
Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies—Current Research and Clinical Approaches


Abstract
1. Introduction
2. Multiple Myeloma Is the Second Most Common Hematological Malignancy: Current Treatment Strategies
3. Pharmacokinetics and Pharmacodynamics of Melphalan
4. Autologous Stem Cell Transplantation (ASCT) in Combination with High Doses of Melphalan (HDM) as the Standard Treatment for Newly Diagnosed Patients with Multiple Myeloma

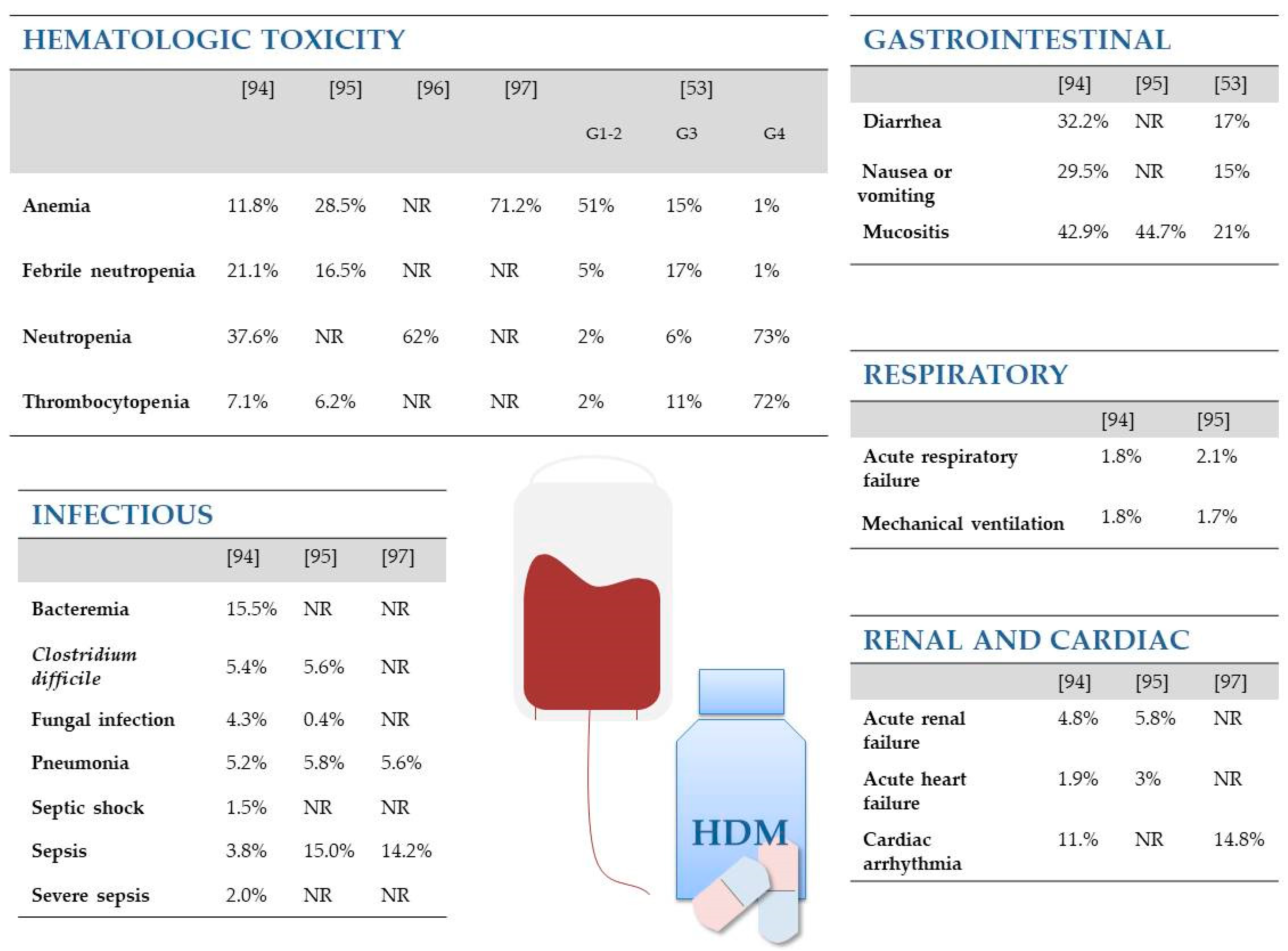{kind=link}
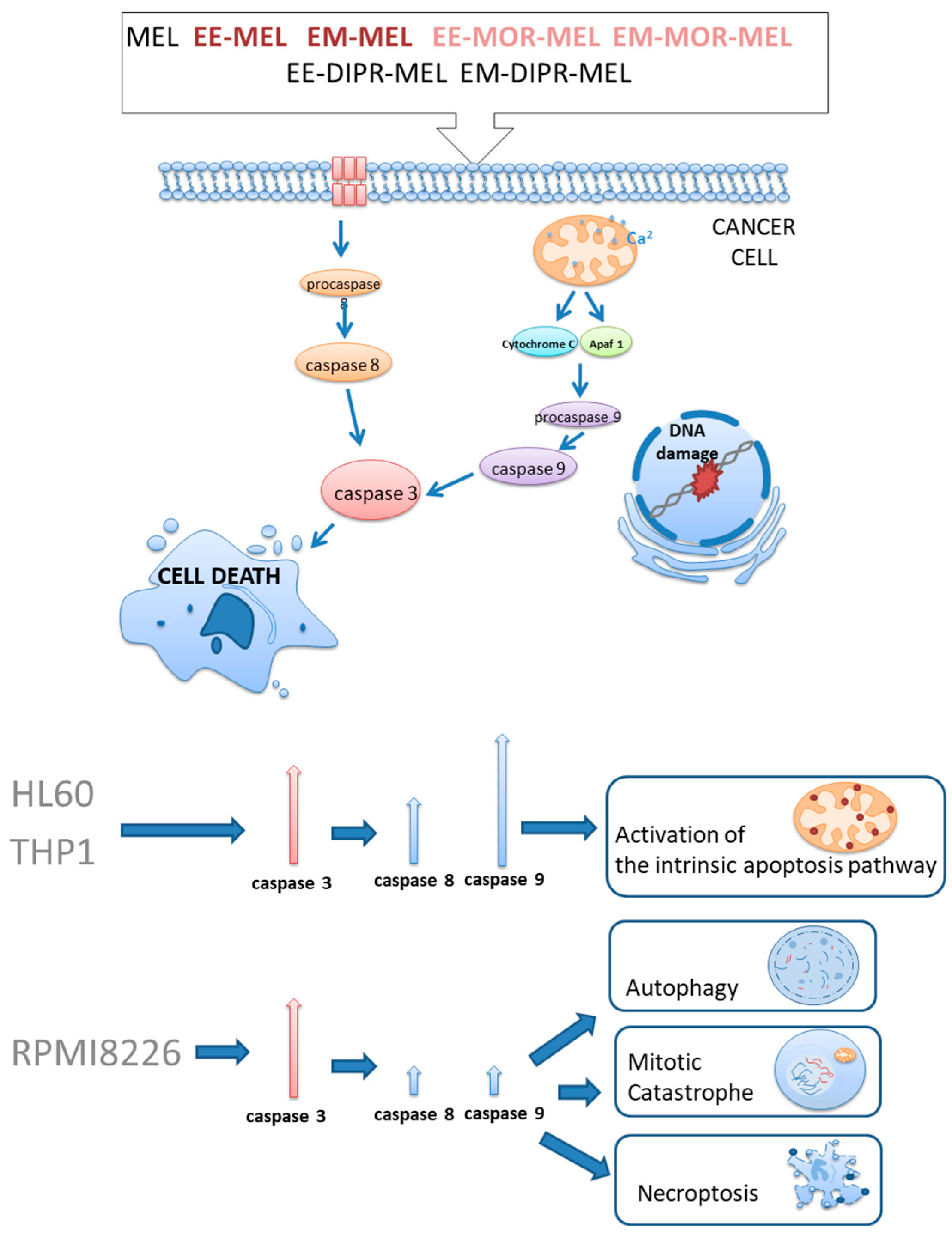{kind=link}
| Ref. | Type of Study | No. of Patients | Treatment Regimen | Results | |||
|---|---|---|---|---|---|---|---|
| Response | PFS | OS | MRD Negativity | ||||
| [53,61] | Multicenter, randomized, open-label, phase III study | 1503 | I: MEL (200 mg/m2) + ASCT (intensification therapy) + RVD/no cons. | VGPR: 84% | 56.7 months (95% CI 49.3–64.5) | NA | 36% (10-5) |
| II: VMP (intensification therapy) + RVD/no cons. | VGPR: 75% | 41·9 months (95% CI 37.5–46.9) | NA | 64% (10-5) | |||
| HR for PFS of ASCT compared with VMP: 0.73, 0.62–0.85; p = 0.0001. | |||||||
| [55,61] | Open-label, randomized, phase III study | 402 | I: MEL (200 mg/m2) + ASCT (consolidation therapy) ± Rm. | CR (post-consolidation): 23% | 43.0 months | 4-year 81.6% | NA |
| II: MPR (consolidation therapy) ± Rm. | CR (post-consolidation): 18% | 22.4 months | 4-year 65.3% | NA | |||
| HR for PFS: 0.44; 95% CI: 0.32–0.61; p < 0.001. HR for OS: 0.55; 95% CI, 0.32–0.93; p = 0.02. | |||||||
| [54,61] | Multicenter, randomized, open-label, phase III study | 389 | I: MEL (200 mg/m2) +ASCT (consolidation therapy) +Rm./RPm. | CR: 33% (MEL-ASCT +Rm.) CR: 37% (MEL-ASCT +RPm.) | 43.3 months (95% CI 33.2–52.2); | 4-year OS: 75% (MEL-ASCT + Rm.) 4-year OS: 77% (MEL-ASCT + RPm.) | NA |
| II: CRD (consolidation therapy) +Rm./RPm. | CR: 27% (CRD + Rm.)CR: 23% (CRD + RPm.) | 28.6 months (95% CI 20.6–36.7) | 4-year OS: 77% (CRD + Rm.) 4-year OS:76% (CRD + RPm.) | NA | |||
| HR for the first 24 months 2.51, 95% CI 1.60–3.94; p < 0.0001 | |||||||
| [59] | Prospective, randomized, phase III study | 758 | I: MEL+ ASCT (consolidation therapy) + Rm. | 1-year ORR: 47.1% (n = 208) | 53.9% (95% CI: 47.4–60%) | 38-month OS: 83.7% (95% CI: 78.4–87.8%) | NA |
