
Morusin Enhances Temozolomide Efficiency in GBM by Inducing Cytoplasmic Vacuolization and Endoplasmic Reticulum Stress

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Cell Viability Assay
2.3. Real-Time Quantitative PCR
2.4. Western Blot
2.5. Scanning Electron Microscope (SEM)
2.6. Cell Cycle Assay
2.7. RNA-Sequencing Analysis
2.8. Generation of Orthotopic Xenograft Models
2.9. Immunohistochemical (IHC) Staining
2.10. Statistical Analysis
3. Results
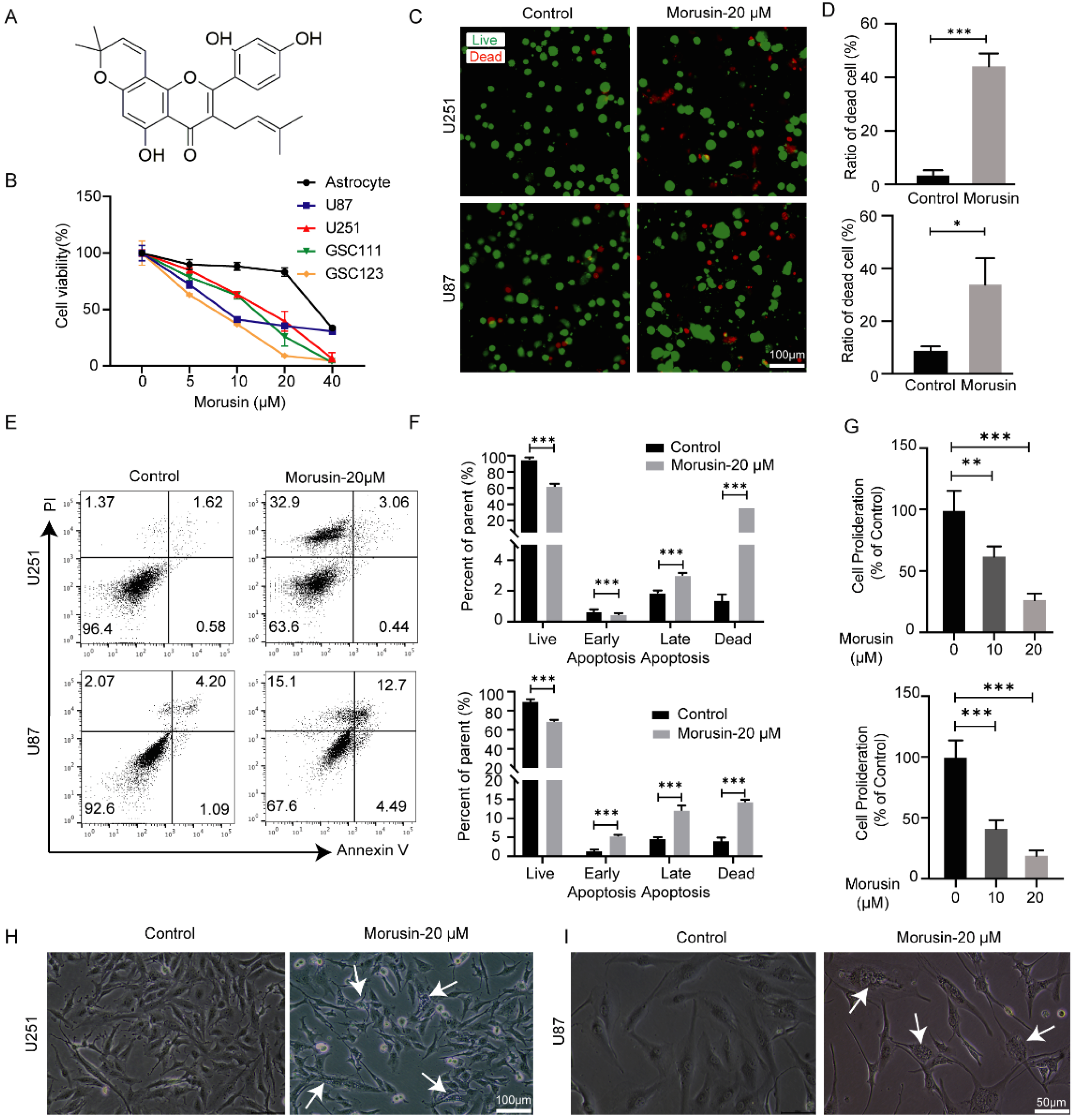
3.1. Selective Cytotoxicity of Morusin toward GBM Cells and GSC Cells
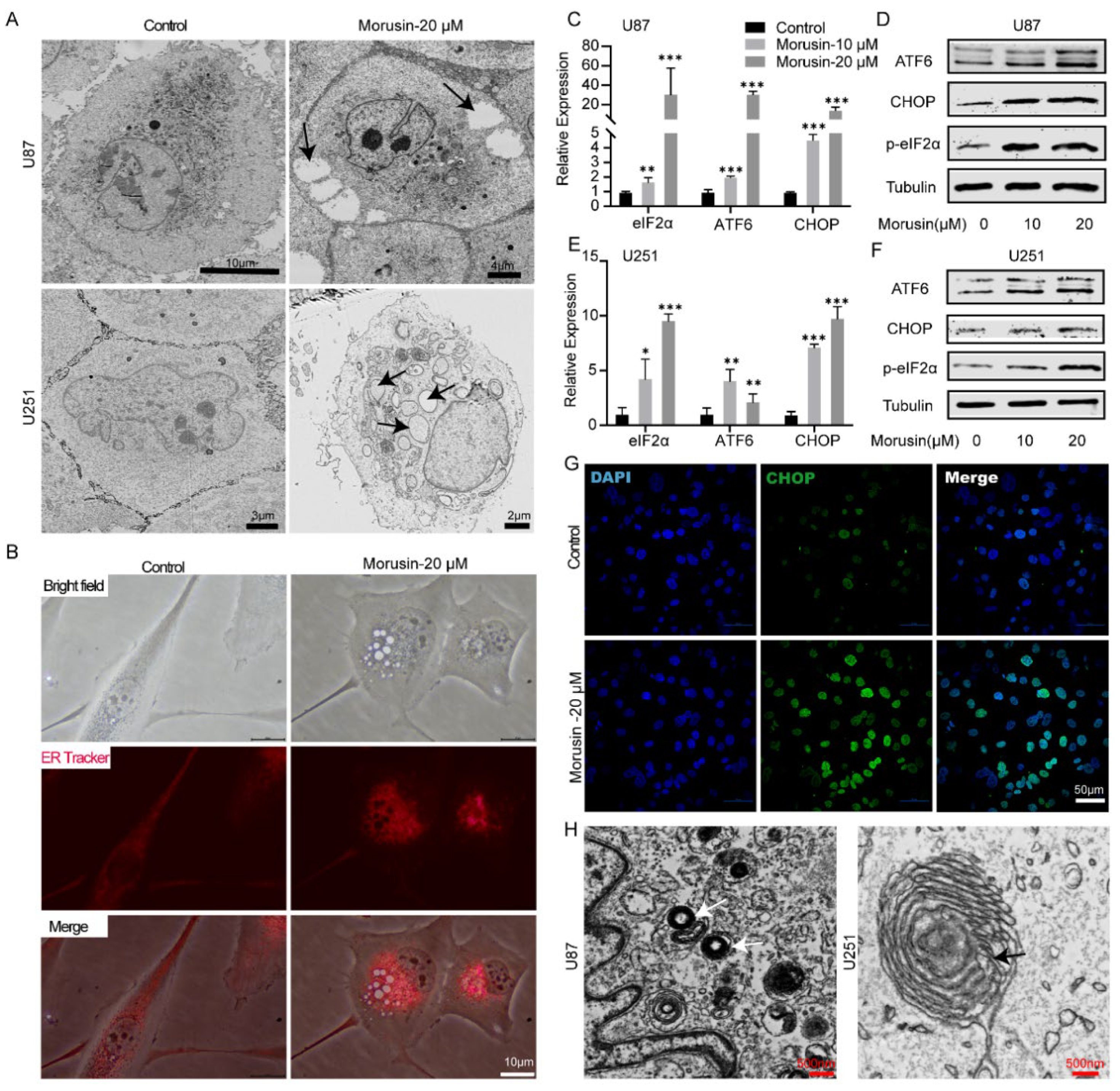
3.2. Morusin-Induced Cytoplasmic Vacuolation and Activated ER Stress in Human GBM Cells
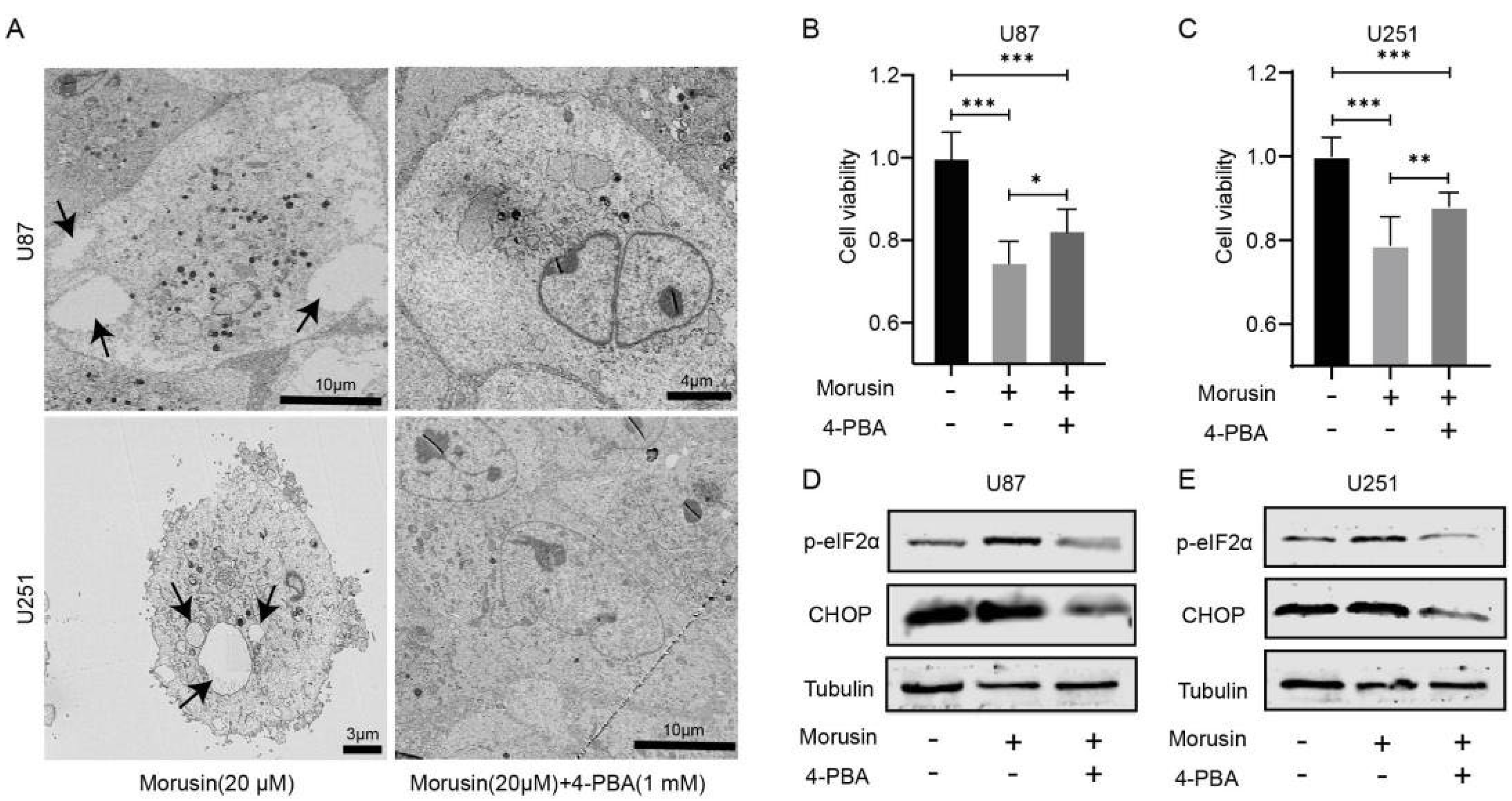
3.3. 4-PBA Rescued the Cytotoxicity of Morusin in GBM Cells
3.4. Morusin Caused Cell Cycle Arrest and Inhibited Cell Proliferation through Akt–mTOR–p70S6K Pathway
3.5. RNA-seq Analysis Revealed the Dysregulation of ERs and Cell Cycle Pathways in GBM Cells after Morusin Treatment
3.6. Combination of Morusin and Temozolomide Remarkably Induced ER Stress and Exhibited an Anti-GBM Effect Both In Vitro and In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, C.; Li, X.; Yang, J.; Yuan, D.; Zhou, Y.; Zhang, Y.; Shi, G.; Zhang, R.; Liu, J.; Fu, P.; et al. PPFIBP1 induces glioma cell migration and invasion through FAK/Src/JNK signaling pathway. Cell Death Dis. 2021, 12, 827. [Google Scholar] [CrossRef] [PubMed]
- Tivnan, A.; McDonald, K.L. Current Progress for the Use of miRNAs in Glioblastoma Treatment. Mol. Neurobiol. 2013, 48, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Fuchs, S.Y.; Koumenis, C. The Cell Biology of the Unfolded Protein Response. Gastroenterology 2011, 141, 38–41.e2. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Ohoka, N.; Nagai, K.; Shibata, N.; Hattori, T.; Nara, H.; Cho, N.; Naito, M. SNIPER(TACC3) induces cytoplasmic vacuolization and sensitizes cancer cells to Bortezomib. Cancer Sci. 2017, 108, 1032–1041. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival fol-lowing endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef]
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef]
- Gargiulo, G.; Cesaroni, M.; Serresi, M.; de Vries, N.; Hulsman, D.; Bruggeman, S.W.; van Lohuizen, M. In vivo RNAi screen for BMI1 targets identifies TGF-beta/BMP-ER stress pathways as key regulators of neural- and malignant glioma-stem cell homeostasis. Cancer Cell 2013, 23, 660–676. [Google Scholar] [CrossRef] [PubMed]
- Auf, G.; Jabouille, A.; Guérit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1α is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef] [PubMed]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [PubMed]
- Zoofishan, Z.; Hohmann, J.; Hunyadi, A. Phenolic antioxidants of Morus nigra roots, and antitumor potential of morusin. Phytochem. Rev. 2018, 17, 1031–1045. [Google Scholar] [CrossRef]
- Lee, J.C.; Won, S.J.; Chao, C.L.; Wu, F.L.; Liu, H.S.; Ling, P.; Su, C.L. Morusin induces apoptosis and suppresses NF-kappaB activity in human colorectal cancer HT-29 cells. Biochem. Biophys. Res. Commun. 2008, 372, 236–242. [Google Scholar] [CrossRef]
- Wang, L.; Guo, H.; Yang, L.; Dong, L.; Lin, C.; Zhang, J.; Wang, X. Morusin inhibits human cervical cancer stem cell growth and migration through attenuation of NF-kappaB activity and apoptosis induction. Mol. Cell Biochem. 2013, 379, 7–18. [Google Scholar] [CrossRef]
- Lim, S.-L.; Park, S.-Y.; Kang, S.; Park, D.; Kim, S.-H.; Um, J.-Y.; Jang, H.-J.; Lee, J.-H.; Jeong, C.-H.; Jang, J.-H.; et al. Morusin induces cell death through inactivating STAT3 signaling in prostate cancer cells. Am. J. Cancer Res. 2014, 5, 289–299. [Google Scholar]
