
Combined Inhibition of TGF-β1-Induced EMT and PD-L1 Silencing Re-Sensitizes Hepatocellular Carcinoma to Sorafenib Treatment

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. RNA Extraction and cDNA Synthesis
2.4. Quantitative Reverse Transcription-PCR (qRT-PCR)
2.5. Western Blot Analysis
2.6. Immunofluorescence
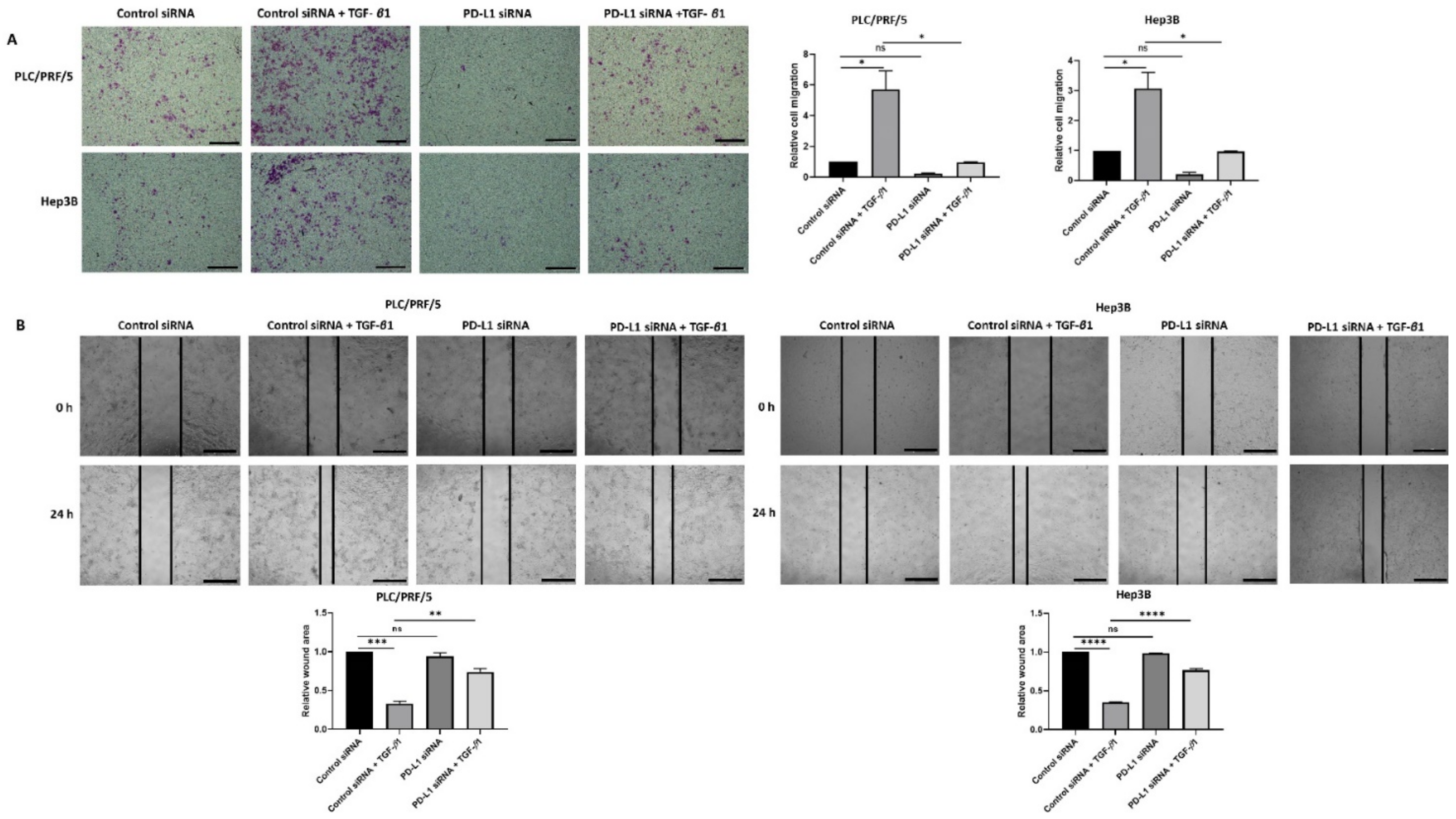
2.7. Wound Healing Assay
2.8. Transwell Migration Assay
2.9. Cell Viability Assay
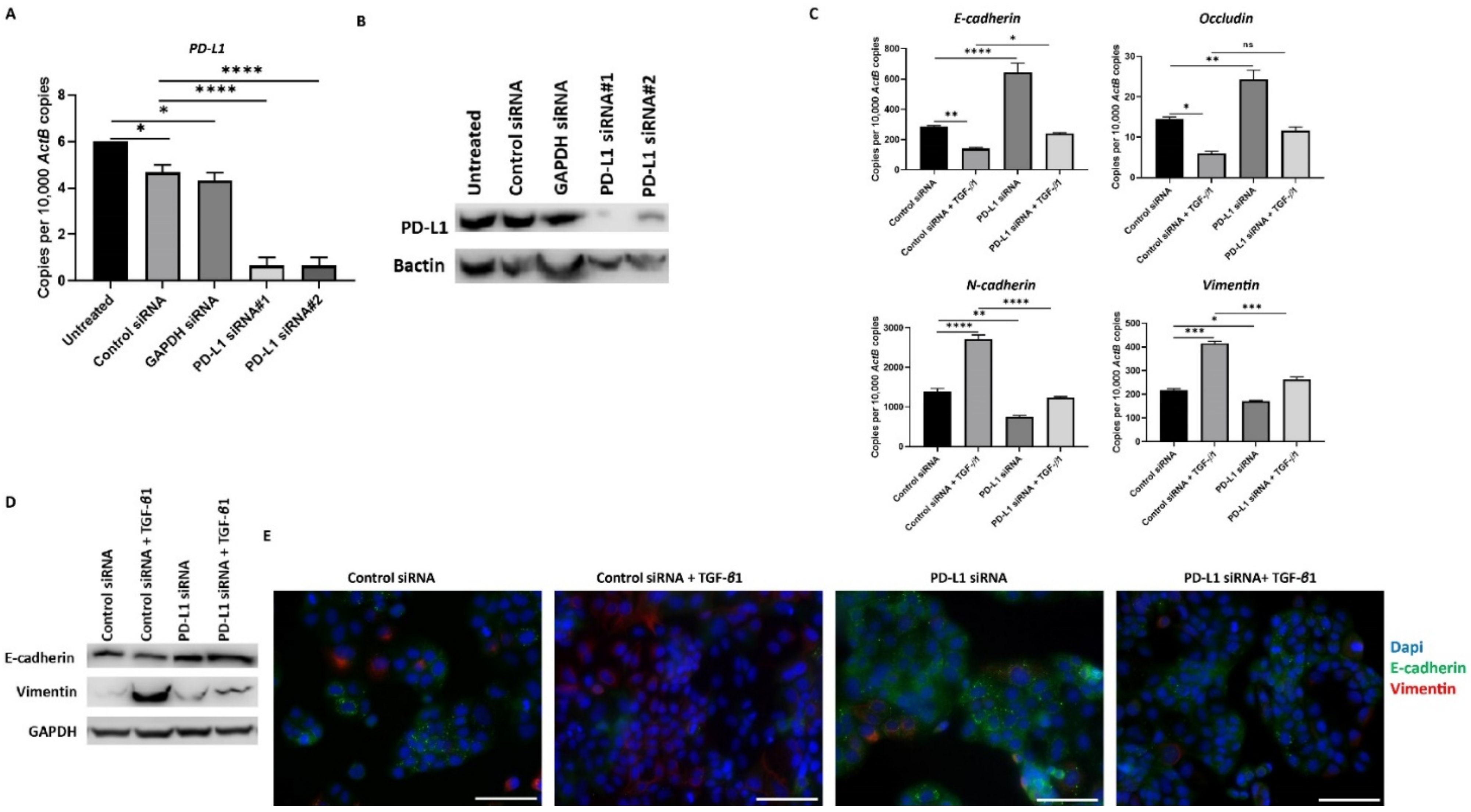
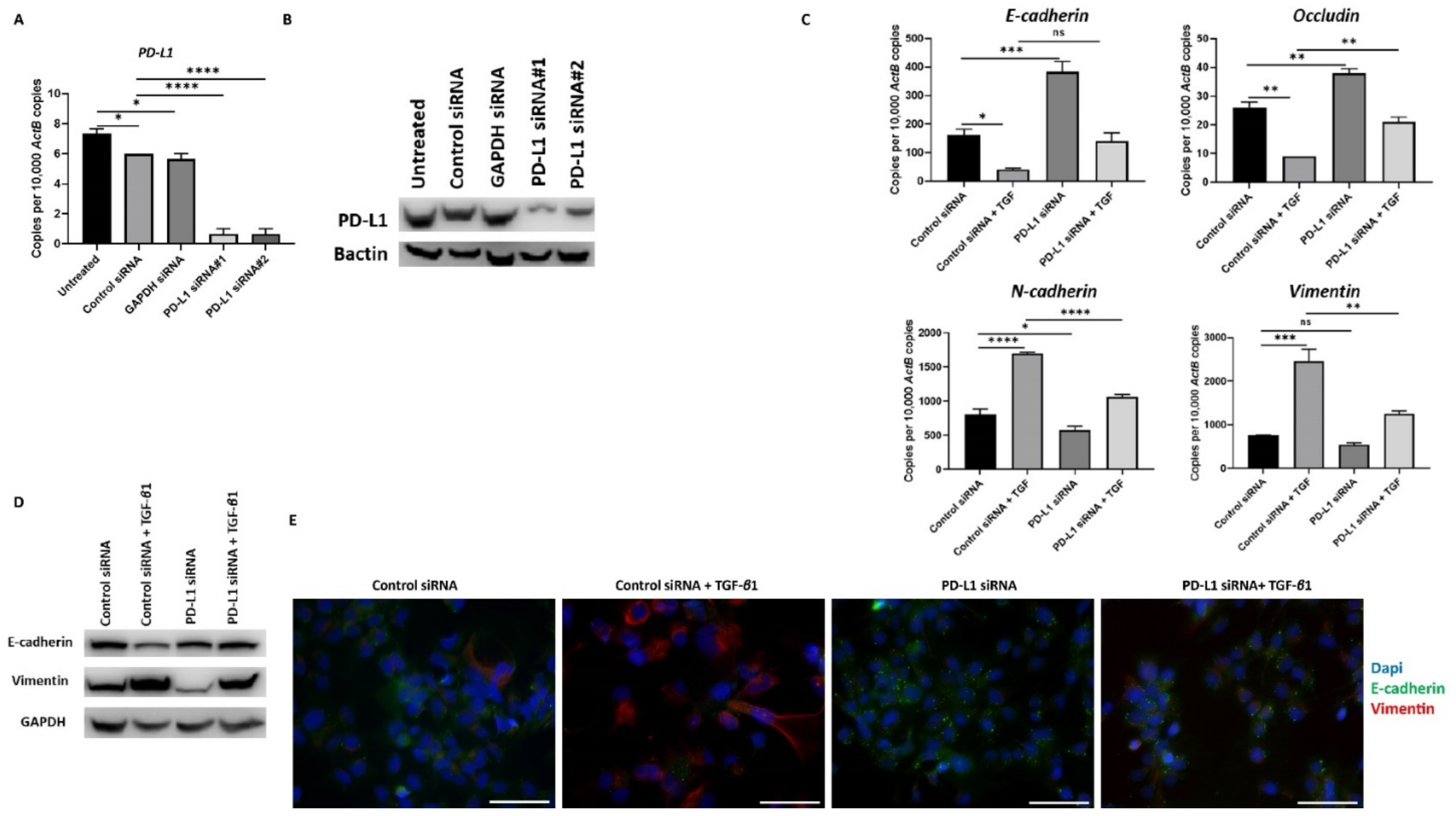
2.10. PDL-1 Knockdown
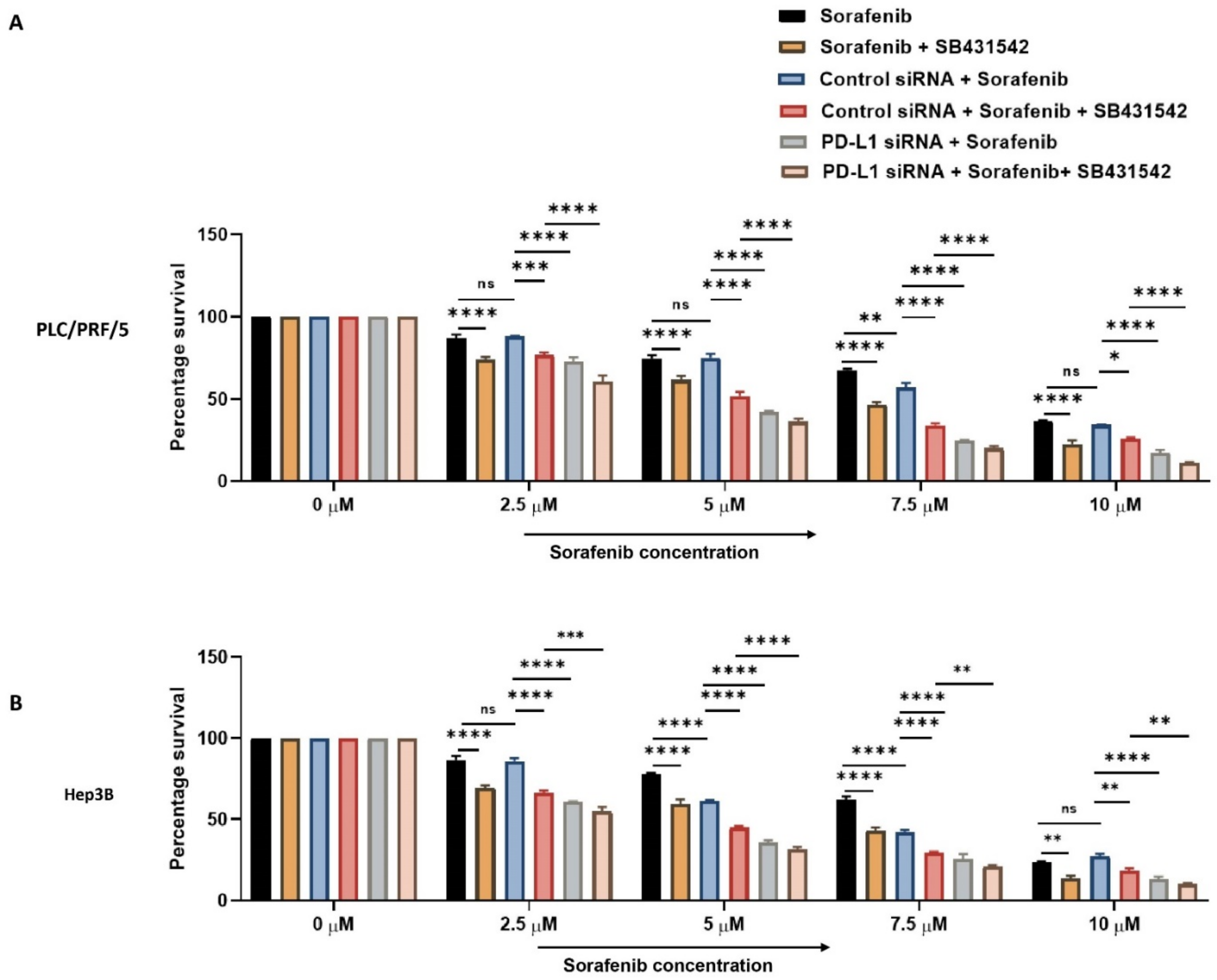
2.11. Drug Combination Analysis
2.12. Statistical Analysis
3. Results
3.1. HCC Cells Undergo EMT upon Sorafenib Exposure
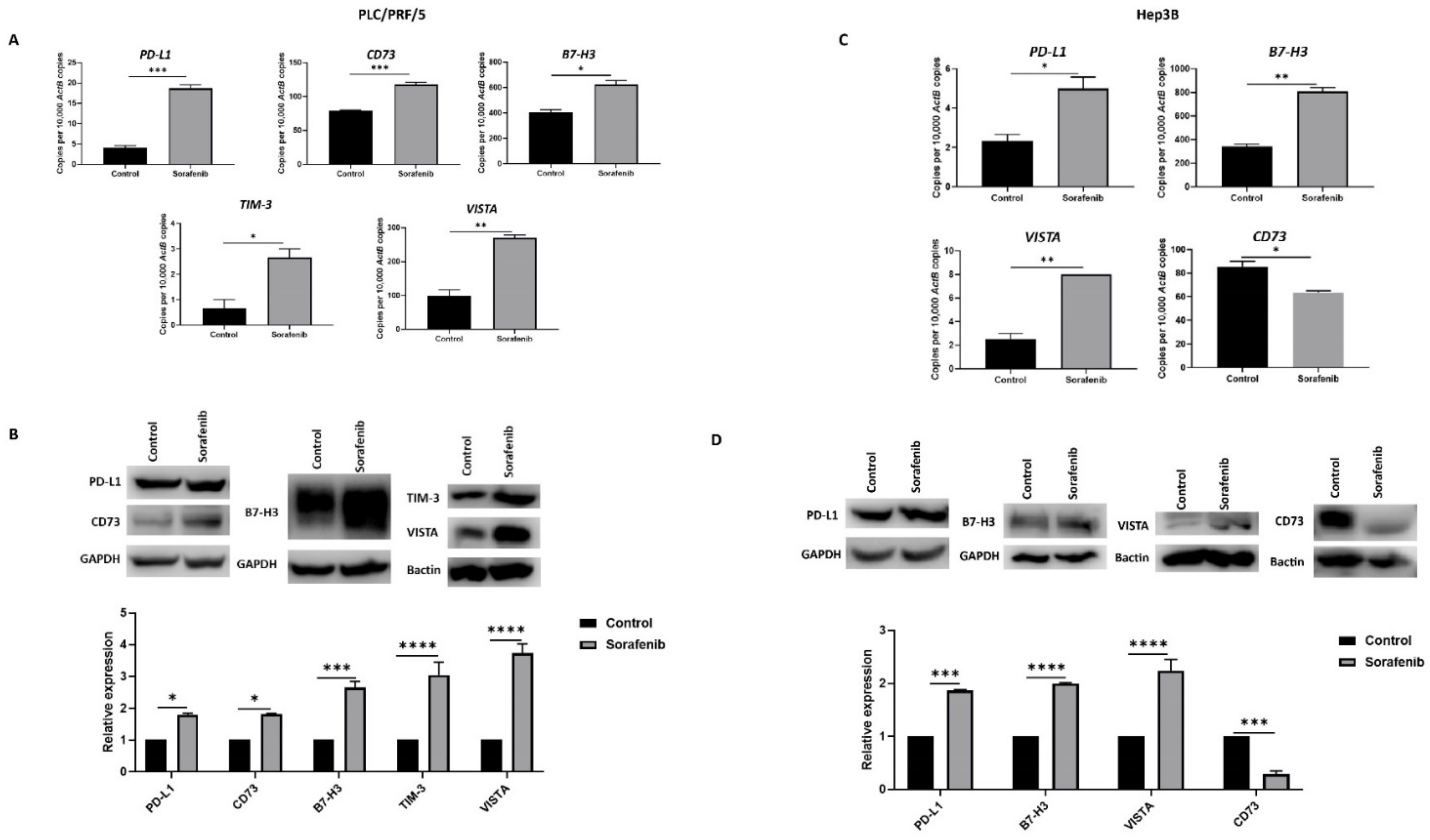
3.2. Sorafenib Treatment Induces Immune Checkpoint Molecules Expression in HCC Cells
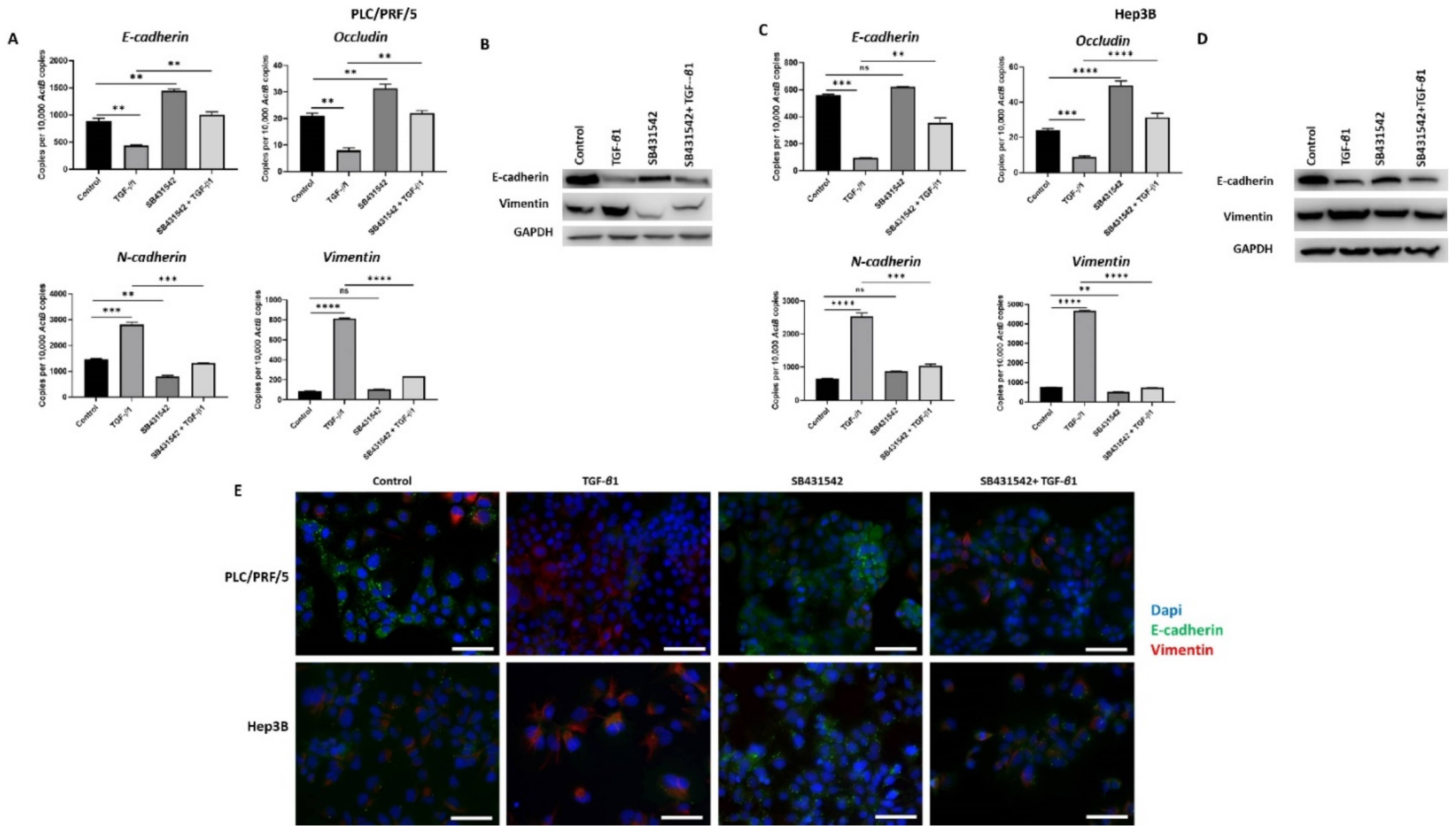
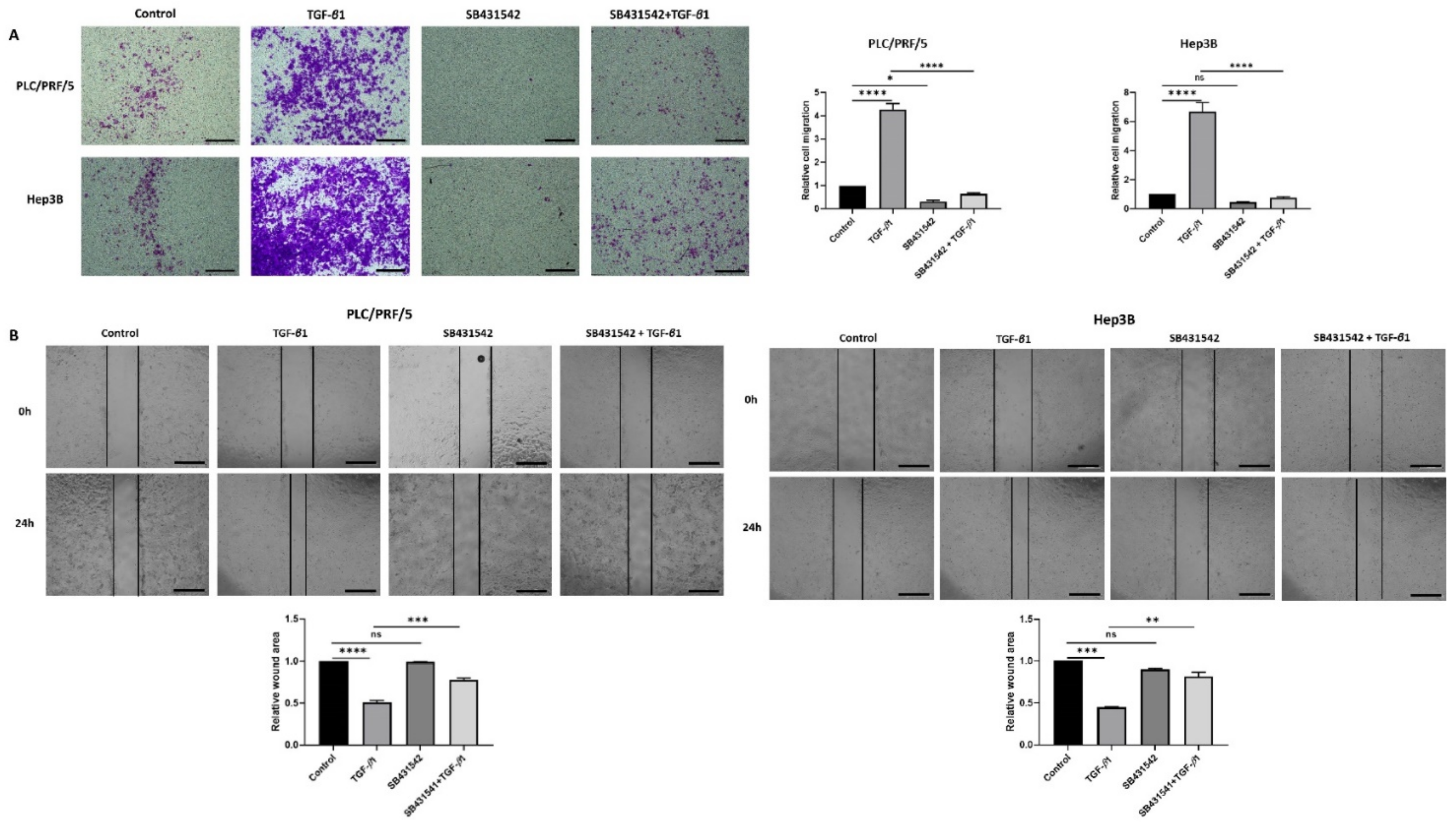
3.3. SB431542 Inhibits TGF-β1-Mediated EMT in HCC Cells
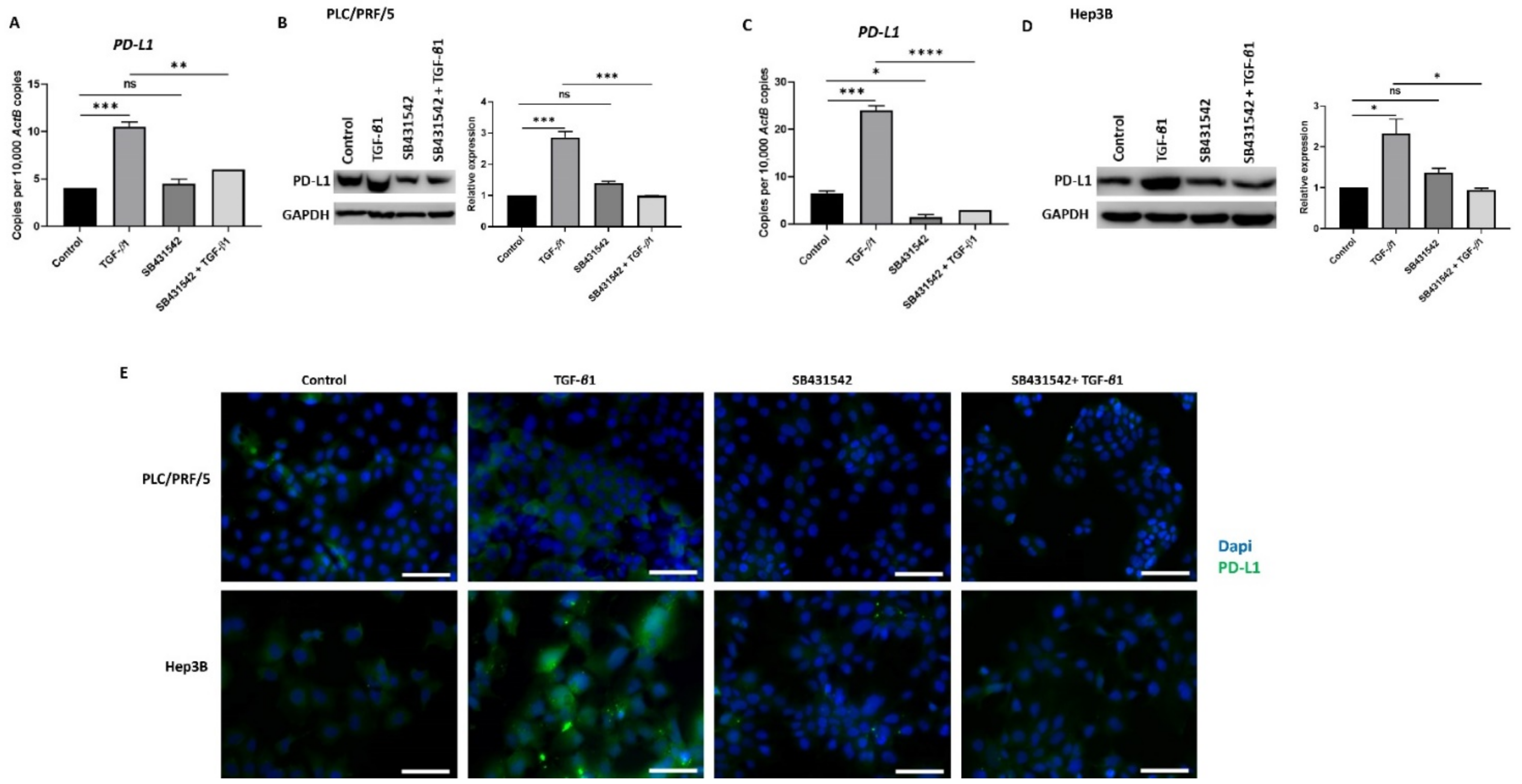
3.4. SB431542 Attenuates TGF-β1-Induced PD-L1 Expression
3.5. Knockdown of PD-L1 in HCC Cells Can Reverse TGF-β1-Mediated EMT
3.6. HCC Cells Can Overcome Sorafenib Resistance by Combined Targeting of PD-L1 and TGF-β1 Signalling
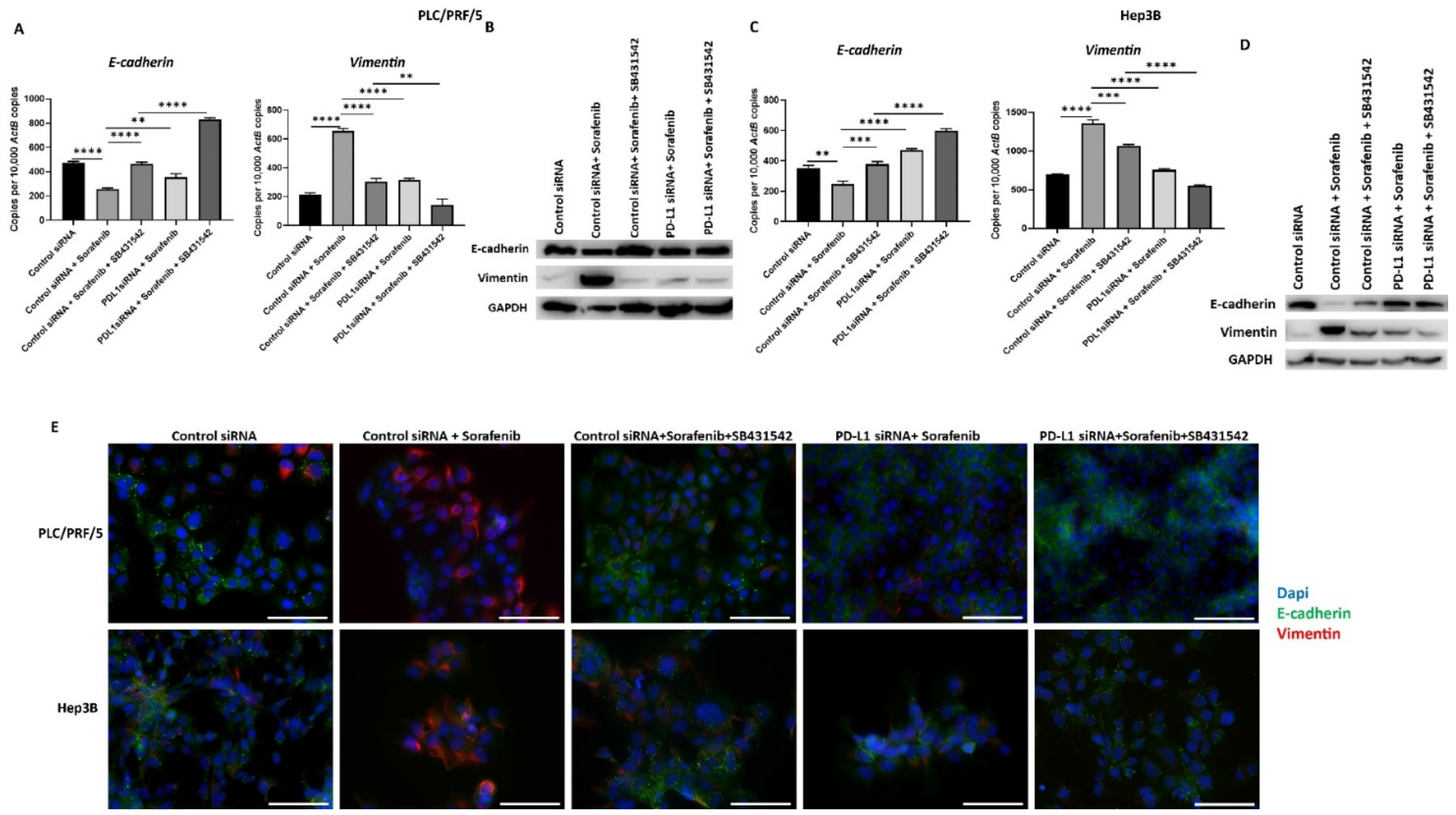
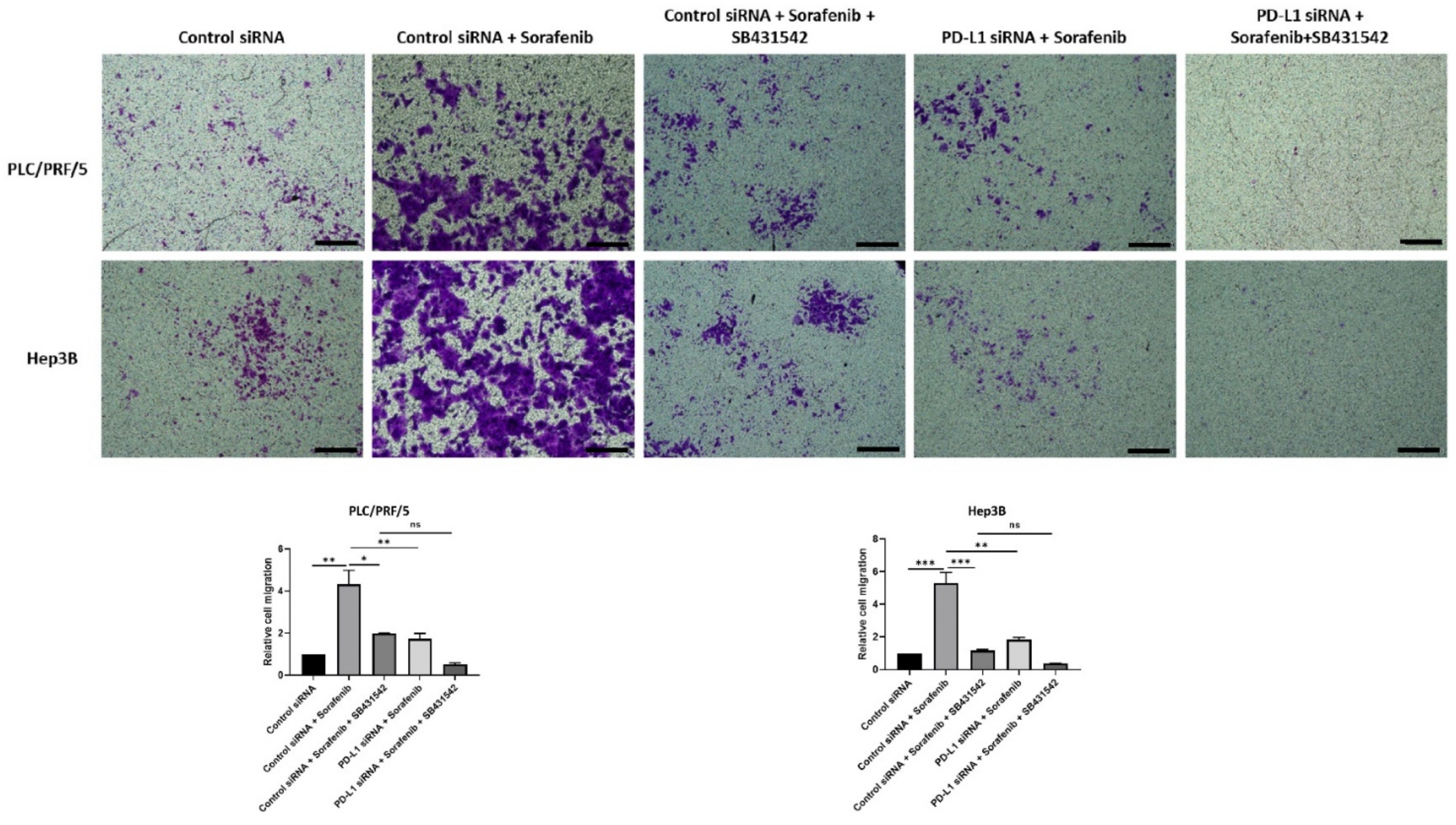
3.7. Combining PD-L1 Knockdown with SB431542 Can Reverse Sorafenib-Induced EMT in HCC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Chacko, S.; Samanta, S. Hepatocellular carcinoma: A life-threatening disease. Biomed. Pharmacother. 2016, 84, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Mir, N.; Jayachandran, A.; Dhungel, B.; Shrestha, R.; Steel, J.C. Epithelial-to-Mesenchymal Transition: A Mediator of Sorafenib Resistance in Advanced Hepatocellular Carcinoma. Curr. Cancer Drug Targets 2017, 17, 698–706. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Gao, Y.; Fan, X.; Li, N.; Du, C.; Yang, B.; Qin, W.; Fu, J.; Markowitz, G.J.; Wang, H.; Ma, J.; et al. CCL22 signaling contributes to sorafenib resistance in hepatitis B virus-associated hepatocellular carcinoma. Pharmacol. Res. 2020, 157, 104800. [Google Scholar] [CrossRef]