| II: MEL+ ASCT/ASCT (consolidation therapy) + Rm. | 1-year ORR: 50.5% (n = 192) | 58.5% (95% CI: 51.7–64.6%) | 38-month OS: 81.8% (95% CI: 76.2–86.2%) | NA | |||
| III: MEL+ ASCT +RVD (consolidation therapy) +Rm. | 1-year ORR: 58.4% (n = 209) | 57.8% (95% CI: 51.4–63.7%) | 38-month OS: 85.4% (95% CI: 80.4–89.3%) | NA | |||
| Patients with high-risk disease experienced higher rates of treatment failure (progression or death; HR, 1.66; 95% CI: 1.30–2.11) and overall mortality (HR, 1.49; 95% CI: 1.01– 2.20) compared with patients with standard-risk disease. | |||||||
| [52,61] | Open-label, randomized, phase III study | 700 | I: MEL (200 mg/m2) + ASCT+ RVD (consolidation therapy) + Rm. | CR: 59% | 50 months | 4-year OS: 81% | 79% (10-4) |
| II: RVD (consolidation therapy) +Rm. | CR: 48% | 36 months | 4-year OS: 82% | 65% (10-4) | |||
| HR for disease progression or death, 0.65; p < 0.001 | |||||||
| [58,63] | Open-label, randomized, phase III study | 297 | I: MEL (200 mg/m2) + sASCT (consolidation therapy) | CR: 92.1% | 19 months (95% CI 16–26) | 67 months (95% CI 55–not estimable) | NA |
| II: cyclophosphamine (consolidation therapy) | CR: 94.1% | 11 months (95% CI: 9–12) | 52 months (95% CI 42–60) | NA | |||
| HR for PFS: 0.45 (95% CI 0.31–0.64), p < 0.0001 HR for OS: 0.56 (0.35–0.90), p = 0.0169 | |||||||
| Ref. | Type of Study | No. of Patients | Treatment Regimen | Results |
|---|---|---|---|---|
| [64] | Randomized, a double-blind, placebo-controlled phase III trial | 656 | I: ixazomib maintenance therapy II: placebo both groups had undergone standard induction therapy with MEL (200 mg/m2) conditioning and a single ASCT | There was a 28% reduction in the risk of PFS with ixazomib vs. placebo (26.5 months (95% CI 23.7–33.8) vs. 21.3 months (18.0–24.7); HR 0.72, 95% CI 0.58–0.89; p = 0.0023). At the time of this analysis no increase in secondary malignancies was observed with ixazomib therapy (3% patients) compared with placebo (3% patients). |
