
Detection and Prediction of Macrophage Activation Syndrome in Still’s Disease

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Patients and Methods
2.1. Study Design and Data Collection
2.2. Definitions
2.3. Statistical Analysis
2.4. Ethics
3. Results
3.1. Description of the Study Population
3.2. Specific Biological Parameters Are Useful for Distinguishing between MAS and SD Flare
3.3. The Detection of MAS Is Improved with Optimized Thresholds of Ferritin, LDH, and Fibrinogen
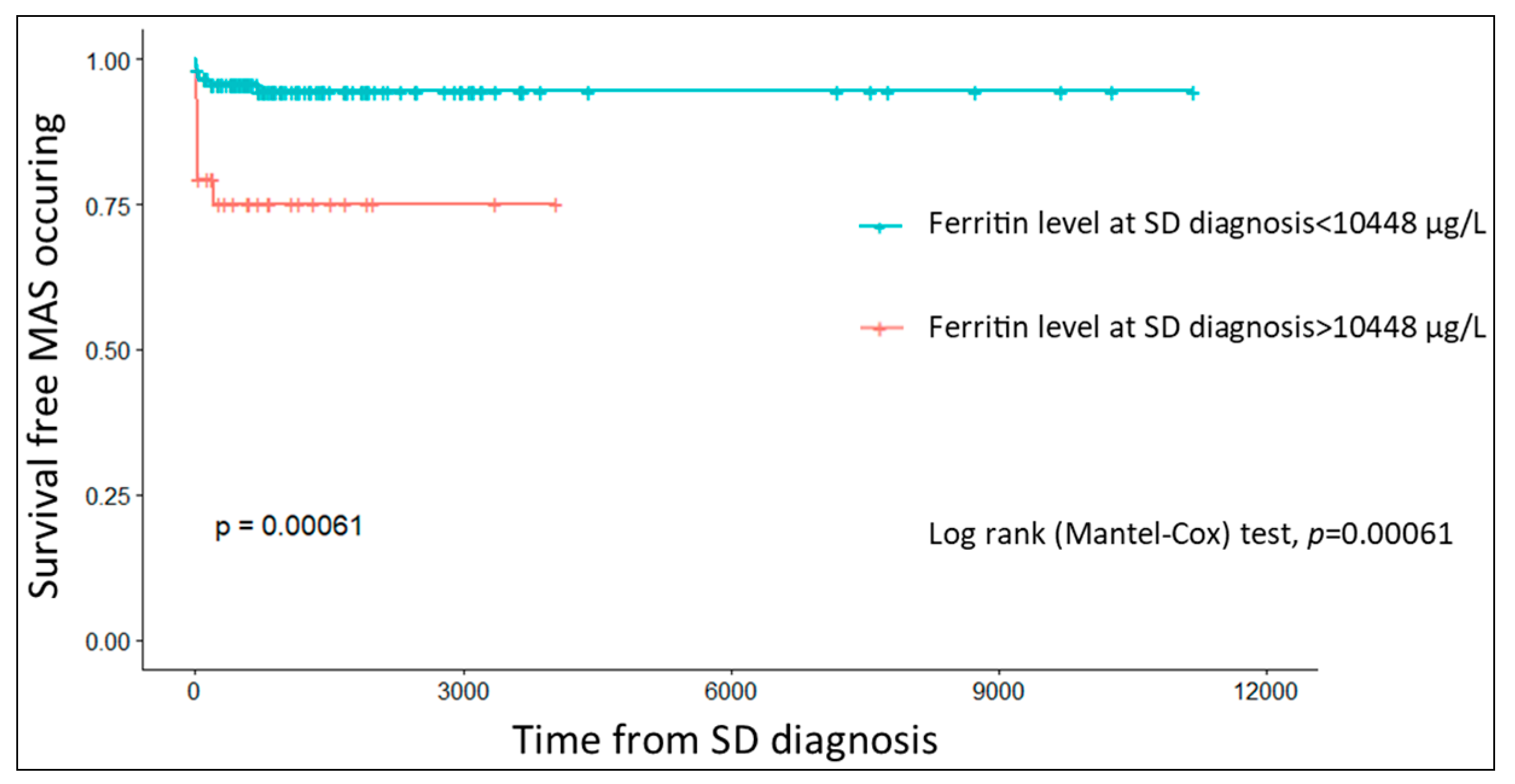
3.4. Predictive Factors of MAS Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bywaters, E.G. Still’s disease in the adult. Ann. Rheum. Dis. 1971, 30, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Gerfaud-Valentin, M.; Maucort-Boulch, D.; Hot, A.; Iwaz, J.; Ninet, J.; Durieu, I.; Broussolle, C.; Sève, P. Adult-Onset Still Disease: Manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine 2014, 93, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Nirmala, N.; Brachat, A.; Feist, E.; Blank, N.; Specker, C.; Witt, M.; Zernicke, J.; Martini, A.; Junge, G. Gene-expression analysis of adult-onset Still’s disease and systemic juvenile idiopathic arthritis is consistent with a continuum of a single disease entity. Pediatr. Rheumatol. 2015, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jamilloux, Y.; Gerfaud-Valentin, M.; Martinon, F.; Belot, A.; Henry, T.; Sève, P. Pathogenesis of adult-onset Still’s disease: New insights from the juvenile counterpart. Immunol. Res. 2015, 61, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Pay, S.; Türkçapar, N.; Kalyoncu, M.; Şimşek, I.; Beyan, E.; Ertenli, I.; Öztürk, M.A.; Düzgün, N.; Erdem, H.; Ankara Rheumatology Study Group; et al. A multicenter study of patients with adult-onset Still’s disease compared with systemic juvenile idiopathic arthritis. Clin. Rheumatol. 2006, 25, 639–644. [Google Scholar] [CrossRef]
- Magadur-Joly, G.; Billaud, E.; Barrier, J.H.; Pennec, Y.L.; Masson, C.; Renou, P.; Prost, A. Epidemiology of adult Still’s disease: Estimate of the incidence by a retrospective study in west France. Ann. Rheum. Dis. 1995, 54, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Wakai, K.; Ohta, A.; Tamakoshi, A.; Ohno, Y.; Kawamura, T.; Aoki, R.; Kojima, M.; Lin, Y.; Hashimoto, S.; Inaba, Y.; et al. Estimated Prevalence and Incidence of Adult Still’s Disease: Findings by a Nationwide Epidemiological Survey in Japan. J. Epidemiol. 1997, 7, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, M.; Nitsch-Osuch, A.; Samel-Kowalik, P.; Goryński, P.; Tyszko, P.; Kanecki, K. Adult-onset Still’s disease in Poland—A nationwide population-based study. Ann. Agric. Environ. Med. 2021, 28, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Evensen, K.J.; Nossent, J. Epidemiology and outcome of adult-onset Still’s disease in Northern Norway. Scand. J. Rheumatol. 2006, 35, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Feist, E.; Mitrovic, S.; Fautrel, B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat. Rev. Rheumatol. 2018, 14, 603–618. [Google Scholar] [CrossRef]
