
Daytime Neuromuscular Electrical Therapy of Tongue Muscles in Improving Snoring in Individuals with Primary Snoring and Mild Obstructive Sleep Apnea

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Recruitment
2.2. Procedures
2.3. Statistical Considerations
3. Results
3.1. Demographics
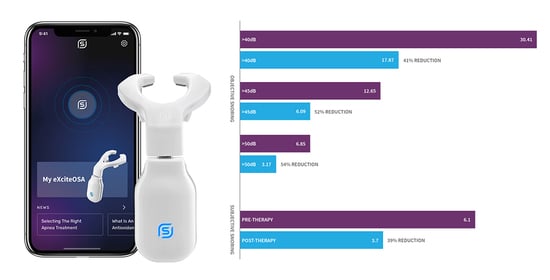
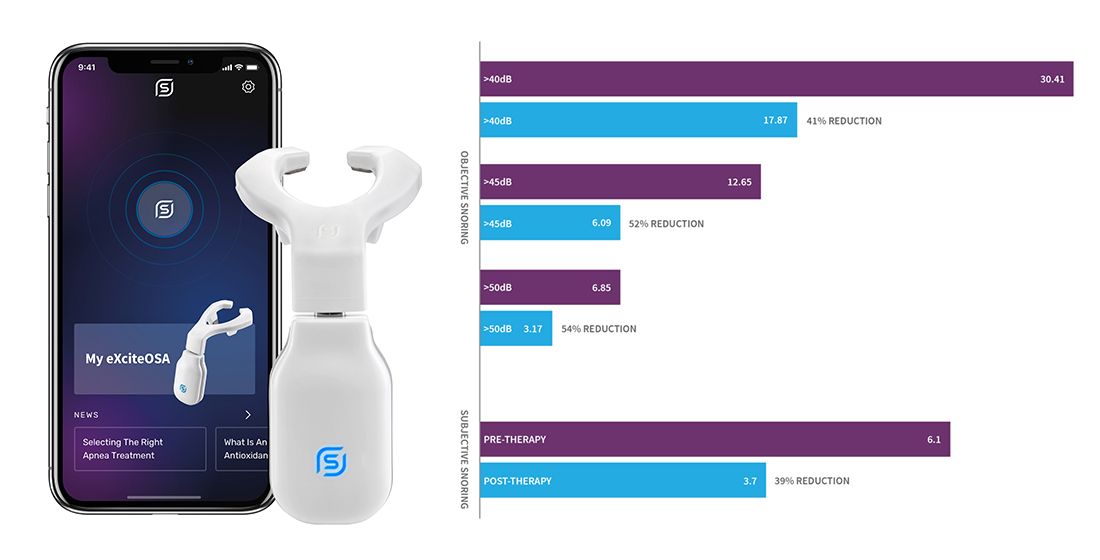
3.2. Change in Objective. Snoring-Sleep Study-% of Sleep Time Snoring
3.3. Change in Bed Partner Reported Snoring—Visual Analog Scale
3.4. Change in Sleep Quality and Daytime Somnolence Parameters
3.5. Correlating Factors
3.6. Stimulation Intensity
3.7. Side Effects and Adverse Events
4. Discussion
4.1. Tolerability of eXciteOSA®
4.2. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.; Morrell, M.J.; Nunez, C.M.; Patel, S.R.; Penzel, T.; Pépin, J.-L.; et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: A literature-based analysis. Lancet Respir. Med. 2019, 7, 687–698. [Google Scholar] [CrossRef]
- Maimon, N.; Hanly, P.J. Does snoring intensity correlate with the severity of obstructive sleep apnea? J. Clin. Sleep Med. 2010, 6, 475–478. [Google Scholar] [CrossRef]
- Teculescu, D.; Benamghar, L.; Hannhart, B.; Michaely, J.-P. Habitual Loud Snoring. A Study of Prevalence and Associations in 850 Middle-Aged French Males. Respiration 2006, 73, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Palta, M.; Dempsey, J.; Skatrud, J.; Weber, S.; Badr, S. The Occurrence of Sleep-Disordered Breathing among Middle-Aged Adults. N. Engl. J. Med. 1993, 328, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Durán, J.; Esnaola, S.; Rubio, R.; Iztueta, Á. Obstructive Sleep Apnea–Hypopnea and Related Clinical Features in a Population-based Sample of Subjects Aged 30 to 70 Yr. Am. J. Respir. Crit. Care Med. 2001, 163, 685–689. [Google Scholar] [CrossRef]
- Kalchiem-Dekel, O.; Westreich, R.; Regev, A.; Novack, V.; Goldberg, M.; Maimon, N. Snoring intensity and excessive daytime sleepiness in subjects without obstructive sleep apnea. Laryngoscope 2016, 126, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, D.; Wang, X.; He, D. Self-reported habitual snoring and risk of cardiovascular disease and all-cause mortality. Atherosclerosis 2014, 235, 189–195. [Google Scholar] [CrossRef]
- Loke, Y.K.; Brown, J.W.L.; Kwok, C.S.; Niruban, A.; Myint, P.K. Association of Obstructive Sleep Apnea with Risk of Serious Cardiovascular Events. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 720–728. [Google Scholar] [CrossRef]
- Yaggi, H.K.; Concato, J.; Kernan, W.N.; Lichtman, J.H.; Brass, L.M.; Mohsenin, V. Obstructive Sleep Apnea as a Risk Factor for Stroke and Death. N. Engl. J. Med. 2005, 353, 2034–2041. [Google Scholar] [CrossRef]
- Valham, F.; Mooe, T.; Rabben, T.; Stenlund, H.; Wiklund, U.; Franklin, K.A. Increased Risk of Stroke in Patients With Coronary Artery Disease and Sleep Apnea. Circulation 2008, 118, 955–960. [Google Scholar] [CrossRef]
