
Myeloid-Derived Suppressor Cells: New Insights into the Pathogenesis and Therapy of MDS



{kind=link}
Abstract
1. Introduction
1.1. Myeloid-Derived Suppressor Cells (MDSCs)
1.2. MDS Molecular Pathogenesis
2. Expansion and Significance of MDSCs Derived from Nonmalignant Clones in Patients with MDS
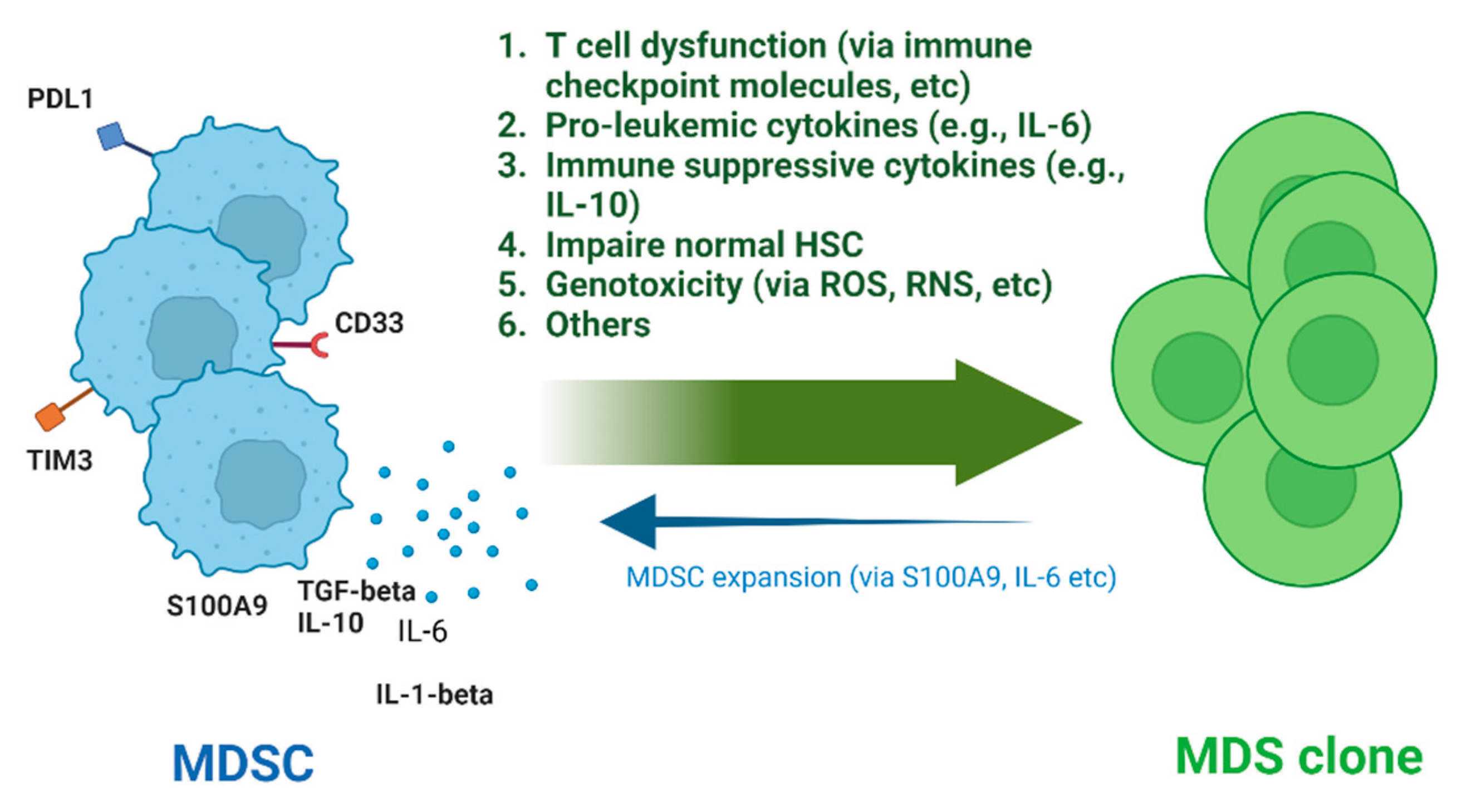
3. MDSCs as Inducers of Myelodysplasia
4. MDSCs and Immune Dysregulation in the MDS
5. MDSCs as Therapeutic Targets in MDS
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Zea, A.H.; Rodriguez, P.C.; Atkins, M.B.; Hernandez, C.; Signoretti, S.; Zabaleta, J.; McDermott, D.; Quiceno, D.; Youmans, A.; O’Neill, A.; et al. Arginase-Producing Myeloid Suppressor Cells in Renal Cell Carcinoma Patients: A Mechanism of Tumor Evasion. Cancer Res. 2005, 65, 3044–3048. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y.; Nagaraj, S.; Rosenbauer, A.; Muro-Cacho, C.; Sebti, S.M.; Gabrilovich, D.I. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005, 65, 9525–9535. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Ostrand-Rosenberg, S. Interleukin-13–regulated M2 Macrophages in Combination with Myeloid Suppressor Cells Block Immune Surveillance against Metastasis. Cancer Res. 2005, 65, 11743–11751. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.-H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-Derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Mandruzzato, S.; Brandau, S.; Britten, C.M.; Bronte, V.; Damuzzo, V.; Gouttefangeas, C.; Maurer, D.; Ottensmeier, C.; Van Der Burg, S.H.; Welters, M.J.P.; et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: Results from an interim study. Cancer Immunol. Immunother. 2016, 65, 161–169. [Google Scholar] [CrossRef]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5, eaay6017. [Google Scholar] [CrossRef]
- Khan, A.N.H.; Emmons, T.R.; Wong, J.T.; Alqassim, E.; Singel, K.L.; Mark, J.; Smith, B.E.; Tario, J.D.; Eng, K.H.; Moysich, K.B.; et al. Quantification of Early-Stage Myeloid-Derived Suppressor Cells in Cancer Requires Excluding Basophils. Cancer Immunol. Res. 2020, 8, 819–828. [Google Scholar] [CrossRef]
- Mastio, J.; Condamine, T.; Dominguez, G.; Kossenkov, A.V.; Donthireddy, L.; Veglia, F.; Lin, C.; Wang, F.; Fu, S.; Zhou, J.; et al. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J. Exp. Med. 2019, 216, 2150–2169. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Jia, Q.; Wang, L.; Fang, W.; Wang, Z.; Jiang, T.; Zhou, F.; Jin, Z.; Huang, J.; Zhou, L.; et al. Tumor-Induced erythroid precursor-differentiated myeloid cells mediate immunosuppression and curtail anti-PD-1/PD-L1 treatment efficacy. Cancer Cell 2022, 40, 674–693.e7. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Bozkus, C.C.; Elzey, B.D.; Crist, S.A.; Ellies, L.G.; Ratliff, T.L. Expression of Cationic Amino Acid Transporter 2 Is Required for Myeloid-Derived Suppressor Cell-Mediated Control of T Cell Immunity. J. Immunol. 2015, 195, 5237–5250. [Google Scholar] [CrossRef] [PubMed]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.J.F.H.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro-Oncology 2016, 18, 1253–1264. [Google Scholar] [CrossRef]
