizes glucocorticoid-resistant acute lymphoblastic leukemia cells to dexamethasone via an Akt-dependent pathway. Blood 2007, 110, 2084–2091. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.-H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef]
- Gu, L.; Yi, Z.; Zhang, Y.; Ma, Z.; Zhu, Y.; Gao, J. Low dose of 2-deoxy-D-glucose kills acute lymphoblastic leukemia cells and reverses glucocorticoid resistance via N-linked glycosylation inhibition under normoxia. Oncotarget 2017, 8, 30978–30991. [Google Scholar] [CrossRef]
- Pui, C.-H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef]
- Lomelino, C.L.; Andring, J.T.; McKenna, R.; Kilberg, M.S. Asparagine synthetase: Function, structure, and role in disease. J. Biol. Chem. 2017, 292, 19952–19958. [Google Scholar] [CrossRef]
- Ueno, T.; Ohtawa, K.; Mitsui, K.; Kodera, Y.; Hiroto, M.; Matsushima, A.; Inada, Y.; Nishimura, H. Cell cycle arrest and apoptosis of leukemia cells induced by L-asparaginase. Leukemia 1997, 11, 1858–1861. [Google Scholar] [CrossRef]
- Sugimoto, K.; Suzuki, H.I.; Fujimura, T.; Ono, A.; Kaga, N.; Isobe, Y.; Sasaki, M.; Taka, H.; Miyazono, K.; Komatsu, N. A clinically attainable dose of L-asparaginase targets glutamine addiction in lymphoid cell lines. Cancer Sci. 2015, 106, 1534–1543. [Google Scholar] [CrossRef]
- Zhang, L.; Marrano, P.; Wu, B.; Kumar, S.; Thorner, P.; Baruchel, S. Combined Antitumor Therapy with Metronomic Topotecan and Hypoxia-Activated Prodrug, Evofosfamide, in Neuroblastoma and Rhabdomyosarcoma Preclinical Models. Clin. Cancer Res. 2016, 22, 2697–2708. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Horton, J.K.; Wilson, S.H. Predicting enhanced cell killing through PARP inhibition. Mol. Cancer Res. 2013, 11, 13–18. [Google Scholar] [CrossRef]
- Smith, M.A.; Reynolds, C.P.; Kang, M.H.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.B.; Keir, S.T.; Maris, J.M.; Billups, C.A.; et al. Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 819–832. [Google Scholar] [CrossRef]
- Pawlik, C.A.; Houghton, P.J.; Stewart, C.F.; Cheshire, P.J.; Richmond, L.B.; Danks, M.K. Effective schedules of exposure of medulloblastoma and rhabdomyosarcoma xenografts to topotecan correlate with in vitro assays. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1998, 4, 1995–2002. [Google Scholar]
- Zhou, Q.; Guo, P.; Wang, X.; Nuthalapati, S.; Gallo, J.M. Preclinical pharmacokinetic and pharmacodynamic evaluation of metronomic and conventional temozolomide dosing regimens. J. Pharmacol. Exp. Ther. 2007, 321, 265–275. [Google Scholar] [CrossRef]
- van Geel, R.M.J.M.; Beijnen, J.H.; Schellens, J.H.M. Concise drug review: Pazopanib and axitinib. Oncologist 2012, 17, 1081–1089. [Google Scholar] [CrossRef]
- Hartmann, J.T.; Lipp, H.-P. Camptothecin and podophyllotoxin derivatives: Inhibitors of topoisomerase I and II—mechanisms of action, pharmacokinetics and toxicity profile. Drug Saf. 2006, 29, 209–230. [Google Scholar] [CrossRef]
- Roux, C.; Revon-Rivière, G.; Gentet, J.C.; Verschuur, A.; Scavarda, D.; Saultier, P.; Appay, R.; Padovani, L.; André, N. Metronomic Maintenance with Weekly Vinblastine after Induction with Bevacizumab-Irinotecan in Children with Low-grade Glioma Prevents Early Relapse. J. Pediatr. Hematol. Oncol. 2021, 43, E630–E634. [Google Scholar] [CrossRef]