- Lin, W.L.; Lai, D.Y.; Lee, Y.J.; Chen, N.F.; Tseng, T.H. Antitumor progression potential of morusin suppressing STAT3 and NFkappaB in human hepatoma SK-Hep1 cells. Toxicol. Lett. 2015, 232, 490–498. [Google Scholar] [CrossRef]
- Wan, L.Z.; Ma, B.; Zhang, Y.Q. Preparation of morusin from Ramulus mori and its effects on mice with transplanted H22 hepatocarcinoma. Biofactors 2014, 40, 636–645. [Google Scholar] [CrossRef]
- Li, H.; Wang, Q.; Dong, L.; Liu, C.; Sun, Z.; Gao, L.; Wang, X. Morusin suppresses breast cancer cell growth in vitro and in vivo through C/EBPbeta and PPARgamma medi-ated lipoapoptosis. J. Exp. Clin. Cancer Res. 2015, 34, 137. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, C.; Yang, L.; Dong, L.; Wang, L.; Wang, Q.; Li, H.; Zhang, J.; Lin, P.; Wang, X. Morusin inhibits glioblastoma stem cell growth in vitro and in vivo through stemness attenuation, adipocyte transdifferentiation, and apoptosis induction. Mol. Carcinog. 2014, 55, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Ha, I.J.; Park, S.-Y.; Choi, M.; Lim, S.-L.; Kim, S.-H.; Lee, J.-H.; Ahn, K.S.; Yun, M.; Lee, S.-G. Morusin Induces TRAIL Sensitization by Regulating EGFR and DR5 in Human Glioblastoma Cells. J. Nat. Prod. 2016, 79, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, J.H.; Oh, E.Y.; Nam, D.; Lee, S.G.; Lee, J.; Ahn, K.S. Blockage of STAT3 Signaling Pathway by Morusin Induces Apoptosis and Inhibits Invasion in Human Pan-creatic Tumor Cells. Pancreas 2016, 45, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, L.; Sun, Z.; Li, H.; Wang, Q.; Yi, C.; Wang, X. Morusin shows potent antitumor activity for human hepatocellular carcinoma in vitro and in vivo through apoptosis induction and angiogenesis inhibition. Drug Des. Dev. Ther. 2017, 11, 1789–1802. [Google Scholar] [CrossRef]