- Dopp, J.M.; Reichmuth, K.J.; Morgan, B.J. Obstructive sleep apnea and hypertension: Mechanisms, evaluation, and management. Curr. Hypertens. Rep. 2007, 9, 529–534. [Google Scholar] [CrossRef]
- Parish, J.M.; Somers, V.K. Obstructive Sleep Apnea and Cardiovascular Disease. Mayo Clin. Proc. 2004, 79, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Vgontzas, A.N.; Li, Y.; He, F.; Fernandez-Mendoza, J.; Gaines, J.; Liao, D.; Basta, M.; O Bixler, E. Mild-to-moderate sleep apnea is associated with incident hypertension: Age effect. Sleep 2018, 42, zsy265. [Google Scholar] [CrossRef] [PubMed]
- Shahar, E.; Whitney, C.W.; Redline, S.; Lee, E.T.; Newman, A.B.; Nieto, F.J.; O’Connor, G.T.; Boland, L.L.; Schwartz, J.E.; Samet, J.M. Sleep-disordered Breathing and Cardiovascular Disease. Am. J. Respir. Crit. Care Med. 2001, 163, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Saletu, M.; Nosiska, D.; Kapfhammer, G.; Lalouschek, W.; Saletu, B.; Benesch, T.; Zeitlhofer, J. Structural and serum surrogate markers of cerebrovascular disease in obstructive sleep apnea (OSA). J. Neurol. 2006, 253, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Weaver, T.E.; Kribbs, N.B.; Pack, A.I.; Kline, L.R.; Chugh, D.K.; Maislin, G.; Smith, P.L.; Schwartz, A.R.; Schubert, N.M.; Gillen, K.A.; et al. Night-To-Night Variability in CPAP Use Over the First Three Months of Treatment. Sleep 1997, 20, 278–283. [Google Scholar] [CrossRef]
- Ramar, K.; Dort, L.C.; Katz, S.G.; Lettieri, C.J.; Harrod, C.G.; Thomas, S.M.; Chervin, R.D. Clinical Practice Guideline for the Treatment of Obstructive Sleep Apnea and Snoring with Oral Appliance Therapy: An Update for 2015. J. Clin. Sleep Med. 2015, 11, 773–827. [Google Scholar] [CrossRef]
- Jennum, P.; Ibsen, R.; Kjellberg, J. Social consequences of sleep disordered breathing on patients and their partners: A controlled national study. Eur. Respir. J. 2013, 43, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Isono, S.; Remmers, J.E.; Tanaka, A.; Sho, Y.; Sato, J.; Nishino, T. Anatomy of pharynx in patients with obstructive sleep apnea and in normal subjects. J. Appl. Physiol. 1997, 82, 1319–1326. [Google Scholar] [CrossRef]
- Schwartz, A.R.; Smith, P.L.; Wise, R.A.; Gold, A.R.; Permutt, S. Induction of upper airway occlusion in sleeping individuals with subatmospheric nasal pressure. J. Appl. Physiol. 1988, 64, 535–542. [Google Scholar] [CrossRef]
- Sillen, M.J.H.; Franssen, F.M.E.; Gosker, H.R.; Wouters, E.F.M.; Spruit, M.A. Metabolic and Structural Changes in Lower-Limb Skeletal Muscle Following Neuromuscular Electrical Stimulation: A Systematic Review. PLoS ONE 2013, 8, e69391. [Google Scholar] [CrossRef] [PubMed]
- Wessolleck, E.; Bernd, E.; Dockter, S.; Lang, S.; Sama, A.; Stuck, B.A. Intraoral electrical muscle stimulation in the treatment of snoring. Somnologie 2018, 22, 47–52. [Google Scholar] [CrossRef]
- Kotecha, B.; Wong, P.Y.; Zhang, H.; Hassaan, A. A novel intraoral neuromuscular stimulation device for treating sleep-disordered breathing. Sleep Breath 2021, 1–8. [Google Scholar] [CrossRef]