- Zapletalova, D.; Andr, N.; Deak, L.; Kyr, M.; Bajciova, V.; Mudry, P.; Dubska, L.; Demlova, R.; Pavelka, Z.; Zitterbart, K.; et al. Metronomic chemotherapy with the COMBAT regimen in advanced pediatric malignancies: A multicenter experience. Oncology 2012, 82, 249–260. [Google Scholar] [CrossRef]
- Carcamo, B.; Francia, G. Cyclic Metronomic Chemotherapy for Pediatric Tumors: Six Case Reports and a Review of the Literature. J. Clin. Med. 2022, 11, 2849. [Google Scholar] [CrossRef]
- Štěrba, J.; Pavelka, Z.; Šlampa, P. Concomitant radiotherapy and metronomic temozolomide in pediatric high-risk brain tumors. Neoplasma 2002, 49, 117–120. [Google Scholar]
- Fousseyni, T.; Diawara, M.; Pasquier, E.; André, N. Children treated with metronomic chemotherapy in a low-income country: METRO-MALI-01. J. Pediatr. Hematol. Oncol. 2011, 33, 31–34. [Google Scholar] [CrossRef]
- Stempak, D.; Gammon, J.; Halton, J.; Moghrabi, A.; Koren, G.; Baruchel, S. A pilot pharmacokinetic and antiangiogenic biomarker study of celecoxib and low-dose metronomic vinblastine or cyclophosphamide in pediatric recurrent solid tumors. J. Pediatr. Hematol. Oncol. 2006, 28, 720–728. [Google Scholar] [CrossRef]
- Traore, F.; Togo, B.; Pasquier, E.; Dembélé, A.; André, N. Preliminary evaluation of children treated with metronomic chemotherapy and valproic acid in a low-income country: Metro-Mali-02. Indian J. Cancer 2013, 50, 250–253. [Google Scholar] [CrossRef]
- Kieran, M.W.; Turner, C.D.; Rubin, J.B.; Chi, S.N.; Zimmerman, M.A.; Chordas, C.; Klement, G.; Laforme, A.; Gordon, A.; Thomas, A.; et al. A feasibility trial of antiangiogenic (metronomic) chemotherapy in pediatric patients with recurrent or progressive cancer. J. Pediatr. Hematol. Oncol. 2005, 27, 573–581. [Google Scholar] [CrossRef]
- Manji, A.; Samson, Y.; Deyell, R.J.; Johnston, D.L.; Lewis, V.A.; Zorzi, A.P.; Berman, J.N.; Brodeur-Robb, K.; Morrison, E.; Kee, L.; et al. Low-Dose Metronomic Topotecan and Pazopanib (TOPAZ) in Children with Relapsed or Refractory Solid Tumors: A C17 Canadian Phase I Clinical Trial. Cancers 2022, 14, 2985. [Google Scholar] [CrossRef]
- Ali, A.M.; El-Sayed, M.I. Metronomic chemotherapy and radiotherapy as salvage treatment in refractory or relapsed pediatric solid tumours. Curr. Oncol. 2016, 23, e253–e259. [Google Scholar] [CrossRef][Green Version]
- Pasqualini, C.; Rubino, J.; Brard, C.; Cassard, L.; André, N.; Rondof, W.; Scoazec, J.Y.; Marchais, A.; Nebchi, S.; Boselli, L.; et al. Phase II and biomarker study of programmed cell death protein 1 inhibitor nivolumab and metronomic cyclophosphamide in paediatric relapsed/refractory solid tumours: Arm G of AcSé-ESMART, a trial of the European Innovative Therapies for Children With Cance. Eur. J. Cancer 2021, 150, 53–62. [Google Scholar] [CrossRef]
- Robison, N.J.; Campigotto, F.; Chi, S.N.; Manley, P.E.; Turner, C.D.; Zimmerman, M.A.; Chordas, C.A.; Werger, A.M.; Allen, J.C.; Goldman, S.; et al. A phase II trial of a multi-agent oral antiangiogenic (metronomic) regimen in children with recurrent or progressive cancer. Pediatr. Blood Cancer 2014, 61, 636–642. [Google Scholar] [CrossRef]
- Verschuur, A.; Heng-Maillard, M.-A.; Dory-Lautrec, P.; Truillet, R.; Jouve, E.; Chastagner, P.; Leblond, P.; Aerts, I.; Honoré, S.; Entz-Werle, N.; et al. Metronomic Four-Drug Regimen Has Anti-tumor Activity in Pediatric Low-Grade Glioma; The Results of a Phase II Clinical Trial. Front. Pharmacol. 2018, 9, 00950. [Google Scholar] [CrossRef]