- Si, Y.; Merz, S.F.; Jansen, P.; Wang, B.; Bruderek, K.; Altenhoff, P.; Mattheis, S.; Lang, S.; Gunzer, M.; Klode, J.; et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci. Immunol. 2019, 4, eaaw9159. [Google Scholar] [CrossRef]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef]
- Lu, T.; Ramakrishnan, R.; Altiok, S.; Youn, J.-I.; Cheng, P.; Celis, E.; Pisarev, V.; Sherman, S.; Sporn, M.B.; Gabrilovich, D. Tumor-Infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J. Clin. Investig. 2011, 121, 4015–4029. [Google Scholar] [CrossRef]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Xing, Y.-F.; Hui-Min, D.; Tang, J.-X.; Dong, H.-M.; He, Y.-M.; Ruan, D.-Y.; Ye, Q.-J.; Cai, J.-R.; Ma, X.-K.; et al. Endoplasmic reticulum stress induced LOX-1+ CD15+ polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunolog 2018, 154, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- Zheng, W.; Song, H.; Luo, Z.; Wu, H.; Chen, L.; Wang, Y.; Cui, H.; Zhang, Y.; Wang, B.; Li, W.; et al. Acetylcholine ameliorates colitis by promoting IL-10 secretion of monocytic myeloid-derived suppressor cells through the nAChR/ERK pathway|PNAS. Proc. Natl. Acad. Sci. 2021, 118, e2017762118. [Google Scholar] [CrossRef]
- Gonzalez-Junca, A.; Driscoll, K.E.; Pellicciotta, I.; Du, S.; Lo, C.H.; Roy, R.; Parry, R.; Tenvooren, I.; Marquez, D.M.; Spitzer, M.H.; et al. Autocrine TGFβ Is a Survival Factor for Monocytes and Drives Immunosuppressive Lineage Commitment. Cancer Immunol. Res. 2019, 7, 306–320. [Google Scholar] [CrossRef]
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.C.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer 2013, 134, 2853–2864. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stangé, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; Veld, P.I.; De Baetselier, P.; et al. Different Tumor Microenvironments Contain Functionally Distinct Subsets of Macrophages Derived from Ly6C (high) Monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- Bizymi, N.; Bjelica, S.; Kittang, A.O.; Mojsilovic, S.; Velegraki, M.; Pontikoglou, C.; Roussel, M.; Ersvær, E.; Santibañez, J.F.; Lipoldová, M.; et al. Myeloid-Derived Suppressor Cells in Hematologic Diseases: Promising Biomarkers and Treatment Targets. HemaSphere 2019, 3, e168. [Google Scholar] [CrossRef] [PubMed]
- Gunes, E.G.; Rosen, S.T.; Querfeld, C. The role of myeloid-derived suppressor cells in hematologic malignancies. Curr. Opin. Oncol. 2020, 32, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Nimer, S.D. Myelodysplastic syndromes. Blood 2008, 111, 4841–4851. [Google Scholar] [CrossRef]
- Tefferi, A. Myelodysplastic Syndromes. N. Engl. J. Med. 2009, 361, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, L.; Chlon, T.; Starczynowski, D.T. Chronic immune response dysregulation in MDS pathogenesis. Blood 2018, 132, 1553–1560. [Google Scholar] [CrossRef]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef]