| [65,66,67] | Open-label, randomized, phase III study | 458 | RVD (induction therapy) + BU (12 mg/kg)- MEL (140 mg/m2) + ASCT /MEL (200 mg/m2) +ASCT + RVD (consolidation therapy) | Conditioning with BU-MEL in comparison to MEL was associated with longer PFS (41 vs. 31 months; p = 0.009), although OS was similar to that in the melphalan 200 mg/m2 group. This should be counterbalanced against the higher frequency of veno-occlusive disease-related deaths. Access to novel agents as a salvage therapy after relapse/progression was decreased for patients receiving BU-MEL (43%) vs. MEL (58%; p = 0.01). |
| [68] | Prospective, investigator-initiated, nonrandomized, multicenter, open-label, phase II study | 100 | RVD (induction therapy) + MEL (200 mg/m2) + ASCT + Rm ± PCD | PCD was an effective therapy after first relapse with RVD. Responses were obtained in 85% of patients evaluated: CR (1%), VGPR (33%).After 4 cycles, the rate of PR (or better) was 85%. 94% of planned ASCTs were performed. |
| [69] | Single-arm, prospective phase II study | 125 | I: MEL (200 mg/m2) + ASCT + Lipegfilgrastim (LIP) II: MEL (200 mg/m2) + ASCT + Filgrastim (FIL) | The median duration of grade 4 neutropenia was 5 days in both LIP and FIL groups. The incidence of FN was significantly lower in the LIP than in the FIL group (29% vs. 49%, respectively, p = 0.024). The HR of ANC ≥ 0.5 × 10(9)/L was 3.5 times higher in patients treated with LIP than in those treated with FIL (HR 3.50, 95% CI 2.28–5.38, p < 0.001), indicating that the response was faster in LIP treated patients than in those treated with FIL. |
| Clinical Trial Identifier | Trial Phase | Treatment Regimen | Objective of Trial |
|---|---|---|---|
| NCT03829371 | 1 | VMP, MPT and lenalidomide with low-dose dexamethasone | Comparison of treatment regimens in an autologous stem cell transplantation ineligible population affected by MM. |
| NCT03346135 | 2 | melphalan, daratumumab | Daratumumab after stem cell transplant for the treatment of MM. |
| NCT03481556 | 2 | melphalan, dexamethasone, bortezomib, daratumumab | Assessing patients with relapsed or relapsed-refractory MM following 1–4 lines of prior therapy. |
| NCT04466475 | 1 | astatine at 211 anti-cd38 monoclonal antibody okt10-b10, melphalan | Radioimmunotherapy and chemotherapy before stem cell transplantation. Therapy based on 211At-OKT10-B10 in combination with melphalan before a stem cell transplant may be more effective than melphalan monotherapy in MM. |