- Fransson, A.M.C.; Tegelberg, Å.; Leissner, L.; Wenneberg, B.; Isacsson, G. Effects of a Mandibular Protruding Device on the Sleep of Patients with Obstructive Sleep Apnea and Snoring Problems: A 2-Year Follow-Up. Sleep Breath 2003, 7, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Battagel, J.M. Non-apneic snoring and the orthodontist: The effectiveness of mandibular advancement splints. J. Orthod. 2004, 31, 115–123. [Google Scholar] [CrossRef]
- Koutsourelakis, I.; Keliris, A.; Minaritzoglou, A.; Zakynthinos, S. Nasal steroids in snorers can decrease snoring frequency: A randomized placebo-controlled crossover trial. J. Sleep Res. 2015, 24, 160–166. [Google Scholar] [CrossRef]
- Brietzke, S.E.; Mair, E.A. Injection snoreplasty: Extended follow-up and new objective data. Otolaryngol. Head Neck Surg. 2003, 128, 605–615. [Google Scholar] [CrossRef]
- Johnson, J.T.; Vates, J.; Wagner, R.L. Reduction of snoring with a plasma-mediated radiofrequency-based ablation (Coblation) device. Ear Nose Throat J. 2008, 87, 40–43. [Google Scholar] [CrossRef]
- Walker, R.P.; Gatti, W.M.; Poirier, N.; Davis, J.S. Objective Assessment of Snoring Before and After Laser-Assisted Uvulopalatoplasty. Laryngoscope 1996, 106, 1372–1377. [Google Scholar] [CrossRef]
- Hukins, C.A.; Mitchell, I.C.; Hillman, D.R. Radiofrequency tissue volume reduction of the soft palate in simple snoring. Arch. Otolaryngol. Head Neck Surg. 2000, 126, 602–606. [Google Scholar] [CrossRef]
- Cartwright, R.; Venkatesan, T.K.; Caldarelli, D.; Diaz, F. Treatments for Snoring: A Comparison of Somnoplasty and an Oral Appliance. Laryngoscope 2000, 110, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, G.; Vatts, A.; Whinney, D.; Kotecha, B.; Croft, C. SomnoplastyR for simple snoring-a pilot study. Clin. Otolaryngol. 2003, 28, 425–429. [Google Scholar] [CrossRef]
- Prichard, A.J.N.; Smithson, A.J.; White, J.E.S.; Close, P.R.; Drinnan, M.J.; Griffiths, C.J.; Marshall, H.F.; Gibson, G.J. Objective measurement of the results of uvulopalatopharyngoplasty. Clin. Otolaryngol. 1995, 20, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Puhan, M.A.; Suarez, A.; Cascio, C.L.; Zahn, A.; Heitz, M.; Braendli, O. Didgeridoo playing as alternative treatment for obstructive sleep apnoea syndrome: Randomised controlled trial. BMJ 2005, 332, 266–270. [Google Scholar] [CrossRef]
- Guimarães, K.C.; Drager, L.F.; Genta, P.R.; Marcondes, B.F.; Lorenzi-Filho, G. Effects of Oropharyngeal Exercises on Patients with Moderate Obstructive Sleep Apnea Syndrome. Am. J. Respir. Crit. Care Med. 2009, 179, 962–966. [Google Scholar] [CrossRef]
- Pae, E.-K.; Hyatt, J.-P.K.; Wu, J.; Chien, P. Short-term electrical stimulation alters tongue muscle fibre type composition. Arch. Oral Biol. 2007, 52, 544–551. [Google Scholar] [CrossRef]