- Haase, D. Cytogenetic features in myelodysplastic syndromes. Ann. Hematol. 2008, 87, 515–526. [Google Scholar] [CrossRef]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstöcker, M.; Nösslinger, T.; Hildebrandt, B.; Kundgen, A.; Lübbert, M.; Kunzmann, R.; Giagounidis, A.A.N.; et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: Evidence from a core dataset of 2124 patients. Blood 2007, 110, 4385–4395. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.L.T.M.; Van Der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; De Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Kato, M.; Kawahata, R.; Matsubara, A.; Takita, J.; Shih, L.-Y.; Mori, H.; Koeffler, H.P.; Ogawa, S. A nonsense mutation of IDH1 in myelodysplastic syndromes and related disorders. Leukemia 2010, 25, 184–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; LaFave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef]
- Dolatshad, H.; Pellagatti, A.; Fernandez-Mercado, M.; Yip, B.H.; Malcovati, L.; Attwood, M.; Przychodzen, B.; Sahgal, N.; Kanapin, A.; E Lockstone, H.; et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 2014, 29, 1092–1103. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Graubert, T.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.N.; Jankowska, A.M.; Mahfouz, R.Z.; Dunbar, A.J.; Sugimoto, Y.; Hosono, N.; Hu, Z.; Cheriyath, V.; Vatolin, S.; Przychodzen, B.; et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia 2013, 27, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, X.; Cai, C.-L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.-C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef]
- Javier, J.; Hinge, A.; Bartram, J.; Xu, J.; Filippi, M.-D. Transforming growth factor-beta signaling modifies the hematopoietic acute inflammatory response to drive bone marrow failure. Haematologica 2022, 107, 1323–1334. [Google Scholar]
- Hinge, A.; Filippi, M.D. Deconstructing the Complexity of TGFbeta Signaling in Hematopoietic Stem Cells: Quiescence and Beyond. Curr. Stem. Cell Rep. 2016, 2, 388–397. [Google Scholar] [CrossRef]
- Dimicoli, S.; Wei, Y.; Bueso-Ramos, C.; Yang, H.; Dinardo, C.; Jia, Y.; Zheng, H.; Fang, Z.; Nguyen, M.; Pierce, S.; et al. Overexpression of the Toll-Like Receptor (TLR) Signaling Adaptor MYD88, but Lack of Genetic Mutation, in Myelodysplastic Syndromes. PLoS ONE 2013, 8, e71120. [Google Scholar] [CrossRef]
- Maratheftis, C.I.; Andreakos, E.; Moutsopoulos, H.M.; Voulgarelis, M. Toll-like Receptor-4 Is Up-Regulated in Hematopoietic Progenitor Cells and Contributes to Increased Apoptosis in Myelodysplastic Syndromes. Clin. Cancer Res. 2007, 13, 1154–1160. [Google Scholar] [CrossRef]
- Velegraki, M.; Papakonstanti, E.; Mavroudi, I.; Psyllaki, M.; Tsatsanis, C.; Oulas, A.; Iliopoulos, I.; Katonis, P.; Papadaki, H.A. Impaired clearance of apoptotic cells leads to HMGB1 release in the bone marrow of patients with myelodysplastic syndromes and induces TLR4-mediated cytokine production. Haematologica 2013, 98, 1206–1215. [Google Scholar] [CrossRef]
- Wei, Y.; Dimicoli, S.; Bueso-Ramos, C.; Chen, R.; Yang, H.; Neuberg, N.; Pierce, S.; Jia, Y.; Zheng, H.; Wang, H.; et al. Toll-Like receptor alterations in myelodysplastic syndrome. Leukemia 2013, 27, 1832–1840. [Google Scholar] [CrossRef]
