
Lipoprotein(a): Insights for the Practicing Clinician


Abstract
:1. Introduction
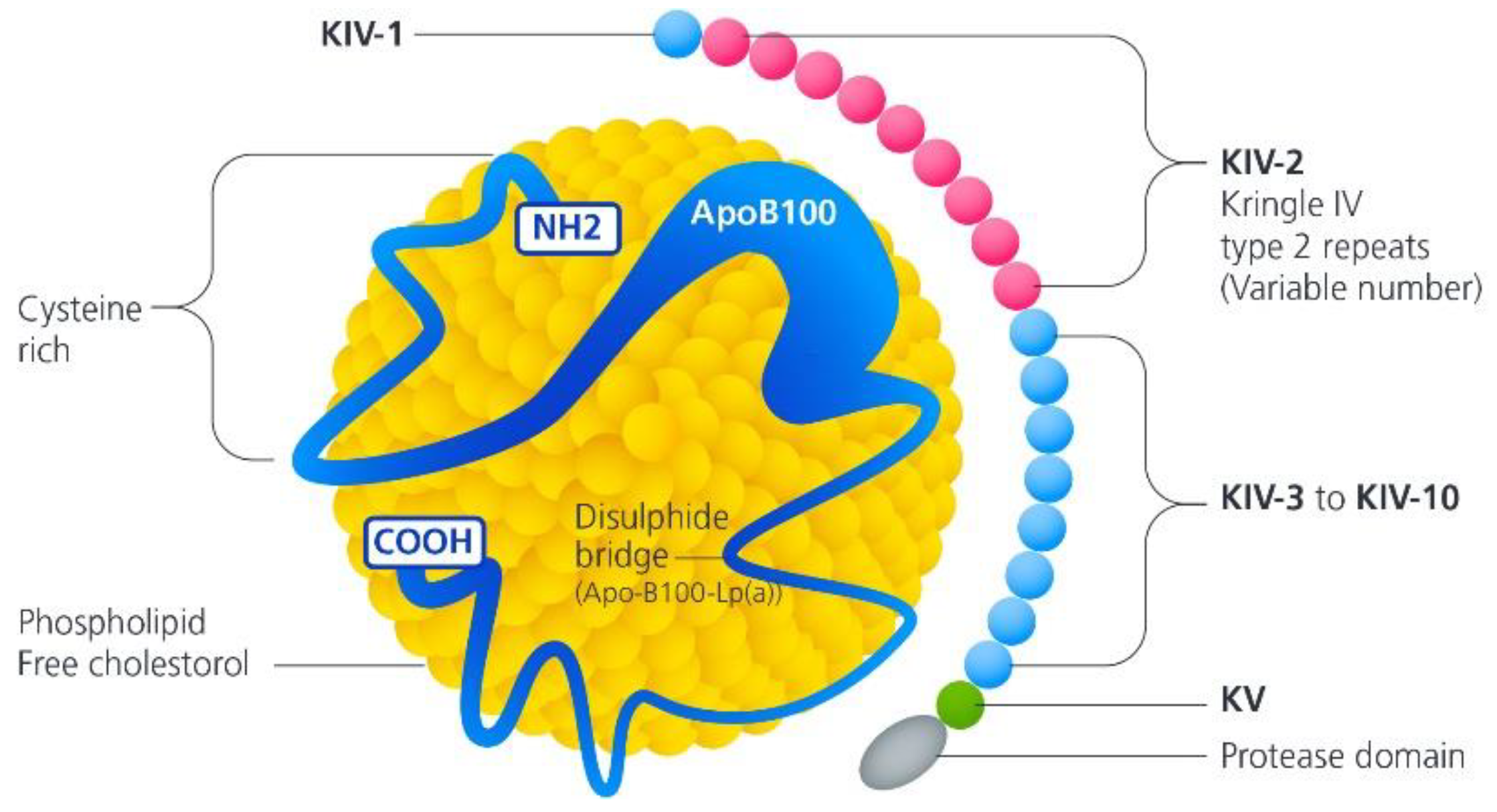
2. Lipoprotein(a) Genetic Inheritance and Molecular Structure
3. Measurement of Lipoprotein(a)
- 32–90 nmol/L—minor risk of CV disease;
- 90–200 nmol/L—moderate risk;
- 200–400 nmol/L—high risk;
- >400 nmol/L—very high risk.
