
Involvement of DNA Damage Response via the Ccndbp1–Atm–Chk2 Pathway in Mice with Dextran-Sodium-Sulfate-Induced Colitis

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. DSS-Induced Colitis
2.3. Histological Analysis
2.4. Terminal-Deoxynucleotidyl-Transferase-Mediated Dutp-Biotin Nick End Labeling (TUNEL) Staining
2.5. Whole-Transcriptome Sequencing
2.6. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.7. Statistical Analyses
3. Results
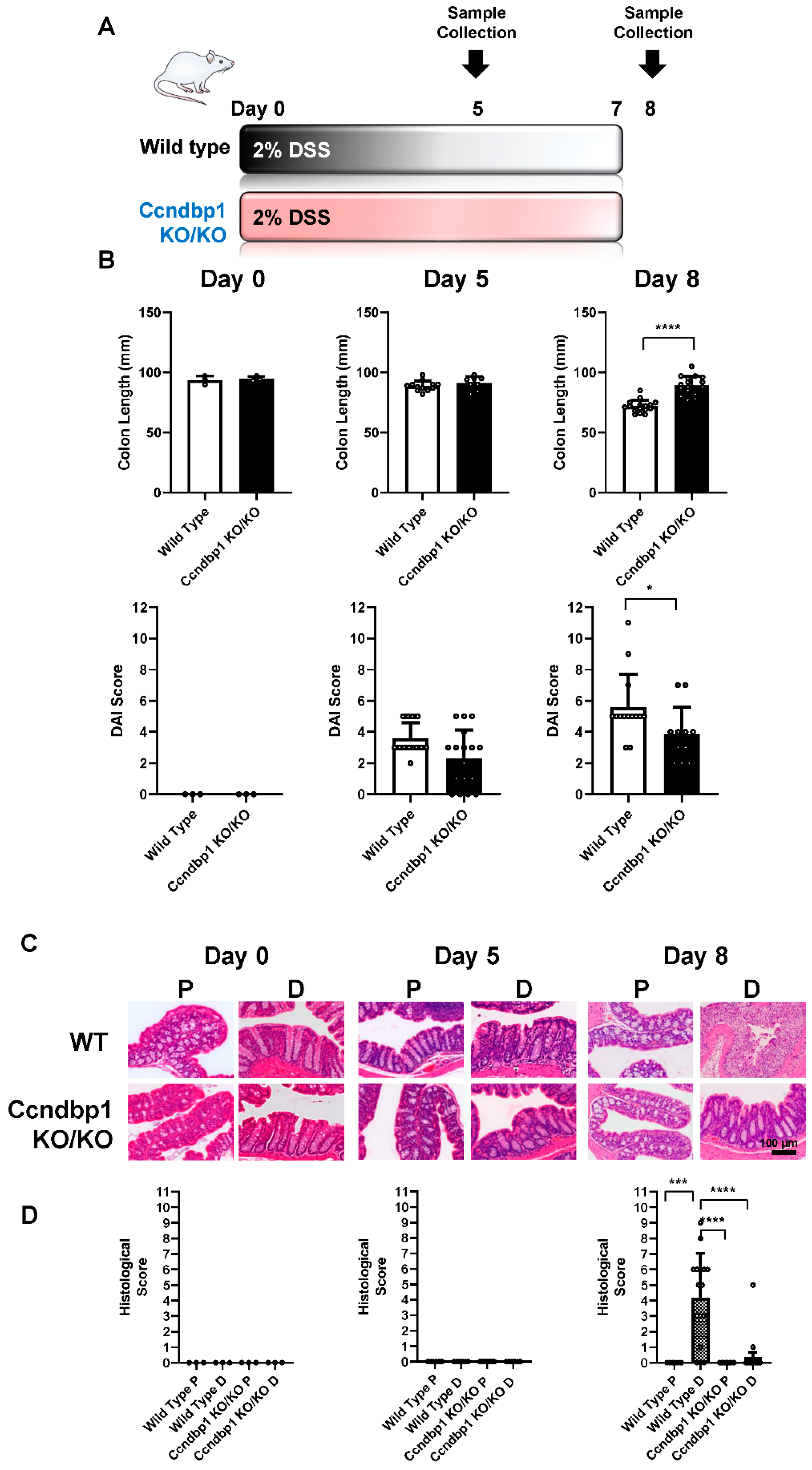
3.1. Effect of DSS-Induced Colitis on Ccndbp1-Knockout Mice
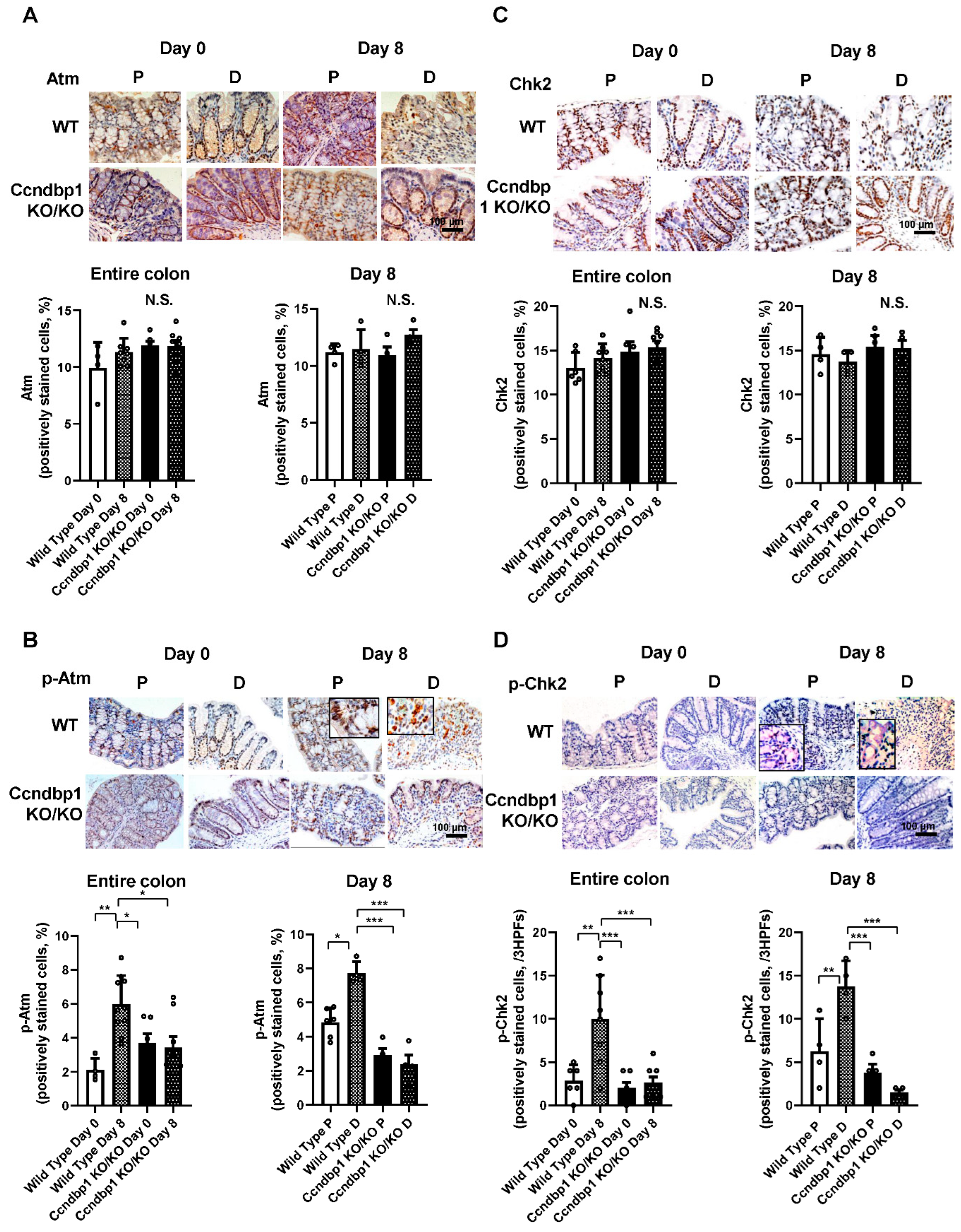
3.2. Effect of DSS-Induced Colitis on the Atm–Chk2 Pathway in the Colon
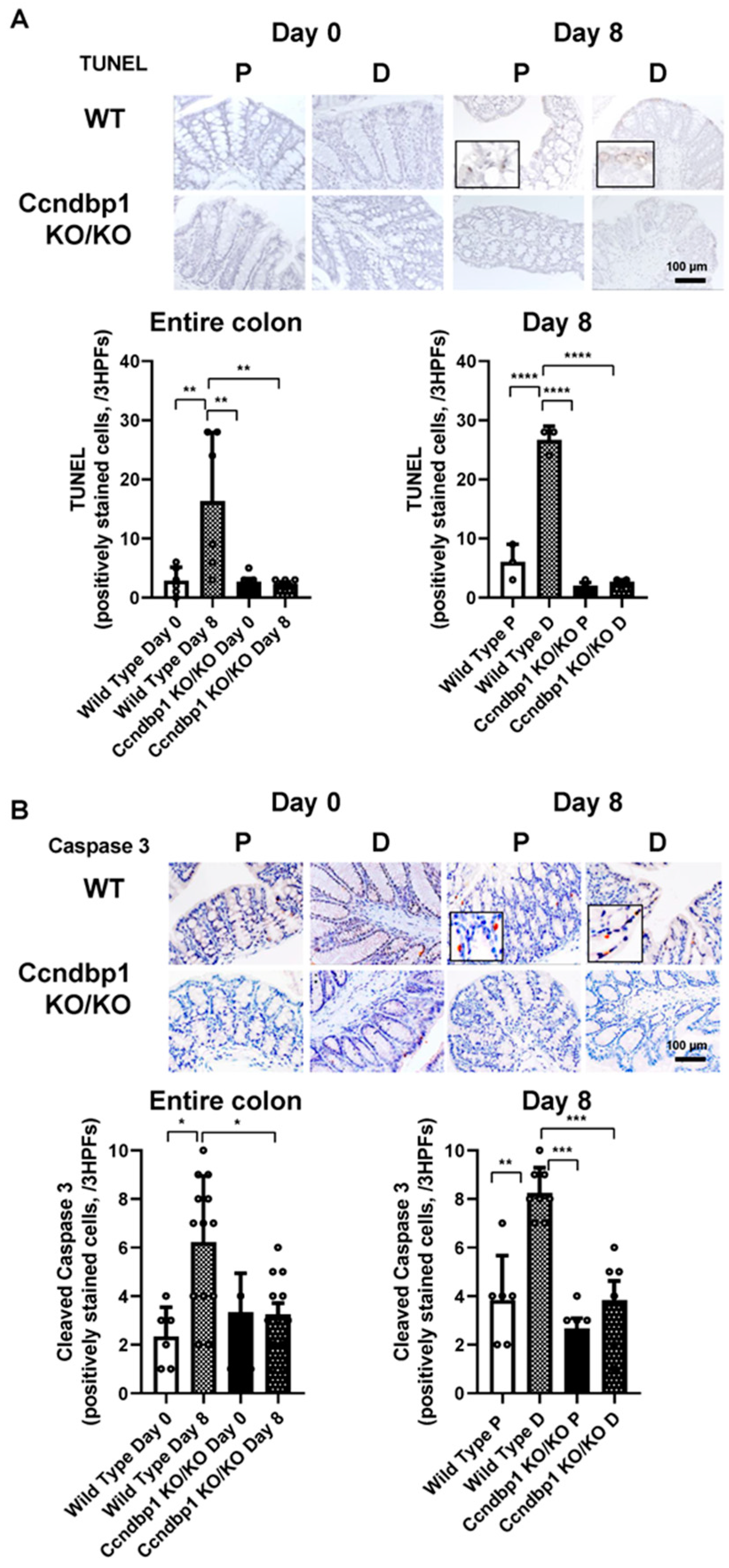
3.3. Effect of Ccndbp1 on DSS-Induced Apoptosis in the Colonic Mucosa
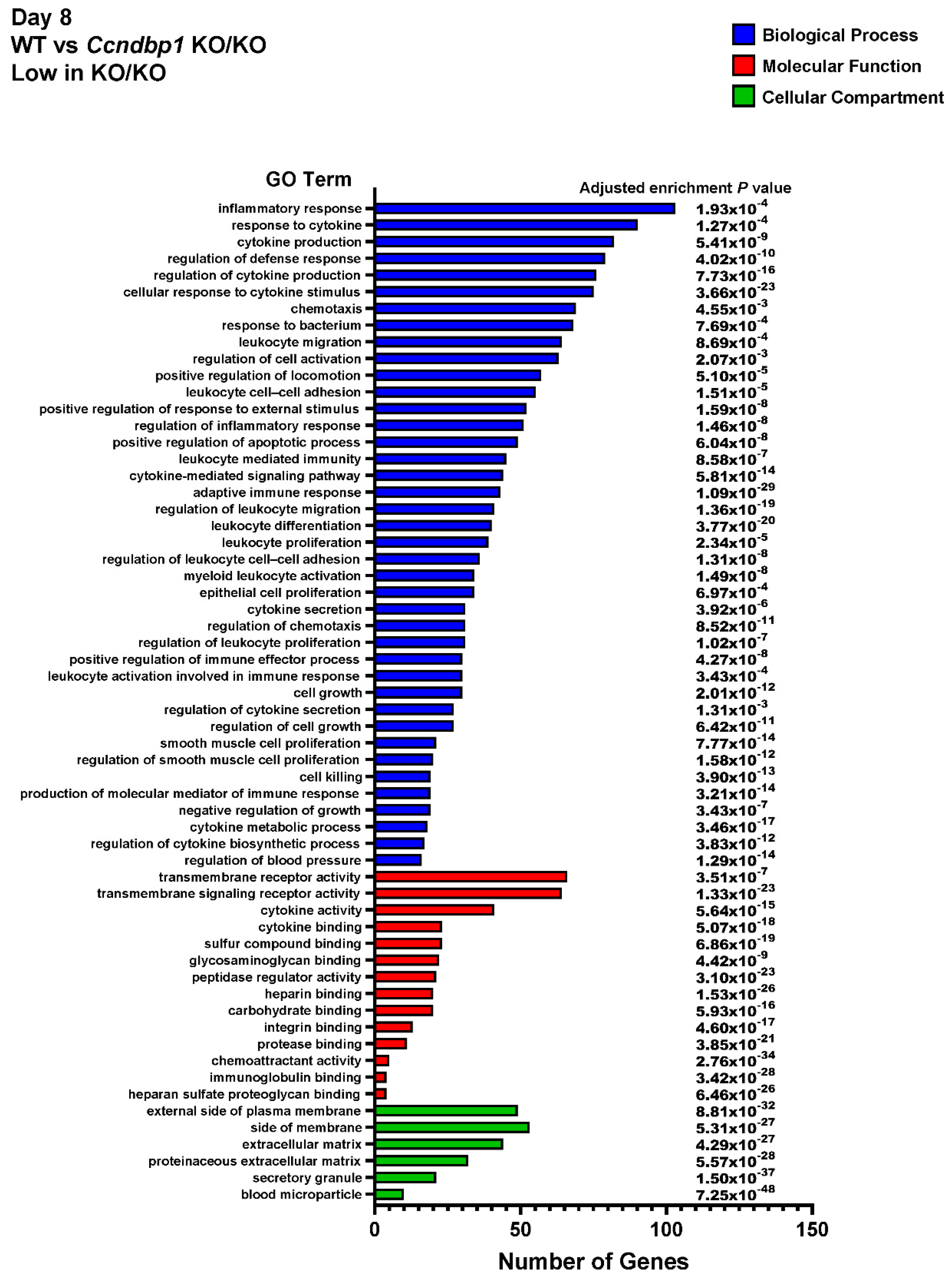
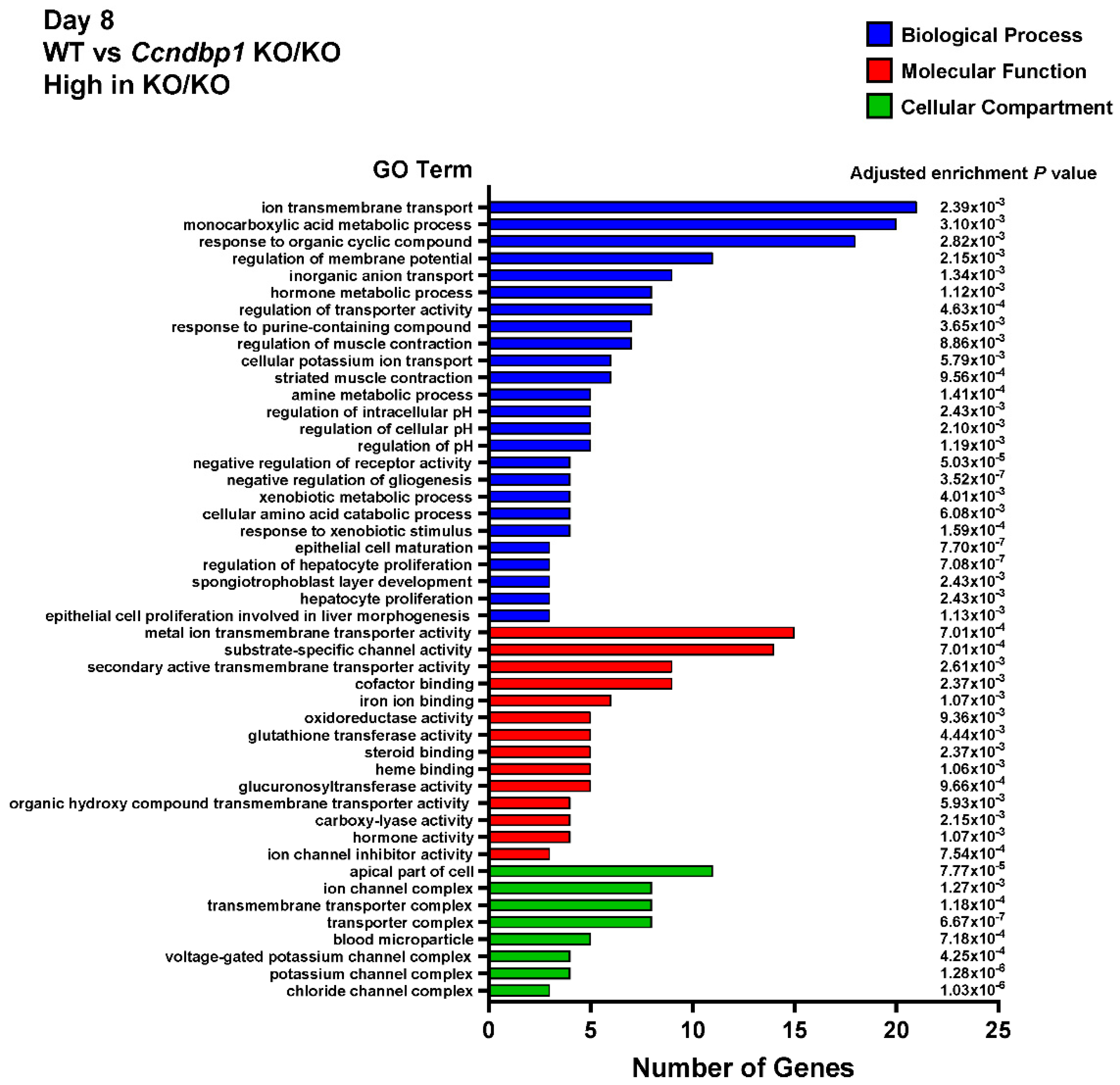
3.4. Effect of DSS on the Gene Expression in the Colon Tissue of Ccndbp1 KO/KO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef] [PubMed]