- Winter, S.; Shoaie, S.; Kordasti, S.Y.; Platzbecker, U. Integrating the “Immunome” in the Stratification of Myelodysplastic Syndromes and Future Clinical Trial Design. J. Clin. Oncol. 2020, 38, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.-C.; Cesaro, A.; Chapeton-Montes, J.; Tardif, M.; Antoine, F.; Girard, D.; Tessier, P.A. S100A8 and S100A9 induce cytokine expression and regulate the NLRP3 inflammasome via ROS-Dependent activation of NF-κB1. PLoS ONE 2013, 8, e72138. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Gondek, L.P.; Tiu, R.; O’Keefe, C.L.; Sekeres, M.; Theil, K.S.; Maciejewski, J.P. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood 2008, 111, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, A.; Qian, Z.; Fernald, A.A.; Bergerson, R.J.; Wang, J.; Karrison, T.; Anastasi, J.; Bartom, E.T.; Sarver, A.L.; McNerney, M.E.; et al. Retroviral insertional mutagenesis identifies the del(5q) genes, CXXC5, TIFAB and ETF1, as well as the Wnt pathway, as potential targets in del (5q) myeloid neoplasms. Haematologica 2016, 101, e232–e236. [Google Scholar] [CrossRef]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; A Wells, R.; et al. Identification of miR-145 and miR-146a as mediators of the 5q– syndrome phenotype. Nat. Med. 2009, 16, 49–58. [Google Scholar] [CrossRef]
- E Varney, M.; Choi, K.; Bolanos, L.; Christie, S.; Fang, J.; Grimes, H.L.; Maciejewski, J.P.; Inoue, J.-I.; Starczynowski, D.T. Epistasis between TIFAB and miR-146a: Neighboring genes in del (5q) myelodysplastic syndrome. Leukemia 2016, 31, 491–495. [Google Scholar] [CrossRef]
- Varney, M.E.; Niederkorn, M.; Konno, H.; Matsumura, T.; Gohda, J.; Yoshida, N.; Akiyama, T.; Christie, S.; Fang, J.; Miller, D.; et al. Loss of Tifab, a del (5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor-TRAF6 signaling. J. Exp. Med. 2015, 212, 1967–1985. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56–70.e13. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 Proteins Regulate the Accumulation of Myeloid-Derived Suppressor Cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef] [PubMed]
- Tengesdal, I.W.; Dinarello, A.; Powers, N.E.; Burchill, M.A.; Joosten, L.A.B.; Marchetti, C.; Dinarello, C.A. Tumor NLRP3-Derived IL-1β Drives the IL-6/STAT3 Axis Resulting in Sustained MDSC-Mediated Immunosuppression. Front. Immunol. 2021, 12, 661323. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of Interleukin-1β Induces Gastric Inflammation and Cancer and Mobilizes Myeloid-Derived Suppressor Cells in Mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Weber, R.; Riester, Z.; Hüser, L.; Sticht, C.; Siebenmorgen, A.; Groth, C.; Hu, X.; Altevogt, P.; Utikal, J.S.; Umansky, V. IL-6 regulates CCR5 expression and immunosuppressive capacity of MDSC in murine melanoma. J. Immunother. Cancer 2020, 8, e000949. [Google Scholar] [CrossRef]
- Zhao, X.; Rong, L.; Zhao, X.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef]