- Kang, S.; Kim, E.-O.; Kim, S.-H.; Lee, J.; Ahn, K.S.; Yun, M.; Lee, S.-G. Morusin induces apoptosis by regulating expression of Bax and Survivin in human breast cancer cells. Oncol. Lett. 2017, 13, 4558–4562. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, D.; Mao, J.; Ke, X.-X.; Zhang, R.; Yin, C.; Gao, N.; Cui, H. Morusin inhibits cell proliferation and tumor growth by down-regulating c-Myc in human gastric cancer. Oncotarget 2017, 8, 57187–57200. [Google Scholar] [CrossRef]
- Cho, S.W.; Na, W.; Choi, M.; Kang, S.J.; Lee, S.-G.; Choi, C.Y. Autophagy inhibits cell death induced by the anti-cancer drug morusin. Am. J. Cancer Res. 2017, 7, 518–530. [Google Scholar]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to ge-nomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, N.M.P.; Meijer, C.; Kruyt, F.A. The endoplasmic reticulum stress/unfolded protein response in glio-magenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharmacol. 2016, 118, 1–8. [Google Scholar] [CrossRef]
- Bisol, A.; De Campos, P.S.; Lamers, M.L. Flavonoids as anticancer therapies: A systematic review of clinical trials. Phytotherapy Res. 2019, 34, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, T.D.; Call, T.G.; Zent, C.S.; Leis, J.F.; LaPlant, B.; Bowen, D.A.; Roos, M.; Laumann, K.; Ghosh, A.K.; Lesnick, C.; et al. Phase 2 trial of daily, oral polyphenon E in patients with asymptomatic, Rai stage 0 to II chronic lymphocytic leukemia. Cancer 2013, 119, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Clavel, M.; Dodion, P.; Monfardini, S.; ten Bokkel-Huinink, W.; Wagener, D.T.; Cavalli, F. Phase II trials with flavone acetic acid (NCS. 347512, LM975) in patients with advanced carcinoma of the breast, colon, head and neck and melanoma. Investig. New Drugs 1990, 8 (Suppl. 1), S95–S99. [Google Scholar] [CrossRef]