| NCT03556332 | 1 | carfilzomib, lenalidomide, dexamethasone, daratumumab, Procedure: autologous hematopoietic cell transplantation (melphalan) | Assessing patients with relapsed or refractory myeloma with re-administration of ASCT to a patient with symptoms of disease progression. The effect of the drugs in combinations will be compared before and after ASCT in MM. |
| NCT02581007 | 2 | fludarabine, melphalan, cyclophosphamide | Evaluation of the safety and efficacy of a reduced intensity allogeneic HSCT from partially HLA-mismatched first-degree relatives utilizing PBSC as the stem cell source. |
| NCT04008888 | 1 | melphalan, fludarabine, PI and dexamethasone as maintenance therapy, PI + IMids + dexamethasone as consolidated chemotherapy | Assessing efficacy and safety of the holistic treatment of young high-risk MM patients who were designed to receive a combination of high-dose chemotherapy with allogeneic or autologous HSCT. |
| NCT01453088 | 3 | melphalan, bortezomib | Assessing a standard regimen and the newly established melphalan and bortezomib regimen in patients with MM 65 years or older. |
| NCT02780609 | ½ | selinexor, melphalan, dexamethasone, fosaprepitant | Determination of the maximum tolerated dose of selinexor in combination with high-dose melphalan as a conditioning regimen for hematopoietic cell transplant in MM. |
| NCT03570983 | 2 | allopurinol, carmustine, etoposide, cytarabine, melphalan | Comparing melphalan to carmustine, etoposide, cytarabine, and melphalan (beam) as a conditioning regimen for patients with MM undergoing high dose therapy followed by autologous stem cell reinfusion. |
| NCT02043847 | 1 | radiation: total marrow irradiation drug:melphalan, filgrastim (g-csf) | Assessing patients with relapsed or refractory MM will receive high dose melphalan with autologous stem cell rescue. The pre-transplant conditioning is based on total marrow irradiation. |
5. Clinical Usage of Combination Treatment with Melphalan to Improve the Effectiveness of Cancer Therapy
6. “Weak Side” of Melphalan