- Villanueva, A.; Llovet, J.M. Second-line therapies in hepatocellular carcinoma: Emergence of resistance to sorafenib. Clin. Cancer Res. 2012, 18, 1824–1826. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Chen, X.; Lingala, S.; Khoobyari, S.; Nolta, J.; Zern, M.A.; Wu, J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J. Hepatol. 2011, 55, 838–845. [Google Scholar] [CrossRef]
- Ezzoukhry, Z.; Louandre, C.; Trécherel, E.; Godin, C.; Chauffert, B.; Dupont, S.; Diouf, M.; Barbare, J.C.; Mazière, J.C.; Galmiche, A. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int. J. Cancer 2012, 131, 2961–2969. [Google Scholar] [CrossRef]
- Haga, Y.; Kanda, T.; Nakamura, M.; Nakamoto, S.; Sasaki, R.; Takahashi, K.; Wu, S.; Yokosuka, O. Overexpression of c-Jun contributes to sorafenib resistance in human hepatoma cell lines. PLoS ONE 2017, 12, e0174153. [Google Scholar] [CrossRef]
- Hikita, H.; Takehara, T.; Shimizu, S.; Kodama, T.; Shigekawa, M.; Iwase, K.; Hosui, A.; Miyagi, T.; Tatsumi, T.; Ishida, H.; et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology 2010, 52, 1310–1321. [Google Scholar] [CrossRef]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef]
- Tan, W.; Zhu, S.; Cao, J.; Zhang, L.; Li, W.; Liu, K.; Zhong, J.; Shang, C.; Chen, Y. Inhibition of MMP-2 Expression Enhances the Antitumor Effect of Sorafenib in Hepatocellular Carcinoma by Suppressing the PI3K/AKT/mTOR Pathway. Oncol. Res. 2017, 25, 1543–1553. [Google Scholar] [CrossRef]
- Van Malenstein, H.; Dekervel, J.; Verslype, C.; Van Cutsem, E.; Windmolders, P.; Nevens, F.; van Pelt, J. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013, 329, 74–83. [Google Scholar] [CrossRef]
- Zhao, D.; Zhai, B.; He, C.; Tan, G.; Jiang, X.; Pan, S.; Dong, X.; Wei, Z.; Ma, L.; Qiao, H.; et al. Upregulation of HIF-2α induced by sorafenib contributes to the resistance by activating the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014, 26, 1030–1039. [Google Scholar] [CrossRef]
- Chao, T.I.; Tai, W.T.; Hung, M.H.; Tsai, M.H.; Chen, M.H.; Chang, M.J.; Shiau, C.W.; Chen, K.F. A combination of sorafenib and SC-43 is a synergistic SHP-1 agonist duo to advance hepatocellular carcinoma therapy. Cancer Lett. 2016, 371, 205–213. [Google Scholar] [CrossRef]