- Thatcher, N.; Dazzi, H.; Mellor, M.; Ghosh, A.; Carrington, B.; Johnson, R.; Loriaux, E.; Craig, R. Recombinant interleukin-2 (rIL-2) with flavone acetic acid (FAA) in advanced malignant melanoma: A phase II study. Br. J. Cancer 1990, 61, 618–621. [Google Scholar] [CrossRef]
- Fotopoulou, C.; Vergote, I.; Mainwaring, P.; Bidzinski, M.; Vermorken, J.B.; Ghamande, S.A.; Gabra, H. Weekly AUC2 carboplatin in acquired platinum resistant ovarian cancer with or without oral phe-noxodiol, a sensitizer of platinum cytotoxicity: The phase Ill OVATURE multicenter randomized study. Ann. Oncol. 2014, 25, 160–165. [Google Scholar] [CrossRef]
- Kelly, M.G.; Mor, G.; Husband, A.; O’Malley, D.M.; Baker, L.; Azodi, M.; Rutherford, T.J. Phase II evaluation of phenoxodiol in combination with cisplatin or paclitaxel in women with plati-num/taxane-refractory/resistant epithelial ovarian, fallopian tube, or primary peritoneal cancers. Int. J. Gynecol. Cancer 2011, 21, 633–639. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Fathallah-Shaykh, H.M. Genomic discovery reveals a molecular system for resistance to oxidative and endoplasmic reticu-lum stress in cultured glioma. Arch. Neurol. 2005, 62, 233–236. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pyrko, P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef] [PubMed]
- Epple, L.M.; Dodd, R.D.; Merz, A.L.; Dechkovskaia, A.M.; Herring, M.; Winston, B.A.; Graner, M.W. Induction of the Unfolded Protein Response Drives Enhanced Metabolism and Chemoresistance in Glioma Cells. PLoS ONE 2013, 8, e73267. [Google Scholar]
- Hou, X.; Liu, Y.; Liu, H.; Chen, X.; Liu, M.; Che, H.; Guo, F.; Wang, C.; Zhang, D.; Wu, J.; et al. PERK silence inhibits glioma cell growth under low glucose stress by blockage of p-AKT and subsequent HK2’s mitochondria translocation. Sci. Rep. 2015, 5, srep09065. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2015, 283, 2640–2652. [Google Scholar] [CrossRef]