- Jiang, H.-J.; Fu, R.; Wang, H.-Q.; Li, L.-J.; Qu, W.; Liang, Y.; Wang, G.-J.; Wang, X.-M.; Wu, Y.-H.; Liu, H.; et al. Increased circulating of myeloid-derived suppressor cells in myelodysplastic syndrome. Chin. Med. J. 2013, 126, 2582–2584. [Google Scholar]
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. OncoImmunology 2015, 5, e1062208. [Google Scholar] [CrossRef]
- Qi, X.; Jiang, H.; Liu, P.; Xie, N.; Fu, R.; Wang, H.; Liu, C.; Zhang, T.; Wang, H.; Shao, Z. Increased myeloid-derived suppressor cells in patients with myelodysplastic syndromes suppress CD8+ T lymphocyte function through the STAT3-ARG1 pathway. Leuk. Lymphoma 2020, 62, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Montes, P.; Bernal, M.; Campo, L.N.; González-Ramírez, A.R.; Jiménez, P.; Garrido, P.; Jurado, M.; Garrido, F.; Ruiz-Cabello, F.; Hernández, F. Tumor genetic alterations and features of the immune microenvironment drive myelodysplastic syndrome escape and progression. Cancer Immunol. Immunother. 2019, 68, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Behbehani, G.K.; Finck, R.; Samusik, N.; Sridhar, K.; Fantl, W.J.; Greenberg, P.L.; Nolan, G.P. Profiling myelodysplastic syndromes by mass cytometry demonstrates abnormal progenitor cell phenotype and differentiation. Cytom. Part B Clin. Cytom. 2020, 98, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ren, X.; Meng, F.; Guo, X.; Tao, J.; Zhang, W.; Liu, Z.; Fu, R.; Li, L. TIM3/CEACAM1 pathway involves in myeloid-derived suppressor cells induced CD8(+) T cells exhaustion and bone marrow inflammatory microenvironment in myelodysplastic syndrome. Immunology 2022. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Zhao, B.; A Basiorka, A.; Yang, J.; Cao, L.; Zhang, J.; List, A.; Ji, P. Age-Related inflammatory bone marrow microenvironment induces ineffective erythropoiesis mimicking del (5q) MDS. Leukemia 2017, 32, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.-A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Mei, Y.; Liu, Y.; Han, X.; Yang, J.; Ji, M.P. IL-6 Deficiency Reverses Leukemic Transformation in an MDS Mouse Model. Blood 2020, 136, 36. [Google Scholar] [CrossRef]
- Smith, A.D.; Lu, C.; Payne, D.; Paschall, A.V.; Klement, J.D.; Redd, P.S.; Ibrahim, M.L.; Yang, D.; Han, Q.; Liu, Z.; et al. Autocrine IL6-Mediated Activation of the STAT3-DNMT Axis Silences the TNFalpha-RIP1 Necroptosis Pathway to Sustain Survival and Accumulation of Myeloid-Derived Suppressor Cells. Cancer Res. 2020, 80, 3145–3156. [Google Scholar] [CrossRef]
- Cheng, P.; Eksioglu, E.A.; Chen, X.; Kandell, W.; Le Trinh, T.; Cen, L.; Qi, J.; Sallman, D.A.; Zhang, A.; Tu, N.; et al. S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leukemia 2019, 33, 2034–2046. [Google Scholar] [CrossRef]
- Ivy, K.S.; Ferrell, P.B. Disordered Immune Regulation and its Therapeutic Targeting in Myelodysplastic Syndromes. Curr. Hematol. Malign Rep. 2018, 13, 244–255. [Google Scholar] [CrossRef]