- Hajighasemlou, S.; Nikbakht, M.; Pakzad, S.; Muhammadnejad, S.; Gharibzadeh, S.; Mirmoghtadaei, M.; Zafari, F.; Seyhoun, I.; Ai, J.; Verdi, J. Sorafenib and Mesenchymal Stem Cell Therapy: A Promising Approach for Treatment of HCC. Evid. Based Complement. Altern. Med. 2020, 2020, 9602728. [Google Scholar] [CrossRef]
- Ho, V.; Lim, T.S.; Lee, J.; Steinberg, J.; Szmyd, R.; Tham, M.; Yaligar, J.; Kaldis, P.; Abastado, J.P.; Chew, V. TLR3 agonist and Sorafenib combinatorial therapy promotes immune activation and controls hepatocellular carcinoma progression. Oncotarget 2015, 6, 27252–27266. [Google Scholar] [CrossRef]
- Jiang, X.; Feng, K.; Zhang, Y.; Li, Z.; Zhou, F.; Dou, H.; Wang, T. Sorafenib and DE605, a novel c-Met inhibitor, synergistically suppress hepatocellular carcinoma. Oncotarget 2015, 6, 12340–12356. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.; Ducreux, M.; Zhu, A.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. IMbrave150: Efficacy and safety results from a ph III study evaluating atezolizumab (atezo) 1 bevacizumab (bev) vs. sorafenib (Sor) as first treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Li, Z.; Li, N.; Li, F.; Zhou, Z.; Sang, J.; Chen, Y.; Han, Q.; Lv, Y.; Liu, Z. Immune checkpoint proteins PD-1 and TIM-3 are both highly expressed in liver tissues and correlate with their gene polymorphisms in patients with HBV-related hepatocellular carcinoma. Medicine 2016, 95, e5749. [Google Scholar] [CrossRef]
- Shrestha, R.; Prithviraj, P.; Anaka, M.; Bridle, K.R.; Crawford, D.H.G.; Dhungel, B.; Steel, J.C.; Jayachandran, A. Monitoring Immune Checkpoint Regulators as Predictive Biomarkers in Hepatocellular Carcinoma. Front. Oncol. 2018, 8, 269. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Jayachandran, A.; Dhungel, B.; Steel, J.C. Epithelial-to-mesenchymal plasticity of cancer stem cells: Therapeutic targets in hepatocellular carcinoma. J. Hematol. Oncol. 2016, 9, 74. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Chen, J.; Jin, R.; Zhao, J.; Liu, J.; Ying, H.; Yan, H.; Zhou, S.; Liang, Y.; Huang, D.; Liang, X.; et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015, 367, 1–11. [Google Scholar] [CrossRef]