4. Lipoprotein(a) and Cardiovascular Disease
5. Potential Thrombogenic Role for Lp(a)
6. Lp(a) and Universal Testing
7. Therapeutic Developments
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Apo(a) | Apolipoprotein (a) |
| AVS | Aortic Valve Stenosis |
| CHD | Coronary Heart Disease |
| CV | Cardiovascular |
| CVA | Cerebrovascular Accident |
| LDL-C | Low Density Lipoprotein Cholesterol |
| Lp(a) | Lipoprotein a |
| MI | Myocardial Infarction |
References
- Kostner, K.M.; Kostner, G.M. Lipoprotein (a): A historical appraisal. J. Lipid Res. 2017, 58, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerging Risk Factors Collaboration; Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009, 302, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguib, Y.; Suwaidi, J.A. The Copenhagen City Heart Study (Østerbroundersøgelsen). Glob. Cardiol. Sci. Pract. 2015, 2015, 33. [Google Scholar] [CrossRef]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bossé, Y.; Dumesnil, J.G.; Tsimikas, S. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavuranakis, M.A.; Jones, S.R.; Cardoso, R.; Gerstenblith, G.; Leucker, T.M. The role of Lipoprotein(a) in cardiovascular disease: Current concepts and future perspectives. Hell. J. Cardiol. 2020, 61, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Miksenas, H.; Januzzi, J.L.; Natarajan, P. Lipoprotein(a) and Cardiovascular Diseases. JAMA 2021, 326, 352. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Soffer, G.; Ginsberg, H.N.; Berglund, L.; Duell, P.B.; Heffron, S.P.; Kamstrup, P.R. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement from the American Heart Association. Atheroscler. Thromb. Vasc. Biol. 2021, 42, e48–e60. [Google Scholar] [CrossRef]
- Kamstrup, P.R. Lipoprotein(a) and Cardiovascular Disease. Clin. Chem. 2021, 67, 154–166. [Google Scholar] [CrossRef]
- Cegla, J.; Neely, R.D.G.; France, M.; Ferns, G.; Byrne, C.D.; Halcox, J. HEART UK consensus statement on Lipoprotein(a): A call to action. Atherosclerosis 2019, 291, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein (a): Truly a direct prothrombotic factor in cardiovascular disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Cegla, J.; France, M.; Marcovina, S.M.; Neely, R.D.G. Lp(a): When and how to measure it. Ann. Clin. Biochem. Int. J. Lab. Med. 2021, 58, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Trinder, M.; Uddin, M.M.; Finneran, P.; Aragam, K.G.; Natarajan, P. Clinical Utility of Lipoprotein(a) and LPA Genetic Risk Score in Risk Prediction of Incident Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2021, 6, 287. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.; Koskinas, K.C.; Casula, M.; Badimon, L.; Patel, R.S. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2019, 41, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Yeboah, J. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.J.; Thanassoulis, G.; Anderson, T.J.; Barry, A.R.; Couture, P.; Dayan, N.; Wray, W. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can. J. Cardiol. 2021, 37, 1129–1150. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) Concentrations and Incident Atherosclerotic Cardiovascular Disease. Atheroscler. Thromb. Vasc. Biol. 2021, 41, 465–474. [Google Scholar]
- Pare, G.; Caku, A.; McQueen, M.; Anand, S.S.; Enas, E.; Clarke, R. Lipoprotein(a) and the Risk of Myocardial Infarction Among 7 Ethnic Groups. Circulation 2019, 139, 1472–1482. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Rider, D.A.; Yavari, A.; Lindholm, M.W.; Campion, G.V.; Nissen, S.E. Treatment and prevention of lipoprotein(a)-mediated cardiovascular disease: The emerging potential of RNA interference therapeutics. Cardiovasc. Res. 2022, 118, 1218–1231. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Yeang, C.; Chan, K.L.; Pibarot, P.; Tsimikas, S. Association of Mild to Moderate Aortic Valve Stenosis Progression with Higher Lipoprotein(a) and Oxidized Phospholipid Levels. JAMA Cardiol. 2018, 3, 1212. [Google Scholar] [CrossRef]
- Labudovic, D.; Kostovska, I.; Trajkovska, K.T.; Cekovska, S.; Kavrakova, J.B.; Topuzovska, S. Lipoprotein (a)—Link between Atherogenesis and Thrombosis. Prague Med. Rep. 2019, 120, 39–51. [Google Scholar] [CrossRef]
- Undas, A.; Stepien, E.; Tracz, W.; Szczeklik, A. Lipoprotein (a) as a modifier of fibrin clot permeability and susceptibility to lysis. J. Thromb. Haemost. 2006, 4, 973–975. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Thrombogenicity of Lipoprotein A: Laboratory Study Defining the Prothrombotic Effects of Lipoprotein A; Identifier: NCT05330819; National Library of Medicine: Bethesda, MD, USA, 2022.