- Fautrel, B.; Le Moël, G.; Saint-Marcoux, B.; Taupin, P.; Vignes, S.; Rozenberg, S.; Koeger, A.C.; Meyer, O.; Guillevin, L.; Piette, J.C.; et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J. Rheumatol. 2001, 28, 322–329. [Google Scholar] [PubMed]
- Fautrel, B.; Zing, E.; Golmard, J.-L.; LE Moel, G.; Bissery, A.; Rioux, C.; Rozenberg, S.; Piette, J.-C.; Bourgeois, P. Proposal for a New Set of Classification Criteria for Adult-Onset Still Disease. Medicine 2002, 81, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Pouchot, J.; Sampalis, J.S.; Beaudet, F.; Carette, S.; Décary, F.; Salusinsky-Sternbach, M.; Hill, R.O.; Gutkowski, A.; Harth, M.; Myhal, D. Adult Still’s disease: Manifestations, disease course, and outcome in 62 patients. Medicine 1991, 70, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Uppal, S.S.; Al-Mutairi, M.; Hayat, S.; Abraham, M.; Malaviya, A. Ten years of clinical experience with adult onset Still’s disease: Is the outcome improving? Clin. Rheumatol. 2007, 26, 1055–1060. [Google Scholar] [CrossRef]
- Mehta, B.Y.; Ibrahim, S.; Briggs, W.; Efthimiou, P. Racial/Ethnic variations in morbidity and mortality in Adult Onset Still’s Disease: An analysis of national dataset. Semin. Arthritis Rheum. 2019, 49, 469–473. [Google Scholar] [CrossRef]
- Carter, S.J.; Tattersall, R.S.; Ramanan, A.V. Macrophage activation syndrome in adults: Recent advances in pathophysiology, diagnosis and treatment. Rheumatology 2019, 58, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Grom, A.A.; Behrens, E.M.; Cron, R. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: Diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012, 13, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Hot, A.; Toh, M.-L.; Coppéré, B.; Perard, L.; Madoux, M.H.G.; Mausservey, C.; Desmurs-Clavel, H.; Ffrench, M.; Ninet, J. Reactive Hemophagocytic Syndrome in Adult-Onset Still Disease: Clinical features and long-term outcome: A case-control study of 8 patients. Medicine 2010, 89, 37–46. [Google Scholar] [CrossRef]
- Arlet, J.-B.; Huong, D.L.T.; Marinho, A.; Amoura, Z.; Wechsler, B.; Papo, T.; Piette, J.-C. Reactive haemophagocytic syndrome in adult-onset Still’s disease: A report of six patients and a review of the literature. Ann. Rheum. Dis. 2006, 65, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Lenert, A.; Oh, G.; Ombrello, M.J.; Kim, S. Clinical characteristics and comorbidities in adult-onset Still’s disease using a large US administrative claims database. Rheumatology 2020, 59, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, S.; Fautrel, B. Complications of adult-onset Still’s disease and their management. Expert Rev. Clin. Immunol. 2018, 14, 351–365. [Google Scholar] [CrossRef]
- Machaczka, M.; Klimkowska, M. Bone marrow assessment in the diagnosis of acquired hemophagocytic lymphohistiocytosis in adults. Am. J. Clin. Pathol. 2015, 143, 308–309. [Google Scholar] [CrossRef]