- Dirisina, R.; Katzman, R.B.; Goretsky, T.; Managlia, E.; Mittal, N.; Williams, D.B.; Qiu, W.; Yu, J.; Chandel, N.S.; Zhang, L.; et al. p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology 2011, 141, 1036–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Han, G.; Dou, Y.; Wang, Y.; Liu, G.; Wang, R.; Xiao, H.; Li, X.; Hou, C.; Shen, B.; et al. Opposite role of tumor necrosis factor receptors in dextran sulfate sodium-induced colitis in mice. PLoS ONE 2012, 7, e52924. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Han, T.; Hu, X.; Li, K.; Zhang, D.; Zhang, Y.; Li, J. Bifidobacterium infantis Maintains Genome Stability in Ulcerative Colitis via Regulating Anaphase-Promoting Complex Subunit 7. Front. Microbiol. 2021, 12, 761113. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Hirao, A.; Kong, Y.Y.; Matsuoka, S.; Wakeham, A.; Ruland, J.; Yoshida, H.; Liu, D.; Elledge, S.J.; Mak, T.W. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000, 287, 1824–1827. [Google Scholar] [CrossRef]
- Spehlmann, M.E.; Manthey, C.F.; Dann, S.M.; Hanson, E.; Sandhu, S.S.; Liu, L.Y.; Abdelmalak, F.K.; Diamanti, M.A.; Retzlaff, K.; Scheller, J.; et al. Trp53 deficiency protects against acute intestinal inflammation. J. Immunol. 2013, 191, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, Y.; Kamimura, K.; Ogawa, K.; Oda, C.; Tanaka, Y.; Horigome, R.; Ohtsuka, M.; Miura, H.; Fujisawa, K.; Yamamoto, N.; et al. Cyclin D1 Binding Protein 1 Responds to DNA Damage through the ATM-CHK2 Pathway. J. Clin. Med. 2022, 11, 851. [Google Scholar] [CrossRef] [PubMed]
- Seto, A.; Ikushima, H.; Suzuki, T.; Sato, Y.; Fukai, S.; Yuki, K.; Miyazawa, K.; Miyazono, K.; Ishitani, R.; Nureki, O. Crystallization and preliminary X-ray diffraction analysis of GCIP/HHM transcriptional regulator. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 21–24. [Google Scholar] [CrossRef]