- Mann, M.; Brunner, A.M. Emerging immuno-oncology targets in Myelodysplastic Syndromes (MDS). Curr. Probl. Cancer 2021, 46, 100824. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Tao, J.; Fu, R.; Shao, Z. Myeloid-Derived suppressor cell cytokine secretion as prognostic factor in myelodysplastic syndromes. Innate Immun. 2020, 26, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Calvi, L.M.; Becker, M.W.; Liesveld, J.L. Flaming and fanning: The Spectrum of inflammatory influences in myelodysplastic syndromes. Blood Rev. 2019, 36, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Han, D.; Gao, S.; Zhang, W.; Yu, H.; Liu, P.; Fu, R.; Li, L.; Shao, Z. CD8(+) T cells exhaustion induced by myeloid-derived suppressor cells in myelodysplastic syndromes patients might be through TIM3/Gal-9 pathway. J. Cell Mol. Med. 2020, 24, 1046–1058. [Google Scholar] [CrossRef]
- Eksioglu, E.A.; Chen, X.; Heider, K.-H.; Rueter, B.; McGraw, K.L.; A Basiorka, A.; Wei, M.; Burnette, A.; Cheng, P.; Lancet, J.; et al. Novel therapeutic approach to improve hematopoiesis in low risk MDS by targeting MDSCs with the Fc-Engineered CD33 antibody BI 836858. Leukemia 2017, 31, 2172–2180. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Carraway, H.E.; Germing, U.; Wermke, M.; Zeidan, A.M.; Fu, E.; Rüter, B.; Burkard, U.; Osswald, A.; Foran, J.M. A phase I/II multicenter, open-label, dose escalation and randomized trial of BI 836858 in patients with low or intermediate-1 risk myelodysplastic syndrome. Haematologica 2022. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Bourgeois, E.; Lobe, I.; Braun, T.; Visentin, G.; Bourhis, J.-H.; Fenaux, P.; Chouaib, S.; Caignard, A. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia 2006, 20, 463–470. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Bai, F.; Painter, J.S.; Rollison, D.E.; Salih, H.R.; Krusch, M.; Zou, J.; Ku, E.; Zhong, B.; Boulware, D.; et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood 2007, 109, 4816–4824. [Google Scholar] [CrossRef]
- Carlsten, M.; Baumann, B.C.; Simonsson, M.; Jädersten, M.; Forsblom, A.-M.; Hammarstedt, C.; Bryceson, Y.T.; Ljunggren, H.-G.; Hellström-Lindberg, E.; Malmberg, K.-J. Reduced DNAM-1 expression on bone marrow NK cells associated with impaired killing of CD34+ blasts in myelodysplastic syndrome. Leukemia 2010, 24, 1607–1616. [Google Scholar] [CrossRef]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-Cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Chen, X.; Dalton, R.; Calescibetta, A.; So, T.; Gilvary, D.; Ward, G.; Smith, V.; Eckard, S.; Fox, J.A.; et al. Immunodepletion of MDSC by AMV564, a novel bivalent, bispecific CD33/CD3 T cell engager, ex vivo in MDS and melanoma. Mol. Ther. 2022, 30, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G. CD123 as a Therapeutic Target in the Treatment of Hematological Malignancies. Cancers 2019, 11, 1358. [Google Scholar] [CrossRef]
- Uckun, F.M.; Watts, J. CD123-Directed Bispecific Antibodies for Targeting MDS Clones and Immunosuppressive Myeloid-Derived Suppressor Cells (MDSC) in High-Risk Adult MDS Patients. Front. Aging 2021, 2, 757276. [Google Scholar] [CrossRef]
- Stevens, B.M.; Engel, K.L.; Gillen, A.E.; Culp-Hill, R.; Dalessandro, A.; Alper, S.; Pollyea, D.A.; Jordan, C.T. Unique Metabolic Vulnerabilities of Myelodysplastic Syndrome Stem Cells. Blood 2021, 138, 1511. [Google Scholar] [CrossRef]