- Gupta, A.; Weitzman, S.; Abdelhaleem, M. The role of hemophagocytosis in bone marrow aspirates in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2008, 50, 192–194. [Google Scholar] [CrossRef]
- Ho, C.; Yao, X.; Tian, L.; Li, F.-Y.; Podoltsev, N.; Xu, M.L. Marrow Assessment for Hemophagocytic Lymphohistiocytosis Demonstrates Poor Correlation with Disease Probability. Am. J. Clin. Pathol. 2014, 141, 62–71. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Zeron, P.B.; López-Guillermo, A.; Khamashta, M.A.; Bosch, X. Adult haemophagocytic syndrome. Lancet 2014, 383, 1503–1516. [Google Scholar] [CrossRef]
- Emmenegger, U.; Reimers, A.; Frey, U.; Fux, C.; Bihl, F.; Semela, D.; Cottagnoud, P.; Cerny, A.; Spaeth, P.J.; Neftel, K.A. Reactive macrophage activation syndrome: A simple screening strategy and its potential in early treatment initiation. Swiss Med. Wkly. 2002, 132, 230–236. [Google Scholar]
- Maria, A.T.J.; Le Quellec, A.; Jorgensen, C.; Touitou, I.; Rivière, S.; Guilpain, P. Adult onset Still’s disease (AOSD) in the era of biologic therapies: Dichotomous view for cytokine and clinical expressions. Autoimmun. Rev. 2014, 13, 1149–1159. [Google Scholar] [CrossRef]
- Grom, A.A. Natural killer cell dysfunction: A common pathway in systemic-onset juvenile rheumatoid arthritis, macrophage activation syndrome, and hemophagocytic lymphohistiocytosis? Arthritis Rheum. 2004, 50, 689–698. [Google Scholar] [CrossRef]
- Henter, J.-I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef]
- Ravelli, A.; Minoia, F.; Davì, S.; Horne, A.; Bovis, F.; Pistorio, A.; Aricò, M.; Avcin, T.; Behrens, E.M.; De Benedetti, F.; et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016, 68, 566–576. [Google Scholar] [CrossRef]
- Ahn, S.S.; Yoo, B.-W.; Jung, S.M.; Lee, S.-W.; Park, Y.-B.; Song, J.J. Application of the 2016 EULAR/ACR/PRINTO Classification Criteria for Macrophage Activation Syndrome in Patients with Adult-onset Still Disease. J. Rheumatol. 2017, 44, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Inokuchi, S.; Maruyama, A.; Suematsu, R.; Sakai, M.; Sadanaga, Y.; Ono, N.; Arinobu, Y.; Koarada, S. Are the 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome applicable to patients with adult-onset Still’s disease? Rheumatol. Int. 2018, 39, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Tristano, A.G. Macrophage activation syndrome: A frequent but under-diagnosed complication associated with rheumatic diseases. Med. Sci. Monit. 2008, 14, RA27–RA36. [Google Scholar] [PubMed]
- La Rosée, P.; Horne, A.; Hines, M.; von Bahr Greenwood, T.; Machowicz, R.; Berliner, N.; Birndt, S.; Gil-Herrera, J.; Girschikofsky, M.; Jordan, M.B.; et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 2019, 133, 2465–2477. [Google Scholar] [CrossRef]
- Ajeganova, S.; De Becker, A.; Schots, R. Efficacy of high-dose anakinra in refractory macrophage activation syndrome in adult-onset Still’s disease: When dosage matters in overcoming secondary therapy resistance. Ther. Adv. Musculoskelet. Dis. 2020, 12, 1759720X20974858. [Google Scholar] [CrossRef]