- Dazert, E.; Colombi, M.; Boldanova, T.; Moes, S.; Adametz, D.; Quagliata, L.; Roth, V.; Terracciano, L.; Heim, M.H.; Jenoe, P.; et al. Quantitative proteomics and phosphoproteomics on serial tumor biopsies from a sorafenib-treated HCC patient. Proc. Natl. Acad. Sci. USA 2016, 113, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.F.; Li, K.S.; Shen, Y.H.; Gao, P.T.; Dong, Z.R.; Cai, J.B.; Zhang, C.; Huang, X.Y.; Tian, M.X.; Hu, Z.Q.; et al. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 2016, 7, e2201. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Bridle, K.R.; Crawford, D.H.G.; Jayachandran, A. TNF-α-mediated epithelial-to-mesenchymal transition regulates expression of immune checkpoint molecules in hepatocellular carcinoma. Mol. Med. Rep. 2020, 21, 1849–1860. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, A.; Shrestha, R.; Dhungel, B.; Huang, I.T.; Vasconcelos, M.Y.K.; Morrison, B.J.; Ramlogan-Steel, C.A.; Steel, J.C. Murine hepatocellular carcinoma derived stem cells reveal epithelial-to-mesenchymal plasticity. World J. Stem Cells 2017, 9, 159–168. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef]
- Duval, A.P.; Troquier, L.; de Souza Silva, O.; Demartines, N.; Dormond, O. Diclofenac Potentiates Sorafenib-Based Treatments of Hepatocellular Carcinoma by Enhancing Oxidative Stress. Cancers 2019, 11, 1453. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef]
- Dong, J.; Zhai, B.; Sun, W.; Hu, F.; Cheng, H.; Xu, J. Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0185088. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Meng, L.; Liu, K.; Ji, B. Targeting the PD-L1/DNMT1 axis in acquired resistance to sorafenib in human hepatocellular carcinoma. Oncol. Rep. 2017, 38, 899–907. [Google Scholar] [CrossRef]
- Chung, C.L.; Wang, S.W.; Sun, W.C.; Shu, C.W.; Kao, Y.C.; Shiao, M.S.; Chen, C.L. Sorafenib suppresses TGF-β responses by inducing caveolae/lipid raft-mediated internalization/degradation of cell-surface type II TGF-β receptors: Implications in development of effective adjunctive therapy for hepatocellular carcinoma. Biochem. Pharmacol. 2018, 154, 39–53. [Google Scholar] [CrossRef]