- André, N.; Abed, S.; Orbach, D.; Alla, C.A.; Padovani, L.; Pasquier, E.; Gentet, J.C.; Verschuur, A. Pilot study of a pediatric metronomic 4-drug regimen. Oncotarget 2011, 2, 960–965. [Google Scholar] [CrossRef][Green Version]
- Heng-Maillard, M.A.; Verschuur, A.; Aschero, A.; Dabadie, A.; Jouve, E.; Chastagner, P.; Leblond, P.; Aerts, I.; De Luca, B.; André, N. SFCE METRO-01 four-drug metronomic regimen phase II trial for pediatric extracranial tumor. Pediatr. Blood Cancer 2019, 66, e27693. [Google Scholar] [CrossRef]
- El kababri, M.; Benmiloud, S.; Cherkaoui, S.; El houdzi, J.; Maani, K.; Ansari, N.; Khoubila, N.; Kili, A.; El khorassani, M.; Madani, A.; et al. Metro-SMHOP 01: Metronomics combination with cyclophosphamide-etoposide and valproic acid for refractory and relapsing pediatric malignancies. Pediatr. Blood Cancer 2020, 67, 1–6. [Google Scholar] [CrossRef]
- Bisogno, G.; De Salvo, G.L.; Bergeron, C.; Gallego Melcón, S.; Merks, J.H.; Kelsey, A.; Martelli, H.; Minard-Colin, V.; Orbach, D.; Glosli, H.; et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 1566–1575. [Google Scholar] [CrossRef]
- Pramanik, R.; Agarwala, S.; Gupta, Y.K.; Thulkar, S.; Vishnubhatla, S.; Batra, A.; Dhawan, D.; Bakhshi, S. Metronomic chemotherapy vs best supportive care in progressive pediatric solid malignant tumors: A randomized clinical trial. JAMA Oncol. 2017, 3, 1222–1227. [Google Scholar] [CrossRef]
- Pramanik, R.; Agarwala, S.; Sreenivas, V.; Dhawan, D.; Bakhshi, S. Quality of life in paediatric solid tumours: A randomised study of metronomic chemotherapy versus placebo. BMJ Support. Palliat. Care 2021. [Google Scholar] [CrossRef]
- Andre, N.; Cointe, S.; Barlogis, V.; Arnaud, L.; Lacroix, R.; Pasquier, E.; Dignat-George, F.; Michel, G.; Sabatier, F. Maintenance chemotherapy in children with ALL exerts metronomic-like thrombospondin-1 associated anti-endothelial effect. Oncotarget 2015, 6, 23008–23014. [Google Scholar] [CrossRef]
- Vo, K.T.; Karski, E.E.; Nasholm, N.M.; Allen, S.; Hollinger, F.; Gustafson, W.C.; Long-Boyle, J.R.; Shiboski, S.; Matthay, K.K.; DuBois, S.G. Phase 1 study of sirolimus in combination with oral cyclophosphamide and topotecan in children and young adults with relapsed and refractory solid tumors. Oncotarget 2017, 8, 23851–23861. [Google Scholar] [CrossRef]
- Pramanik, R.; Tyagi, A.; Agarwala, S.; Vishnubhatla, S.; Dhawan, D.; Bakhshi, S. Evaluation of Vascular Endothelial Growth Factor (VEGF) and Thrombospondin-1 as Biomarkers of Metronomic Chemotherapy in Progressive Pediatric Solid Malignancies. Indian Pediatr. 2020, 57, 508–511. [Google Scholar] [CrossRef]
- Toh, Y.-M.; Li, T.-K. Mitoxantrone inhibits HIF-1α expression in a topoisomerase II-independent pathway. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5026–5037. [Google Scholar] [CrossRef]
- André, N.; Carré, M.; Pasquier, E. Metronomics: Towards personalized chemotherapy? Nat. Rev. Clin. Oncol. 2014, 11, 413–431. [Google Scholar] [CrossRef]
- Mariotto, A.B.; Enewold, L.; Zhao, J.; Zeruto, C.A.; Yabroff, K.R. Medical Care Costs Associated with Cancer Survivorship in the United States. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- André, N.; Orbach, D.; Pasquier, E. Metronomic Maintenance for High-Risk Pediatric Malignancies: One Size Will Not Fit All. Trends Cancer 2020, 6, 819–828. [Google Scholar] [CrossRef]