- Komiya, Y.; Takenaka, K.; Nagasaka, K. Successful treatment of glucocorticoid and cyclosporine refractory adult-onset Still’s disease complicated with hemophagocytic syndrome with plasma exchange therapy and tocilizumab: A case report. Jpn. J. Clin. Immunol. 2013, 36, 478–483. [Google Scholar] [CrossRef][Green Version]
- Bae, C.-B.; Jung, J.-Y.; Kim, H.-A.; Suh, C.-H. Reactive Hemophagocytic Syndrome in Adult-Onset Still Disease: Clinical features, predictive factors, and prognosis in 21 patients. Medicine 2015, 94, e451. [Google Scholar] [CrossRef]
- Ruscitti, P.; Iacono, D.; Ciccia, F.; Emmi, G.; Cipriani, P.; Grembiale, R.D.; Perosa, F.; Emmi, L.; Triolo, G.; Giacomelli, R.; et al. Macrophage Activation Syndrome in Patients Affected by Adult-onset Still Disease: Analysis of Survival Rates and Predictive Factors in the Gruppo Italiano di Ricerca in Reumatologia Clinica e Sperimentale Cohort. J. Rheumatol. 2018, 45, 864–872. [Google Scholar] [CrossRef]
- Ruscitti, P.; Rago, C.; Breda, L.; Cipriani, P.; Liakouli, V.; Berardicurti, O.; Carubbi, F.; Di Battista, C.; Verrotti, A.; Giacomelli, R. Macrophage activation syndrome in Still’s disease: Analysis of clinical characteristics and survival in paediatric and adult patients. Clin. Rheumatol. 2017, 36, 2839–2845. [Google Scholar] [CrossRef]
- Wang, R.; Li, T.; Ye, S.; Tan, W.; Zhao, C.; Li, Y.; de Bao, C.; Fu, Q. Macrophage activation syndrome associated with adult-onset Still’s disease: A multicenter retrospective analysis. Clin. Rheumatol. 2020, 39, 2379–2386. [Google Scholar] [CrossRef]
- Yang, X.-P.; Wang, M.; Li, T.-F.; Li, W.; Zhang, L.; Liu, S.-Y. Predictive factors and prognosis of macrophage activation syndrome associated with adult-onset Still’s disease. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 121), 83–88. [Google Scholar]
- Fardet, L.; Galicier, L.; Lambotte, O.; Marzac, C.; Aumont, C.; Chahwan, D.; Coppo, P.; Hejblum, G. Development and Validation of the HScore, a Score for the Diagnosis of Reactive Hemophagocytic Syndrome. Arthritis Rheumatol. 2014, 66, 2613–2620. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T. Preliminary criteria for classification of adult Still’s disease. J. Rheumatol. 1992, 19, 424–430. [Google Scholar] [PubMed]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.-M.; et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar]
- Martini, A.; Ravelli, A.; Avcin, T.; Beresford, M.W.; Burgos-Vargas, R.; Cuttica, R.; Ilowite, N.T.; Khubchandani, R.; Laxer, R.M.; Lovell, D.J.; et al. Toward New Classification Criteria for Juvenile Idiopathic Arthritis: First Steps, Pediatric Rheumatology International Trials Organization International Consensus. J. Rheumatol. 2018, 46, 190–197. [Google Scholar] [CrossRef]
- Haute Aut Santé. Maladie de Still de L’adulte. Available online: https://www.has-sante.fr/jcms/c_2867360/fr/maladie-de-still-de-l-adulte (accessed on 27 May 2021).