- Series, F.; Côté, C.; Simoneau, J.A.; Gélinas, Y.; Pierre, S.S.; Leclerc, J.; Ferland, R.; Marc, I. Physiologic, metabolic, and muscle fiber type characteristics of musculus uvulae in sleep apnea hypopnea syndrome and in snorers. J. Clin. Investig. 1995, 95, 20–25. [Google Scholar] [CrossRef]
- Sériès, F.J.; A Simoneau, S.; Pierre, S.S.; Marc, I. Characteristics of the genioglossus and musculus uvulae in sleep apnea hypopnea syndrome and in snorers. Am. J. Respir. Crit. Care Med. 1996, 153, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Eckert, D.J.; Lo, Y.L.; Saboisky, J.P.; Jordan, A.S.; White, D.P.; Malhotra, A. Sensorimotor function of the upper-airway muscles and respiratory sensory processing in untreated obstructive sleep apnea. J. Appl. Physiol. 2011, 111, 1644–1653. [Google Scholar] [CrossRef]
- Randerath, W.J.; Galetke, W.; Domanski, U.; Weitkunat, R.; Ruhle, K.-H. Tongue-muscle training by intraoral electrical neurostimulation in patients with obstructive sleep apnea. Sleep 2004, 27, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Verse, T.; Schwalb, J.; Hörmann, K.; Stuck, B.A.; Maurer, J.T. Transkutane, submentale Elektrostimulationstherapie bei obstruktiver Schlafapnoe. HNO 2003, 51, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Rathi, V.K.; Kondamuri, N.S.; Naunheim, M.R.; Gadkaree, S.K.; Metson, R.B.; Scangas, G.A. Use and Cost of a Hypoglossal Nerve Stimulator Device for Obstructive Sleep Apnea Between 2015 and 2018. JAMA Otolaryngol. Head Neck Surg. 2019, 145, 975–977. [Google Scholar] [CrossRef]
- Wray, C.M.; Thaler, E.R. Hypoglossal nerve stimulation for obstructive sleep apnea: A review of the literature. World J. Otorhinolaryngol. Head Neck Surg. 2016, 2, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Rotenberg, B.W.; Murariu, D.; Pang, K.P. Trends in CPAP adherence over twenty years of data collection: A flattened curve. J. Otolaryngol. Head Neck Surg. 2016, 45, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chowdhuri, S.; Quan, S.F.; Almeida, F.; Ayappa, I.; Batool-Anwar, S.; Budhiraja, R.; Cruse, P.E.; Drager, L.F.; Griss, B.; Marshall, N.; et al. An Official American Thoracic Society Research Statement: Impact of Mild Obstructive Sleep Apnea in Adults. Am. J. Respir. Crit. Care Med. 2016, 193, e37–e54. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Descriptive Statistics | |||||
|---|---|---|---|---|---|
| N | Minimum | Maximum | Mean | Std. Deviation | |
| Age at time of trial | 115 | 24.00 | 79.00 | 45.71 | 13.52 |
| BMI | 115 | 20.40 | 34.00 | 27.02 | 3.26 |
| Pre-AHI | 115 | 0.20 | 15.00 | 6.47 | 4.44 |
| Pre-Patient ESS | 115 | 0.00 | 22.00 | 8.06 | 4.59 |
| Neck Collar (inch) | 115 | 12.50 | 19.00 | 15.23 | 1.49 |
| Intervention Type | Intervention | No. People | Measure | Pre | Post | Significance | % Change | Reference |
|---|---|---|---|---|---|---|---|---|
| Device | MAD (People with OSA) | 22 | Mean Peak Snoring Intensity (dB) | 71.6 dB | 62 dB | Statistical Significance | −14% | Fransson et al. [24] |
| Device | MAD (People with snoring problems) | 13 | Mean Peak Snoring Intensity (dB) | 63.3 dB | 57.5 dB | Statistical Significance | −9% | Fransson et al. [24] |