- Chen, Y.L.; Lv, J.; Ye, X.L.; Sun, M.Y.; Xu, Q.; Liu, C.H.; Min, L.H.; Li, H.P.; Liu, P.; Ding, X. Sorafenib inhibits transforming growth factor β1-mediated epithelial-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology 2011, 53, 1708–1718. [Google Scholar] [CrossRef]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-α is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2019, 40, 446–456. [Google Scholar] [CrossRef]
- Shrestha, R.; Bridle, K.R.; Crawford, D.H.G.; Jayachandran, A. Immune checkpoint blockade therapies for HCC: Current status and future implications. Hepatoma Res. 2019, 2019. [Google Scholar] [CrossRef]
- Buonaguro, L.; Mauriello, A.; Cavalluzzo, B.; Petrizzo, A.; Tagliamonte, M. Immunotherapy in hepatocellular carcinoma. Ann. Hepatol. 2019, 18, 291–297. [Google Scholar] [CrossRef]
- Lu, L.C.; Lee, Y.H.; Chang, C.J.; Shun, C.T.; Fang, C.Y.; Shao, Y.Y.; Liu, T.H.; Cheng, A.L.; Hsu, C.H. Increased Expression of Programmed Death-Ligand 1 in Infiltrating Immune Cells in Hepatocellular Carcinoma Tissues after Sorafenib Treatment. Liver Cancer 2019, 8, 110–120. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Liang, Q.; Liu, B.; Mei, X.; Ma, Y. Combinatorial immunotherapy of sorafenib and blockade of programmed death-ligand 1 induces effective natural killer cell responses against hepatocellular carcinoma. Tumor Biol. 2015, 36, 1561–1566. [Google Scholar] [CrossRef]
- Dong, M.P.; Enomoto, M.; Thuy, L.T.T.; Hai, H.; Hieu, V.N.; Hoang, D.V.; Iida-Ueno, A.; Odagiri, N.; Amano-Teranishi, Y.; Hagihara, A.; et al. Clinical significance of circulating soluble immune checkpoint proteins in sorafenib-treated patients with advanced hepatocellular carcinoma. Sci. Rep. 2020, 10, 3392. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiong, Y.; Li, J.; Zheng, X.; Zhou, Q.; Turner, A.; Wu, C.; Lu, B.; Jiang, J. PD-L1 Expression Promotes Epithelial to Mesenchymal Transition in Human Esophageal Cancer. Cell Physiol. Biochem. 