- Luthi, F.; Zufferey, P.; Hofer, M.F.; So, A.K. “Adolescent-onset Still’s disease”: Characteristics and outcome in comparison with adult-onset Still’s disease. Clin. Exp. Rheumatol. 2002, 20, 427–430. [Google Scholar] [PubMed]
- Sakata, N.; Shimizu, S.; Hirano, F.; Fushimi, K. Epidemiological study of adult-onset Still’s disease using a Japanese administrative database. Rheumatol. Int. 2016, 36, 1399–1405. [Google Scholar] [CrossRef]
- Franchini, S.; Dagna, L.; Salvo, F.; Aiello, P.; Baldissera, E.; Sabbadini, M.G. Adult onset Still’s disease: Clinical presentation in a large cohort of Italian patients. Clin. Exp. Rheumatol. 2010, 28, 41–48. [Google Scholar] [PubMed]
- Ohta, A.; Yamaguchi, M.; Kaneoka, H.; Nagayoshi, T.; Hiida, M. Adult Still’s disease: Review of 228 cases from the literature. J. Rheumatol. 1987, 14, 1139–1146. [Google Scholar]
- Tomaras, S.; Goetzke, C.; Kallinich, T.; Feist, E. Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach. J. Clin. Med. 2021, 10, 733. [Google Scholar] [CrossRef]
- Hu, Q.-Y.; Zeng, T.; Sun, C.-Y.; Luo, C.-N.; Liu, S.; Ding, T.-T.; Ji, Z.-F.; Lu, A.; Yimaiti, K.; Teng, J.-L.; et al. Clinical features and current treatments of adult-onset Still’s disease: A multicentre survey of 517 patients in China. Clin. Exp. Rheumatol. 2019, 52 (Suppl. 121), 52–57. [Google Scholar]
- Behrens, E.M.; Beukelman, T.; Gallo, L.; Spangler, J.; Rosenkranz, M.; Arkachaisri, T.; Ayala, R.; Groh, B.; Finkel, T.H.; Cron, R.Q. Evaluation of the presentation of systemic onset juvenile rheumatoid arthritis: Data from the Pennsylvania Systemic Onset Juvenile Arthritis Registry (PASOJAR). J. Rheumatol. 2007, 35, 343–348. [Google Scholar]
- Lomater, C.; Gerloni, V.; Gattinara, M.; Mazzotti, J.; Cimaz, R.; Fantini, F. Systemic onset juvenile idiopathic arthritis: A retrospective study of 80 consecutive patients followed for 10 years. J. Rheumatol 2000, 27, 491–496. [Google Scholar]
- Uziel, Y.; Pomeranz, A.; Brik, R.; Navon, P.; Mukamel, M.; Press, J.; Barash, J.; Tauber, T.; Harel, L.; Virgilis, D.; et al. Seasonal variation in systemic onset juvenile rheumatoid arthritis in Israel. J. Rheumatol. 1999, 26, 1187–1189. [Google Scholar]
- Sfriso, P.; Priori, R.; Valesini, G.; Rossi, S.; Montecucco, C.; D’Ascanio, A.; Carli, L.; Bombardieri, S.; LaSelva, G.; Iannone, F.; et al. Adult-onset Still’s disease: An Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin. Rheumatol. 2016, 35, 1683–1689. [Google Scholar] [CrossRef]
- Gratton, S.M.; Powell, T.R.; Theeler, B.J.; Hawley, J.S.; Amjad, F.S.; Tornatore, C. Neurological involvement and characterization in acquired hemophagocytic lymphohistiocytosis in adulthood. J. Neurol. Sci. 2015, 357, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-M.; Yum, M.-S.; Choi, H.-W.; Ko, T.-S.; Im, H.J.; Seo, J.-J.; Koh, K.-N. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J. Hematol. 2012, 47, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Horne, A.; Trottestam, H.; Aricò, M.; Egeler, R.M.; Filipovich, A.H.; Gadner, H.; Imashuku, S.; Ladisch, S.; Webb, D.; Janka, G.; et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br. J. Haematol. 2008, 140, 327–335. [Google Scholar] [CrossRef]
- Zhao, M.; Wu, D.; Shen, M. Adult-onset Still’s disease with neurological involvement: A single-centre report. Rheumatology 2021, 60, 4152–4157. [Google Scholar] [CrossRef]