- Suzuki, K.; Gerelchuluun, A.; Hong, Z.; Sun, L.; Zenkoh, J.; Moritake, T.; Tsuboi, K. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro-Oncology 2013, 15, 1186–1199. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer |
|---|---|
| ATF6 | F:AGCAGCACCCAAGACTCAAAC |
| R:GCATAAGCGTTGGTACTGTCTGA | |
| eIF2α | F:TCGACCTCCTGAAGGCAGTT |
| R:AGTTGTAGGTTGGGTATCCCAG | |
| CHOP | F:GGAAACAGAGTGGTCATTCCC |
| R:CTGCTTGAGCCGTTCATTCTC | |
| GAPDH | F:AGAAGGCTGGGGCTCATTTG |
| R:AGGGGCCATCCACAGTCTTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, R.; Zhou, Y.; Zhang, H.; Pan, J.; Yang, F.; Zhang, R.; Ahmad, N.; Yang, J.; Sun, M. Morusin Enhances Temozolomide Efficiency in GBM by Inducing Cytoplasmic Vacuolization and Endoplasmic Reticulum Stress. J. Clin. Med. 2022, 11, 3662. https://doi.org/10.3390/jcm11133662
Zhao R, Zhou Y, Zhang H, Pan J, Yang F, Zhang R, Ahmad N, Yang J, Sun M. Morusin Enhances Temozolomide Efficiency in GBM by Inducing Cytoplasmic Vacuolization and Endoplasmic Reticulum Stress. Journal of Clinical Medicine. 2022; 11(13):3662. https://doi.org/10.3390/jcm11133662
Chicago/Turabian StyleZhao, Rongchuan, Yuanshuai Zhou, Hong Zhang, Jinlin Pan, Fan Yang, Ruobing Zhang, Nafees Ahmad, Jiao Yang, and Minxuan Sun. 2022. "Morusin Enhances Temozolomide Efficiency in GBM by Inducing Cytoplasmic Vacuolization and Endoplasmic Reticulum Stress" Journal of Clinical Medicine 11, no. 13: 3662. https://doi.org/10.3390/jcm11133662
APA StyleZhao, R., Zhou, Y., Zhang, H., Pan, J., Yang, F., Zhang, R., Ahmad, N., Yang, J., & Sun, M. (2022). Morusin Enhances Temozolomide Efficiency in GBM by Inducing Cytoplasmic Vacuolization and Endoplasmic Reticulum Stress. Journal of Clinical Medicine, 11(13), 3662. https://doi.org/10.3390/jcm11133662