2017, 42, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; McCampbell, K.K.; Gulley, J.L.; Schlom, J.; Palena, C. A novel bifunctional anti-PD-L1/TGF-β Trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology 2017, 6, e1349589. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Koh, J.; Kim, M.Y.; Kwon, D.; Go, H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum. Pathol. 2016, 58, 7–14. [Google Scholar] [CrossRef]
- Hirai, M.; Kitahara, H.; Kobayashi, Y.; Kato, K.; Bou-Gharios, G.; Nakamura, H.; Kawashiri, S. Regulation of PD-L1 expression in a high-grade invasive human oral squamous cell carcinoma microenvironment. Int. J. Oncol. 2017, 50, 41–48. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef]
- Liu, L.; Li, N.; Zhang, Q.; Zhou, J.; Lin, L.; He, X. Inhibition of ERK1/2 Signaling Impairs the Promoting Effects of TGF-β1 on Hepatocellular Carcinoma Cell Invasion and Epithelial-Mesenchymal Transition. Oncol. Res. 2017, 25, 1607–1616. [Google Scholar] [CrossRef]
- Park, N.R.; Cha, J.H.; Jang, J.W.; Bae, S.H.; Jang, B.; Kim, J.H.; Hur, W.; Choi, J.Y.; Yoon, S.K. Synergistic effects of CD44 and TGF-β1 through AKT/GSK-3β/β-catenin signaling during epithelial-mesenchymal transition in liver cancer cells. Biochem. Biophys. Res. Commun. 2016, 477, 568–574. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Q.; Li, Z.; Zhang, R.; Jia, C.; Yang, Z.; Zhao, H.; Ya, S.; Mao, R.; Ailijiang, T.; et al. GP73 promotes epithelial-mesenchymal transition and invasion partly by activating TGF-β1/Smad2 signaling in hepatocellular carcinoma. Carcinogenesis 2018, 39, 900–910. [Google Scholar] [CrossRef]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.; Heier, A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFβ: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Mitra, M.S.; Lancaster, K.; Adedeji, A.O.; Palanisamy, G.S.; Dave, R.A.; Zhong, F.; Holdren, M.S.; Turley, S.J.; Liang, W.C.; Wu, Y.; et al. A Potent Pan-TGFβ Neutralizing Monoclonal Antibody Elicits Cardiovascular Toxicity in Mice and Cynomolgus Monkeys. Toxicol. Sci. 2020, 175, 24–34. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Huynh, L.K.; Hipolito, C.J.; Ten Dijke, P. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Gueorguieva, I.; Cleverly, A.L.; Stauber, A.; Sada Pillay, N.; Rodon, J.A.; Miles, C.P.; Yingling, J.M.; Lahn, M.M. Defining a therapeutic window for the novel TGF-β inhibitor LY2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br. J. Clin. Pharmacol. 2014, 77, 796–807. [Google Scholar] [CrossRef]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernández, M.S.; Romero, F.L.; Sepulveda-Sánchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac Safety of TGF-β Receptor I Kinase Inhibitor LY2157299 Monohydrate in Cancer Patients in a First-in-Human Dose Study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef]
- Rodon, J.; Carducci, M.A.; Sepulveda-Sánchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Braña, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.L.; et al. First-in-human dose study of the novel transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin. Cancer Res. 2015, 21, 553–560. [Google Scholar] [CrossRef]
- Liu, J.; Yang, X.; Liang, Q.; Yu, Y.; Shen, X.; Sun, G. Valproic acid overcomes sorafenib resistance by reducing the migration of Jagged2-mediated Notch1 signaling pathway in hepatocellular carcinoma cells. Int. J. Biochem. Cell Biol. 2020, 126, 105820. [Google Scholar] [CrossRef]
- Matsuda, Y.; Wakai, T.; Kubota, M.; Osawa, M.; Hirose, Y.; Sakata, J.; Kobayashi, T.; Fujimaki, S.; Takamura, M.; Yamagiwa, S.; et al. Valproic acid overcomes transforming growth factor-β-mediated sorafenib resistance in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 1299–1313. [Google Scholar] [PubMed]
- Zhu, W.; Liang, Q.; Yang, X.; Yu, Y.; Shen, X.; Sun, G. Combination of sorafenib and Valproic acid synergistically induces cell apoptosis and inhibits hepatocellular carcinoma growth via down-regulating Notch3 and pAkt. Am. J. Cancer Res. 2017, 7, 2503–2514. [Google Scholar] [PubMed]
- Xu, Y.; Liu, Y.; Li, Z.; Li, H.; Li, X.; Yan, L.; Mao, J.; Shen, J.; Chen, W.; Xue, F. Long non-coding RNA H19 is involved in sorafenib resistance in hepatocellular carcinoma by upregulating miR-675. Oncol. Rep. 2020, 44, 165–173. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence (5′-3′) |