- Crayne, C.B.; Albeituni, S.; Nichols, K.E.; Cron, R.Q. The Immunology of Macrophage Activation Syndrome. Front. Immunol. 2019, 10, 119. [Google Scholar] [CrossRef]
- Behrens, E.M.; Beukelman, T.; Paessler, M.; Cron, R.Q. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J. Rheumatol. 2007, 34, 1133–1138. [Google Scholar]
- Zeng, P.; Li, F.; Zeng, H. Occult macrophage activation syndrome in systemic-onset juvenile idiopathic arthritic syndrome—A case report. Acta Reumatol. Port. 2013, 38, 196–200. [Google Scholar]
- Coffernils, M.; Soupart, A.; Pradier, O.; Feremans, W.; Nève, P.; Decaux, G. Hyperferritinemia in adult onset Still’s disease and the hemophagocytic syndrome. J. Rheumatol. 1992, 19, 1425–1427. [Google Scholar]
- Lambotte, O.; Cacoub, P.; Costedoat, N.; Le Moel, G.; Amoura, Z.; Piette, J.-C. High ferritin and low glycosylated ferritin may also be a marker of excessive macrophage activation. J. Rheumatol. 2003, 30, 1027–1028. [Google Scholar]
- Fardet, L.; Coppo, P.; Kettaneh, A.; Dehoux, M.; Cabane, J.; Lambotte, O. Low glycosylated ferritin, a good marker for the diagnosis of hemophagocytic syndrome. Arthritis Care Res. 2008, 58, 1521–1527. [Google Scholar] [CrossRef]
- Fauter, M.; Viel, S.; Zaepfel, S.; Pradat, P.; Fiscus, J.; Villard, M.; Garnier, L.; Walzer, T.; Sève, P.; Henry, T.; et al. Low glycosylated ferritin is a sensitive biomarker of severe COVID-19. Cell. Mol. Immunol. 2020, 17, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Assari, R.; Ziaee, V.; Mirmohammadsadeghi, A.; Moradinejad, M.-H. Dynamic Changes, Cut-Off Points, Sensitivity, and Specificity of Laboratory Data to Differentiate Macrophage Activation Syndrome from Active Disease. Dis. Markers 2015, 2015, 424381. [Google Scholar] [CrossRef]
- Kostik, M.M.; Dubko, M.F.; Masalova, V.V.; Snegireva, L.S.; Kornishina, T.L.; Chikova, I.A.; Likhacheva, T.S.; Isupova, E.A.; Glebova, N.I.; Kuchinskaya, E.M.; et al. Identification of the best cutoff points and clinical signs specific for early recognition of macrophage activation syndrome in active systemic juvenile idiopathic arthritis. Semin. Arthritis Rheum. 2015, 44, 417–422. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meng, J.; Chi, H.; Wang, Z.; Zhang, H.; Sun, Y.; Teng, J.; Hu, Q.; Liu, H.; Cheng, X.; Ye, J.; et al. Characteristics and risk factors of relapses in patients with adult-onset Still’s disease: A long-term cohort study. Rheumatology 2021, 60, 4520–4529. [Google Scholar] [CrossRef]
- Rosário, C.; Zandman-Goddard, G.; Meyron-Holtz, E.G.; D’Cruz, D.P.; Shoenfeld, Y. The Hyperferritinemic Syndrome: Macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013, 11, 185. [Google Scholar] [CrossRef]


{kind=link}
| Characteristics | n = 206 |
|---|---|
| Epidemiology | |
| Age at diagnosis, median (IQR), y | 27.0 (11.0–43.0) |
| Sex (male), No. (%) | 86 (41.7%) |
| Caucasian origin, No. (%) | 108/159 (67.9%) |
| AOSD, No. (%) | 128 (62.1%) |
| SJIA, No. (%) | 78 (37.9%) |
| Clinical | |
| Fever, No. (%) | 183/196 (93.4%) |
| Constitutional symptoms, No. (%) | 61/159 (38.4%) |
| Arthritis/arthralgia, No. (%) | 176/199 (88.4%) |
| Skin rash, No. (%) | 135/192 (70.3%) |
| Sore throat, No. (%) | 92/183 (50.3%) |
| Lymphadenopathy, No. (%) | 66/186 (35.5%) |
| Splenomegaly, No. (%) | 22/157 (14.0%) |
| Hepatomegaly, No. (%) | 20/153 (13.1%) |
| Lung involvement, No. (%) | 37/189 (19.6%) |
| Heart involvement, No. (%) | 38/189 (20.1%) |
| Digestive involvement, No. (%) | 35/187 (8.7%) |
| Neurological involvement, No. (%) | 14 (6.8%) |