- Ishii, R.; Isogaya, K.; Seto, A.; Koinuma, D.; Watanabe, Y.; Arisaka, F.; Yaguchi, S.; Ikushima, H.; Dohmae, N.; Miyazono, K.; et al. Structure of a dominant-negative helix-loop-helix transcriptional regulator suggests mechanisms of autoinhibition. EMBO J. 2012, 31, 2541–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.; Bao, Z.; Tabassam, F.; Ma, W.; Qiu, M.; Hua, S.; Liu, M. GCIP, a novel human grap2 and cyclin D interacting protein, regulates E2F-mediated transcriptional activity. J. Biol. Chem. 2000, 275, 20942–20948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terai, S.; Aoki, H.; Ashida, K.; Thorgeirsson, S.S. Human homologue of maid: A dominant inhibitory helix-loop-helix protein associated with liver-specific gene expression. Hepatology 2000, 32, 357–366. [Google Scholar] [CrossRef]
- Takami, T.; Terai, S.; Yokoyama, Y.; Tanimoto, H.; Tajima, K.; Uchida, K.; Yamasaki, T.; Sakaida, I.; Nishina, H.; Thorgeirsson, S.S.; et al. Human homologue of maid is a useful marker protein in hepatocarcinogenesis. Gastroenterology 2005, 128, 1369–1380. [Google Scholar] [CrossRef]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. 1993, 69, 238–249. [Google Scholar]
- Laroui, H.; Ingersoll, S.A.; Liu, H.C.; Baker, M.T.; Ayyadurai, S.; Charania, M.A.; Laroui, F.; Yan, Y.; Sitaraman, S.V.; Merlin, D. Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS ONE 2012, 7, e32084. [Google Scholar] [CrossRef]
- Vrekoussis, T.; Chaniotis, V.; Navrozoglou, I.; Dousias, V.; Pavlakis, K.; Stathopoulos, E.N.; Zoras, O. Image analysis of breast cancer immunohistochemistry-stained sections using ImageJ: An RGB-based model. Anticancer Res. 2009, 29, 4995–4998. [Google Scholar]