- Choi, D.K.; Helenowski, I.; Hijiya, N. Secondary malignancies in pediatric cancer survivors: Perspectives and review of the literature. Int. J. Cancer 2014, 135, 1764–1773. [Google Scholar] [CrossRef] [PubMed]


| Main Key Words | Secondary a Key Words |
|---|---|
| Metronomic | Preclinical study |
| Low dose | Cell culture |
| Chemotherapy | Animal model |
| Pediatric | Clinical study |
| Childhood | Randomized controlled trial |
| Cancer | Observational study |
| Tumor | Efficacy |
| Neoplasia | Toxicity |
| Angiogenesis | |
| Biomarkers |
| Disease | N° of Patients | Type of Study | Metronomic Regimen | Main Results | Reference |
|---|---|---|---|---|---|
| Low-grade glioma (pLGG) | 18 | Retrospective | Bevacizumab with or without irinotecan + vinblastine/vinorelbine | 2-year OS: 94% | Roux et al. [76] |
| Progressive or relapsed solid tumors | 74 | Retrospective | COMBAT I, COMBAT II, COMBAT IIS, COMBAT III | 43.1% (median: 15.4 months) was 2-year OS | Zapletalova et al. [77] |
| Rhabdoid tumor | 6 | Case series | Etoposide + cyclophosphamide + celecoxib + valproic acid | SD (death from bone marrow transplant-related infectious complications) | Carcamo B. and Francia G. [78] |
| Anaplastic ependymoma | Etoposide + cyclophosphamide + celecoxib + valproic acid | CR | |||
| Medulloblastoma | Etoposide + valproic acid | CR | |||
| Medulloblastoma | Temozolomide + cyclophosphamide + celecoxib + valproic acid | PD | |||
| Neuroblastoma | Etoposide + cyclophosphamide + sulindac/celecoxib | Relapse | |||
| Cervicomedullary tumor | Temozolomide + cyclophosphamide + valproic acid + celecoxib + bevacizumab | SD | |||
| Poor prognosis brain tumors | 8 | Pilot | Temozolomide 90 mg/m2/day for 42 days | Six patients responded to treatment | Sterba et al. [79] |
| Refractory solid tumors | 12 | Pilot, prospective | Vincristine, cyclophosphamide, and methotrexate | Disease stabilization occurred in 7 patients (58%) | Fousseyni et al. [80] |
| Recurrent or progressive solid tumor | 33 | Pilot | Celecoxib + vinorelbine or cyclophosphamide | Median time to progression was 8.5 weeks (range: 3 to 62.5 week) Four (13%) patients had a stable disease with durations of 28 to 76 weeks | Stempak et al. [81] |
| Refractory cancer | 7 | Pilot, prospective | Vincristine, cyclophosphamide, methotrexate | Mean duration treatment: 34 ± 31 weeks PR: 2 patients | Traore et al. [82] |
| Recurrent or progressive poor prognosis solid tumors | 20 | Feasibility trial | Four-drug regimen: thalidomide + celecoxib + alternating cyclophosphamide/VP16 every 21 days for 6 months | Eight patients completed the six-month therapy | Kieran et al. [83] |
| Relapsed or refractory solid tumors | 30 | Phase I | Topotecan + pazopanib | The recommended dose was topotecan 0.22 mg/m2/day and pazopanib PfOS 160 mg/m2/day In total, 10 patients (4 neuroblastoma, 3 osteosarcoma, 2 Ewing sarcoma/PNET, and 1 medulloblastoma) had stable disease with median duration of 6.4 months (1.7–45.1) The longest stable disease was 45 months | Manji et al. [84] |