| Laboratory | |
| WBCs, median (IQR), G/L | 15.3 (11.4–19.7) |
| PMNs count, median (IQR), G/L | 12.0 (8.2–15.9) |
| CRP, median (IQR), mg/L | 160.0 (90.0–231.3) |
| Serum ferritin, median (IQR), μg/mL | 2200 (489–8419) |
| Ferritin glycosylated fraction, median (IQR), % | 14.0 (9.0–24.5) |
| Ferritin glycosylated fraction < 20%, No. (%) | 63/97 (64.9%) |
| ANA, No. (%) | 32/160 (20.0%) |
| Treatments | |
| Steroids, No. (%) | 171/202 (84.7%) |
| IL-1-blockers, No. (%) | 85/202 (42.1%) |
| IL-6-blockers, No. (%) | 32/202 (15.8%) |
| Methotrexate, No. (%) | 77/202 (38.1%) |
| Immunosupressive drugs, No. (%) | 12/202 (5.9%) |
| IVIg, No. (%) | 23/202 (11.4%) |
| Disease course | |
| Monocyclic (systemic), No. (%) | 64/176 (36.4%) |
| Polycyclic (systemic), No. (%) | 61/176 (34.7%) |
| Chronic (articular), No. (%) | 51/176 (28.9%) |
| Characteristics | With MAS (n = 20) | Without MAS (n = 186) | p Value | |
|---|---|---|---|---|
| Epidemiology | ||||
| Age at diagnosis, median (IQR), y | 22.0 (12.0–27.0) | 29.0 (11.0–43.8) | 0.139 | |
| Sex (male), No. (%) | 7 (35.0%) | 79 (42.5%) | 0.685 | |
| Caucasian origin, No. (%) | 9/16 (56.2%) | 99/143 (69.2%) | 0.440 | |
| Immunosuppression, No. (%) | 7 (35.0%) | 5/174 (2.7%) | <0.001 | |
| Classification criteria | ||||
| ILAR, No. (%) | 3/8 (37.5%) | 24/55 (43.6%) | 1.000 | |
| PReS, No. (%) | 3/7 (42.9%) | 28/51 (54.9%) | 0.694 | |
| Fautrel, No. (%) | 9/10 (90.0%) | 74/118 (62.7%) | 0.098 | |
| Yamaguchi, No. (%) | 9/10 (90.0%) | 68/118 (57.6%) | 0.050 | |
| Virus tests | ||||
| Positive CMV PCR, No. (%) | 0/13 (0.0%) | 5/48 (10.4%) | 0.574 | |
| Positive EBV PCR, No. (%) | 4/15 (26.7%) | 10/51 (19.6%) | 0.720 | |
| Clinical features | ||||
| Fever, No. (%) | 19/19 (100.0%) | 165/178 (92.7%) | 0.619 | |
| Arthritis/arthralgia, No. (%) | 14/18 (77.8%) | 162/ 181 (89.5%) | 0.137 | |
| Skin rash, No. (%) | 14/19 (73.7%)) | 120/174 (69.0%) | 0.872 | |
| Splenomegaly, No. (%) | 2/19 (10.5%) | 19/140 (13.6%) | 1.000 | |
| Hepatomegaly, No. (%) | 6/19 (31.6%) | 15/136 (11.0%) | 0.025 | |
| Lymphadenopathy, No. (%) | 7/19 (36.8%) | 57/168 (33.9%) | 1.000 | |
| Digestive involvement, No. (%) | 4/19 (21.1%) | 30/169 (17.8%) | 0.754 | |
| Lung involvement, No. (%) | 3/19 (15.8%) | 32/171 (18.7%) | 1.000 | |
| Heart involvement, No. (%) | 6/19 (31.6%) | 32/170 (18.8%) | 0.225 | |
| Neurological involvement, No. (%) | 4 (20.0%) | 10 (5.4%) | 0.034 | |
| Treatments | ||||
| Steroids, No. (%) | 19 (95.0%) | 152/182 (83.5%) | 0.323 | |
| IL-1-blockers, No. (%) | 13 (65.0%) | 72/182 (39.6%) | 0.051 | |
| IL-6-blockers, No. (%) | 8 (40.0%) | 24/182 (13.2%) | 0.005 | |
| Immunosupressive drugs, No. (%) | 5 (25.0%) | 7/182 (3.85%) | 0.003 | |
| IVIg, No. (%) | 8 (40.0%) | 15/182 (8.2%) | <0.001 | |
| Evolution | ||||
| Recovery, No. (%) | 6 (30.0%) | 66/172 (38.4%) | 0.305 | |
| Relapse, No. (%) | 17 (85.0%) | 92/169 (54.4%) | 0.017 | |
| Death, No. (%) | 0 (0.0%) | 5/165 (3.0%) | 1.000 | |
| Disease course | ||||
| Monocyclic (systemic), No. (%) | 6 (30.0%) | 58/156 (37.2%) | 0.530 | |
| Polycyclic (systemic), No. (%) | 11 (55.0%) | 50/156 (32.1%) | 0.042 | |
| Chronic (articular), No. (%) | 3 (15.0%) | 48/156 (30.8%) | 0.193 | |
| Biological Characteristics | With MAS n = 20 | Without MAS n = 186 | p Value |
|---|---|---|---|
| WBCs, median (IQR), G/L | 6.0 (4.1–14.5) | 15.4 (11.7–19.7) | <0.001 |
| PMNs, median (IQR), G/L | 11.2 (8.8–15.8) | 12.0 (8.1–15.8) | 0.919 |