| Device | MAD | 11/35 * | L5–L95 ** | 240 (mV) | 75 (mV) | Statistical Significance | −70% | Smith and Battagel [25] |
| Medicine | Budesonide Nasal Drops | 24 | Mean Snoring Intensity (dB) | 61.2 dB | 60.1 dB | Not Declared | −2% | Koutsourelakis et al. [26] |
| Medicine | Injection Snoreplasty (3% Sotradecol into soft palate) | 17 | Average Loudness (dB) | 13 dB | 7 dB | Not Significant | −46% | Brietzke and Mair [27] |
| Surgical | Coblation, Radiofrequency of Soft Palate | 21 | SNAP Snoring Loudness | 12 dB | 8 dB | Statistical Significance | −33% | Johnson et al. [28] |
| Surgical | Laser-Assisted uvulopalatoplasty (LAUP) | 27 | Average Loudness (dB) | 12.7 dB | 8.7 dB | Statistical Significance | −32% | Walker et al. [29] |
| Surgical | Radiofrequency Soft Palate Tissue Reduction | 20 | Snoring Intensity | 60.2 dB | 64.9 dB | Statistical Significance | 8% | Hukins et al. [30] |
| Surgical | Somnoplasty (Radiofrequency of soft palate) | 10 | % Time Loud Snoring | 10.62% | 8.03% | Not Declared | −24% | Cartwright et al. [31] |
| Surgical | Somnoplasty (Radiofrequency of soft palate) | 10 | % Time Spent Snoring | 3/7 (42%) people showed improvement in duration of snoring of 30%, 38%, and 48%. | Sandhu et. al. [32] | |||
| Surgical | Uvulopalatopharyngoplasty (UVPP) | 32 | L, level above which 5% of the sound occurs | 41 dB | 38.8 dB | Statistical Significance | −12% | Pritchard et al. [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baptista, P.M.; Martínez Ruiz de Apodaca, P.; Carrasco, M.; Fernandez, S.; Wong, P.Y.; Zhang, H.; Hassaan, A.; Kotecha, B. Daytime Neuromuscular Electrical Therapy of Tongue Muscles in Improving Snoring in Individuals with Primary Snoring and Mild Obstructive Sleep Apnea. J. Clin. Med. 2021, 10, 1883. https://doi.org/10.3390/jcm10091883
Baptista PM, Martínez Ruiz de Apodaca P, Carrasco M, Fernandez S, Wong PY, Zhang H, Hassaan A, Kotecha B. Daytime Neuromuscular Electrical Therapy of Tongue Muscles in Improving Snoring in Individuals with Primary Snoring and Mild Obstructive Sleep Apnea. Journal of Clinical Medicine. 2021; 10(9):1883. https://doi.org/10.3390/jcm10091883
Chicago/Turabian StyleBaptista, Peter M., Paula Martínez Ruiz de Apodaca, Marina Carrasco, Secundino Fernandez, Phui Yee Wong, Henry Zhang, Amro Hassaan, and Bhik Kotecha. 2021. "Daytime Neuromuscular Electrical Therapy of Tongue Muscles in Improving Snoring in Individuals with Primary Snoring and Mild Obstructive Sleep Apnea" Journal of Clinical Medicine 10, no. 9: 1883. https://doi.org/10.3390/jcm10091883
APA StyleBaptista, P. M., Martínez Ruiz de Apodaca, P., Carrasco, M., Fernandez, S., Wong, P. Y., Zhang, H., Hassaan, A., & Kotecha, B. (2021). Daytime Neuromuscular Electrical Therapy of Tongue Muscles in Improving Snoring in Individuals with Primary Snoring and Mild Obstructive Sleep Apnea. Journal of Clinical Medicine, 10(9), 1883. https://doi.org/10.3390/jcm10091883