| Relapsed or refractory solid tumors | 64 | Prospective | Celecoxib, cyclophosphamide, vinblastine, and methotrexate | Forty-nine patients (77%) had a favorable response (PR and SD) 1-year OS: 62.3% | Ali A.M. and El-Sayed M.I. [85] |
| High-grade glioma, neuroblastoma, desmoplastic small round cell tumor-DSRCT, alveolar rhabdomyosarcoma, ependymoma, melanoma | 13 | Phase II | Nivolumab + cyclophosphamide +/- radiotherapy at discretion of physician | None of patients had confirmed objective response Stable disease occurred in 5 subjects Median PFS = 1.7 months (95% CI: 1.3–3.4) Median OS = 3.4 months (95% CI: 2.2–13.5) | Pasqualini et al. [86] |
| Recurrent or progressive tumors | 101 | Phase II, prospective | Five drugs: cyclophosphamide, etoposide, thalidomide, celecoxib, and fenofibrate | Twenty-four patients (25%) completed 27 weeks therapy | Robison et al. [87] |
| Relapsed/refractory pediatric brain tumors | 29 | Phase II | Four drugs: celecoxib, vinblastine, cyclophosphamide, and methotrexate | Median number of cycles was 6.8 (range 1–12) Good response in LGG patients | Verschuur et al. [88] |
| Refractory or relapsing tumors | 16 | Pilot | Vinblastine, cyclophosphamide, methotrexate, and celecoxib | Disease stabilizations (25%) that lasted 24 weeks or more | André at al. [89] |
| Neuroblastoma, soft-tissue sarcoma, bone sarcoma, miscellaneous | 50 | Phase II, prospective | Vinblastine, cyclophosphamide/methotrexate, celecoxib | 1-year PFS = 6.8% 1-year OS = 55.3% | Heng-Maillard et al. [90] |
| Refractory/relapsing solid tumors, or advanced disease | 98 | Phase II, prospective | Cyclophosphamide + etoposide + valproic acid | 6-month OS = 40% (95% CI) 1-year OS = 22% 1-year PFS = 19% | El Kababri et al. [91] |
| Rhabdomyosarcoma at high-risk of relapse | 371 | Phase III | G1: vinorelbine and cyclophosphamide G2: placebo | In G1 group: 86.5% (95% CI 80.2–90.9) was 5-year overall survival 77.8% (70.8–83.4) was 5-year disease-free survival | Bisogno et al. [92] |
| Non-hematopoietic primarily extracranial solid tumor progressive after treatment with at least 2 lines of chemotherapy | 108 G1: 56 patients G2: 52 patients | Phase III | G1: thalidomide, celecoxib, and etoposide/cyclophosphamide G2: placebo | In the G1 group: 49 days (95% CI, 43–59 days) was the median PFS 85 days was the OS SD = 8 patients PR = 2 patients Good response in patients without a bone tumor | Pramanik et al. [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banchi, M.; Fini, E.; Crucitta, S.; Bocci, G. Metronomic Chemotherapy in Pediatric Oncology: From Preclinical Evidence to Clinical Studies. J. Clin. Med. 2022, 11, 6254. https://doi.org/10.3390/jcm11216254
Banchi M, Fini E, Crucitta S, Bocci G. Metronomic Chemotherapy in Pediatric Oncology: From Preclinical Evidence to Clinical Studies. Journal of Clinical Medicine. 2022; 11(21):6254. https://doi.org/10.3390/jcm11216254
Chicago/Turabian StyleBanchi, Marta, Elisabetta Fini, Stefania Crucitta, and Guido Bocci. 2022. "Metronomic Chemotherapy in Pediatric Oncology: From Preclinical Evidence to Clinical Studies" Journal of Clinical Medicine 11, no. 21: 6254. https://doi.org/10.3390/jcm11216254
APA StyleBanchi, M., Fini, E., Crucitta, S., & Bocci, G. (2022). Metronomic Chemotherapy in Pediatric Oncology: From Preclinical Evidence to Clinical Studies. Journal of Clinical Medicine, 11(21), 6254. https://doi.org/10.3390/jcm11216254