7. Drug Resistance to Melphalan
8. Attempts to Find a “Better Melphalan”
8.1. Drug Carriers as a Way to Reduce Systemic Toxicity
8.2. Chemical Modifications of the Melphalan Molecule as a Way to Improve Antitumor Activity
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433. [Google Scholar] [CrossRef]
- Liu, H.; Lightfoot, R.; Stevens, J.L. Activation of Heat Shock Factor by Alkylating Agents is Triggered by Glutathione Depletion and Oxidation of Protein Thiols. J. Biol. Chem. 1996, 271, 4805–4812. [Google Scholar] [CrossRef] [PubMed]
- Bergel, F.; Stock, J.A. Cyto-active Amino-acid and Peptide Derivatives. J. Chem. Soc. 1954, 2409–2417. [Google Scholar] [CrossRef]
- Falco, P.; Bringhen, S.; Avonto, I.; Gay, F.; Morabito, F.; Boccadoro, M.; Palumbo, A. Melphalan and its role in the management of patients with multiple myeloma. Expert Rev. Anticancer Ther. 2007, 7, 945–957. [Google Scholar] [CrossRef]
- Saikia, T.K. Developments in the Field of Myeloma in the Last Decade. Indian J. Hematol. Blood Transfus. 2017, 33, 3–7. [Google Scholar] [CrossRef][Green Version]
- Dehghanifard, A.; Kaviani, S.; Abroun, S.; Mehdizadeh, M.; Saiedi, S.; Maali, A.; Ghaffari, S.; Azad, M. Various Signaling Pathways in Multiple Myeloma Cells and Effects of Treatment on These Pathways. Clin. Lymphoma Myeloma Leuk. 2018, 18, 311–320. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef]
- Esma, F.; Salvini, M.; Troia, R.; Boccadoro, M.; LaRocca, A.; Pautasso, C. Melphalan hydrochloride for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2017, 18, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.; Paviglianiti, A.; Gentile, M.; Martinelli, G.; Cerchione, C. Allogenic stem cell transplantation in multiple myeloma: Dead or alive and kicking? Panminerva Medica 2021, 62, 234–243. [Google Scholar] [CrossRef]
- Gentile, M.; Vigna, E.; Recchia, A.G.; Morabito, L.; Mendicino, F.; Giagnuolo, G.; Morabito, F. Bendamustine in multiple myeloma. Eur. J. Haematol. 2015, 95, 377–388. [Google Scholar] [CrossRef]
- Chauhan, D.; Hideshima, T.; Rosen, S.; Reed, J.C.; Kharbanda, S.; Anderson, K.C. Apaf-1/Cytochrome c-independent and Smac-dependent Induction of Apoptosis in Multiple Myeloma (MM) Cells. J. Biol. Chem. 2001, 276, 24453–24456. [Google Scholar] [CrossRef]