- Huang, Z.; Shui, X.; Ling, Y.; Zhou, L.; Shi, W.; Luo, Y.; Liu, J. Serum lipoprotein(a) and risk of periprocedural myocardial injury in patients undergoing percutaneous coronary intervention. Clin. Cardiol. 2021, 44, 176–185. [Google Scholar] [CrossRef]
- Wehinger, A.; Kastrati, A.; Elezi, S. Lipoprotein (a) and Coronary Thrombosis and Restenosis after Stent Placement. JACC 1999, 33, 1005–1012. [Google Scholar] [CrossRef]
- Niccoli, G.; Chin, D.; Scalone, G.; Panebianco, M.; Abbolito, S.; Cosentino, N.; De Biase, L. Data on the lipoprotein (a), coronary atherosclerotic burden and vulnerable plaque phenotype in angiographic obstructive coronary artery disease. Data Brief. 2016, 7, 1409–1412. [Google Scholar] [CrossRef]
- Wohlfahrt, P.; Jenca, D.; Melenovsky, V.; Franeková, J.; Jabor, A.; Šramko, M.; Kautzner, J. Very low lipoprotein(a) and increased mortality risk after myocardial infarction. Eur. J. Intern. Med. 2021, 91, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Furlan, L.; Azcarate, P.; Redberg, R.F. Lipoprotein(a) Measurement in Clinical Practice—Reply. JAMA Intern. Med. 2021, 181, 1138. [Google Scholar] [CrossRef]
- McNeil, J.J.; Nelson, M.R.; Woods, R.L.; Lockery, J.E.; Wolfe, R.; Reid, C.M.; Murray, A.M. Effect of Aspirin on All-Cause Mortality in the Healthy Elderly. NEJM 2018, 379, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.I.; Shiffman, D.; Zee, R.Y.; Louie, J.Z.; Luke, M.M.; Rowland, C.M.; Ridker, P.M. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis 2009, 203, 371–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER trial (justification for the use of statins in prevention: An intervention trial evaluating rosuvastatin). Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Handhle, A.; Viljoen, A.; Wierzbicki, A.S. Elevated Lipoporotein(a): Background, Current Insights and Future Potential Therapies. Vasc. Health Risk Manag. 2021, 17, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.; Nissen, S.E. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Greco, M.F.; Ferri, N.; Corsini, A. Lipoprotein (a) and PCSK9 inhibition: Clinical evidence. Eur. Heart J. Suppl. 2020, 18, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, V.; Chemello, K.; Hollstein, T.; Hong-Fong, C.C.; Schumann, F.; Grenkowitz, T.; Lambert, G. The size of apolipoprotein (a) is an independent determinant of the reduction in lipoprotein (a) induced by PCSK9 inhibitors. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Warden, B.; Minnier, J.; Watts, G.F.; Fazio, S.; Shapiro, M.D. Impact of PCSK9 inhibitors on plasma lipoprotein(a) concentrations with or without a background of niacin therapy. J. Clin. Lipidol. 2019, 13, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Kastelein, J.J. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Witztum, J.L. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Assessing the Impact of Lipoprotein(a) Lowering with TQJ230 on Major Cardiovascular Events in Patients with CVD (Lp(a)HORIZON); Identifier: NCT04023552; National Library of Medicine: Bethesda, MD, USA, 2019.

{kind=link}
| Epidemiological Studies | Patient Cohort | Population | Outcome | Results |
|---|---|---|---|---|
| The Copenhagen City Heart study | 10,855 | General population | Registry-based CV outcomes | Increased risk of MI and AVS |
| The Copenhagen General Population study | 66,877 | General population | Incidence of AVS | 3-fold increased risk of AVS with Lp(a) > 90 mg/dL |
| Danesh J et al.: meta-analysis of 27 prospective studies | 5436 | General population | Incidence of CHD | Increased incidence of CHD |
| Erqou et al.: Lipoprotein(a) concentration and the risk of coronary disease, a meta-analysis of 36 prospective studies | 126,634 | General population | Incidence of CHD and CVA | Increased association of Lp(a) with CHD and CVA |
| Pare et al.: Lipoprotein(a) levels and the risk of MI among 7 ethnic groups, INTERHEART study | 12,943 | General population | Incidence of MI | Increased risk of MI |
| Drug/Intervention | Lp(a) Level Reduction | CV Risk Reduction |
|---|---|---|
| Aspirin | N/A | Yes |
| Statin therapy | Conflicting results suggesting potential increase in Lp(a) concentration | Yes |
| Lipoprotein apheresis | >50% | Yes |
| Niacin | 20–25% | No |
| Bempedoic acid | No | Yes |
| Monoclonal antibodies to PCSK9 inhibitors | 20–30% | Yes |
| Inclisiran | 20% | No |
| Antisense oligonucleotides | 80% | No (trials are ongoing) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telyuk, P.; Austin, D.; Luvai, A.; Zaman, A. Lipoprotein(a): Insights for the Practicing Clinician. J. Clin. Med. 2022, 11, 3673. https://doi.org/10.3390/jcm11133673
Telyuk P, Austin D, Luvai A, Zaman A. Lipoprotein(a): Insights for the Practicing Clinician. Journal of Clinical Medicine. 2022; 11(13):3673. https://doi.org/10.3390/jcm11133673
Chicago/Turabian StyleTelyuk, Pyotr, David Austin, Ahai Luvai, and Azfar Zaman. 2022. "Lipoprotein(a): Insights for the Practicing Clinician" Journal of Clinical Medicine 11, no. 13: 3673. https://doi.org/10.3390/jcm11133673
APA StyleTelyuk, P., Austin, D., Luvai, A., & Zaman, A. (2022). Lipoprotein(a): Insights for the Practicing Clinician. Journal of Clinical Medicine, 11(13), 3673. https://doi.org/10.3390/jcm11133673