- Scabini, M.; Stellari, F.; Cappella, P.; Rizzitano, S.; Texido, G.; Pesenti, E. In vivo imaging of early stage apoptosis by measuring real-time caspase-3/7 activation. Apoptosis 2011, 16, 198–207. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Magro, F.; Rodrigues, A.; Vieira, A.I.; Portela, F.; Cremers, I.; Cotter, J.; Correia, L.; Duarte, M.A.; Tavares, M.L.; Lago, P.; et al. Review of the disease course among adult ulcerative colitis population-based longitudinal cohorts. Inflamm. Bowel Dis. 2012, 18, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horigome, R.; Kamimura, K.; Niwa, Y.; Ogawa, K.; Mizuno, K.-I.; Fujisawa, K.; Yamamoto, N.; Takami, T.; Sugano, T.; Sakamaki, A.; et al. Involvement of DNA Damage Response via the Ccndbp1–Atm–Chk2 Pathway in Mice with Dextran-Sodium-Sulfate-Induced Colitis. J. Clin. Med. 2022, 11, 3674. https://doi.org/10.3390/jcm11133674
Horigome R, Kamimura K, Niwa Y, Ogawa K, Mizuno K-I, Fujisawa K, Yamamoto N, Takami T, Sugano T, Sakamaki A, et al. Involvement of DNA Damage Response via the Ccndbp1–Atm–Chk2 Pathway in Mice with Dextran-Sodium-Sulfate-Induced Colitis. Journal of Clinical Medicine. 2022; 11(13):3674. https://doi.org/10.3390/jcm11133674
Chicago/Turabian StyleHorigome, Ryoko, Kenya Kamimura, Yusuke Niwa, Kohei Ogawa, Ken-Ichi Mizuno, Koichi Fujisawa, Naoki Yamamoto, Taro Takami, Tomoyuki Sugano, Akira Sakamaki, and et al. 2022. "Involvement of DNA Damage Response via the Ccndbp1–Atm–Chk2 Pathway in Mice with Dextran-Sodium-Sulfate-Induced Colitis" Journal of Clinical Medicine 11, no. 13: 3674. https://doi.org/10.3390/jcm11133674
APA StyleHorigome, R., Kamimura, K., Niwa, Y., Ogawa, K., Mizuno, K.-I., Fujisawa, K., Yamamoto, N., Takami, T., Sugano, T., Sakamaki, A., Kamimura, H., Takamura, M., & Terai, S. (2022). Involvement of DNA Damage Response via the Ccndbp1–Atm–Chk2 Pathway in Mice with Dextran-Sodium-Sulfate-Induced Colitis. Journal of Clinical Medicine, 11(13), 3674. https://doi.org/10.3390/jcm11133674