- Uckun, F.M.; Lin, T.L.; Mims, A.S.; Patel, P.; Lee, C.; Shahidzadeh, A.; Shami, P.J.; Cull, E.; Cogle, C.R.; Watts, J. A Clinical Phase 1B Study of the CD3xCD123 Bispecific Antibody APVO436 in Patients with Relapsed/Refractory Acute Myeloid Leukemia or Myelodysplastic Syndrome. Cancers 2021, 13, 4113. [Google Scholar] [CrossRef]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-Derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Wei, S.; Mailloux, A.W.; Zhang, L.; Padron, E.; Sallman, D.; Lancet, J.E.; Tinsley, S.; Nardelli, L.A.; Pinilla-Ibarz, J.; et al. A Phase II Study to Determine the Safety and Efficacy of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase (IDO) Enzyme INCB024360 in Patients with Myelodysplastic Syndromes. Clin. Lymphoma Myeloma Leuk. 2018, 19, 157–161. [Google Scholar] [CrossRef]
- Chavakis, T.; Wielockx, B.; Hajishengallis, G. Inflammatory Modulation of Hematopoiesis: Linking Trained Immunity and Clonal Hematopoiesis with Chronic Disorders. Annu. Rev. Physiol. 2022, 84, 183–207. [Google Scholar] [CrossRef]
- Cook, E.K.; Luo, M.; Rauh, M.J. Clonal hematopoiesis and inflammation: Partners in leukemogenesis and comorbidity. Exp. Hematol. 2020, 83, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, J.J.; Starczynowski, D.T. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J. Exp. Med. 2021, 218, e20201544. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; Van Der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Khan, N.; Downey, J.; Sanz, J.; Kaufmann, E.; Blankenhaus, B.; Pacis, A.; Pernet, E.; Ahmed, E.; Cardoso, S.; Nijnik, A.; et al. M. tuberculosis Reprograms Hematopoietic Stem Cells to Limit Myelopoiesis and Impair Trained Immunity. Cell 2020, 183, 752–770.e22. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161.e12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Y.; Dutta, R.; Benard, B.; Zhao, F.; Yin, R.; Majeti, R. IL-6 blockade reverses bone marrow failure induced by human acute myeloid leukemia. Sci. Transl. Med. 2020, 12, eaax5104. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velegraki, M.; Stiff, A.; Papadaki, H.A.; Li, Z. Myeloid-Derived Suppressor Cells: New Insights into the Pathogenesis and Therapy of MDS. J. Clin. Med. 2022, 11, 4908. https://doi.org/10.3390/jcm11164908
Velegraki M, Stiff A, Papadaki HA, Li Z. Myeloid-Derived Suppressor Cells: New Insights into the Pathogenesis and Therapy of MDS. Journal of Clinical Medicine. 2022; 11(16):4908. https://doi.org/10.3390/jcm11164908
Chicago/Turabian StyleVelegraki, Maria, Andrew Stiff, Helen A. Papadaki, and Zihai Li. 2022. "Myeloid-Derived Suppressor Cells: New Insights into the Pathogenesis and Therapy of MDS" Journal of Clinical Medicine 11, no. 16: 4908. https://doi.org/10.3390/jcm11164908
APA StyleVelegraki, M., Stiff, A., Papadaki, H. A., & Li, Z. (2022). Myeloid-Derived Suppressor Cells: New Insights into the Pathogenesis and Therapy of MDS. Journal of Clinical Medicine, 11(16), 4908. https://doi.org/10.3390/jcm11164908