|---|---|
| Snai1 forward | GCTGCAGGACTCTAATCCAGA |
| Snai1 reverse | ATCTCCGGAGGTGGGATG |
| TGF-β1 forward | TACCTGAACCCGTGTTGCTCTC |
| TGF-β1 reverse | GTTGCTGAGGTATCGCCAGGAA |
| TNF-α forward | CCCAGGGACCTCTCTCTAATC |
| TNF-α reverse | TCTCAGCTCCACGCCATT |
| TIM-3 reverse | GACTCTAGCAGACAGTGGGATC |
| TIM-3 reverse | GGTGGTAAGCATCCTTGGAAAGG |
| Antibodies | Cat. No. | Manufacturer | Antibody Category | Western Blot | Immunofluorescence |
|---|---|---|---|---|---|
| TGF-β1 | MA5-15065 | Thermofisher | Primary | 1:1000 | |
| Ki67 | ab66155 | Abcam | Primary | 1:200 | |
| β-Actin | 4967s | Cell Signaling | Primary | 1:2000 | |
| Snai1 | 3879S | Cell Signaling | Primary | 1:1000 | |
| Snai2 | 9585S | Cell Signaling | Primary | 1:1000 | |
| VISTA | ab235362 | Abcam | Primary | 1:1000 | |
| TIM-3 | ab241332 | Abcam | Primary | 1:1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, R.; Prithviraj, P.; Bridle, K.R.; Crawford, D.H.G.; Jayachandran, A. Combined Inhibition of TGF-β1-Induced EMT and PD-L1 Silencing Re-Sensitizes Hepatocellular Carcinoma to Sorafenib Treatment. J. Clin. Med. 2021, 10, 1889. https://doi.org/10.3390/jcm10091889
Shrestha R, Prithviraj P, Bridle KR, Crawford DHG, Jayachandran A. Combined Inhibition of TGF-β1-Induced EMT and PD-L1 Silencing Re-Sensitizes Hepatocellular Carcinoma to Sorafenib Treatment. Journal of Clinical Medicine. 2021; 10(9):1889. https://doi.org/10.3390/jcm10091889
Chicago/Turabian StyleShrestha, Ritu, Prashanth Prithviraj, Kim R. Bridle, Darrell H. G. Crawford, and Aparna Jayachandran. 2021. "Combined Inhibition of TGF-β1-Induced EMT and PD-L1 Silencing Re-Sensitizes Hepatocellular Carcinoma to Sorafenib Treatment" Journal of Clinical Medicine 10, no. 9: 1889. https://doi.org/10.3390/jcm10091889
APA StyleShrestha, R., Prithviraj, P., Bridle, K. R., Crawford, D. H. G., & Jayachandran, A. (2021). Combined Inhibition of TGF-β1-Induced EMT and PD-L1 Silencing Re-Sensitizes Hepatocellular Carcinoma to Sorafenib Treatment. Journal of Clinical Medicine, 10(9), 1889. https://doi.org/10.3390/jcm10091889