| Hemoglobin, mean (SD), g/L | 107 (20.1) | 113 (17.3) | 0.222 |
| Platelets, median (IQR), G/L | 171 (130–210) | 381 (273–509) | <0.001 |
| Serum ferritin, median (IQR), μg/L | 13,444 (4370–26,369) | 2027 (460–7576) | <0.001 |
| Ferritin glycosylated fraction, median (IQR), % | 12.6 (3.00–16.0) | 15.0 (10.0–25.0) | 0.097 |
| Ferritin glycosylated fraction < 20%, No. (%) | 8/9 (88.9%) | 56/89 (62.9%) | 0.156 |
| CRP, median (IQR), mg/L | 142.0 (102.0–217.0) | 162 (90.0–237.0) | 0.907 |
| ASAT, median (IQR), U/L | 130 (84–242) | 36 (24–59) | <0.001 |
| ALAT, median (IQR), U/L | 131 (71–216) | 27 (15–62) | <0.001 |
| GGT, median (IQR), U/L | 132 (59–246) | 50. (22–120) | 0.024 |
| LDH, median (IQR), U/L | 678 (542–1236) | 344 (242–484) | <0.001 |
| PT, median (IQR), % | 69.5 (60.8–82.2) | 78.0 (69.0–93.2) | 0.066 |
| Fibrinogen, mean (SD), g/L | 3.41 (2.11) | 6.27 (2.06) | <0.001 |
| Triglycerides, median (IQR), mmol/L | 2.25 (1.87–3.08) | 1.51 (1.14–2.07) | 0.001 |
| B2 microglobulin, median (IQR), mg/L | 2.60 (2.40–2.76) | 2.35 (2.16–2.95) | 0.423 |
| Autoimmunity, No. (%) | 0/18 (0.0%) | 45/145 (31.0%) | 0.004 |
| ANA, No. (%) | 0/18 (0.0%) | 32/142 (22.5%) | 0.025 |
| Hemophagocytosis, No. (%) | 11/14 (78.6%) | 4/79 (5.1%) | <0.001 |
| Threshold | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | |
|---|---|---|---|---|---|
| HScore > 169 | >169 | 65.0 | 96.2 | 65.0 | 96.2 |
| HScore > 135 | >135 | 90.0 | 85.0 | 39.0 | 98.8 |
| Ferritin (μg/L) | >3500 | 85.0 | 61.7 | 23.9 | 96.6 |
| Ferritin glycosylated fraction (%) | <20 | 88.9 | 36.0 | 12.3 | 97.0 |
| <21 | 100.0 | 33.7 | 13.2 | 100.0 | |
| <25 | 100.0 | 24.7 | 11.8 | 100.0 | |
| LDH (U/L) | >459 | 89.5 | 72.3 | 35.4 | 97.5 |
| Platelet count (/mm3) | <230 | 80.0 | 86.2 | 38.5 | 92.0 |
| Fibrinogen (g/L) | <4.39 | 72.2 | 80.9 | 57.0 | 94.0 |
| Triglycerides (mmol/L) | >1.95 | 75.0 | 70.4 | 61.5 | 91.9 |
| ASAT (U/L) | >84 | 80.0 | 83.3 | 41.0 | 96.6 |
| ALAT (U/L) | >76 | 75.0 | 79.4 | 34.8 | 95.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javaux, C.; El-Jammal, T.; Neau, P.-A.; Fournier, N.; Gerfaud-Valentin, M.; Perard, L.; Fouillet-Desjonqueres, M.; Le Scanff, J.; Vignot, E.; Durupt, S.; et al. Detection and Prediction of Macrophage Activation Syndrome in Still’s Disease. J. Clin. Med. 2022, 11, 206. https://doi.org/10.3390/jcm11010206
Javaux C, El-Jammal T, Neau P-A, Fournier N, Gerfaud-Valentin M, Perard L, Fouillet-Desjonqueres M, Le Scanff J, Vignot E, Durupt S, et al. Detection and Prediction of Macrophage Activation Syndrome in Still’s Disease. Journal of Clinical Medicine. 2022; 11(1):206. https://doi.org/10.3390/jcm11010206
Chicago/Turabian StyleJavaux, Clément, Thomas El-Jammal, Pierre-Antoine Neau, Nicolas Fournier, Mathieu Gerfaud-Valentin, Laurent Perard, Marine Fouillet-Desjonqueres, Julie Le Scanff, Emmanuelle Vignot, Stéphane Durupt, and et al. 2022. "Detection and Prediction of Macrophage Activation Syndrome in Still’s Disease" Journal of Clinical Medicine 11, no. 1: 206. https://doi.org/10.3390/jcm11010206
APA StyleJavaux, C., El-Jammal, T., Neau, P.-A., Fournier, N., Gerfaud-Valentin, M., Perard, L., Fouillet-Desjonqueres, M., Le Scanff, J., Vignot, E., Durupt, S., Hot, A., Belot, A., Durieu, I., Henry, T., Sève, P., & Jamilloux, Y. (2022). Detection and Prediction of Macrophage Activation Syndrome in Still’s Disease. Journal of Clinical Medicine, 11(1), 206. https://doi.org/10.3390/jcm11010206