- Chauhan, D.; Pandey, P.; Ogata, A.; Teoh, G.; Treon, S.; Urashima, M.; Kharbanda, S.; Anderson, K.C. Dexamethasone induces apoptosis of multiple myeloma cells in a JNK/SAP kinase independent mechanism. Oncogene 1997, 15, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Pandey, P.; Ogata, A.; Teoh, G.; Krett, N.; Halgren, R.; Rosen, S.; Kufe, D.; Kharbanda, S.; Anderson, K. Cytochrome c-dependent and -independent Induction of Apoptosis in Multiple Myeloma Cells. J. Biol. Chem. 1997, 272, 29995–29997. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Frost, P.; Shi, Y.; Hoang, B.; Sharma, S.; Fisher, M.; Gera, J.; Lichtenstein, A. Mechanism by Which Mammalian Target of Rapamycin Inhibitors Sensitize Multiple Myeloma Cells to Dexamethasone-Induced Apoptosis. Cancer Res. 2006, 66, 2305–2313. [Google Scholar] [CrossRef]
- Burwick, N.; Sharma, S. Glucocorticoids in multiple myeloma: Past, present, and future. Ann. Hematol. 2019, 98, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Okazuka, K.; Ishida, T. Proteasome inhibitors for multiple myeloma. Jpn. J. Clin. Oncol. 2018, 48, 785–793. [Google Scholar] [CrossRef] [PubMed]
- McKeage, K.; Lyseng-Williamson, K.A. Daratumumab in multiple myeloma: A guide to its use as monotherapy in the EU. Drugs Ther. Perspect. 2016, 32, 463–469. [Google Scholar] [CrossRef]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef]
- Vaxman, I.; Sidiqi, M.H.; Gertz, M. Venetoclax for the treatment of multiple myeloma. Expert Rev. Hematol. 2018, 11, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2009, 24, 22–32. [Google Scholar] [CrossRef]
- Tachita, T.; Kinoshita, S.; Ri, M.; Aoki, S.; Asano, A.; Kanamori, T.; Yoshida, T.; Totani, H.; Ito, A.; Kusumoto, S.; et al. Expression, mutation, and methylation of cereblon-pathway genes at pre- and post-lenalidomide treatment in multiple myeloma. Cancer Sci. 2020, 111, 1333–1343. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G., Jr. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Li, C.; Mei, H.; Hu, Y.; Guo, T.; Liu, L.; Jiang, H.; Tang, L.; Wu, Y.; Ai, L.; Deng, J.; et al. A Bispecific CAR-T Cell Therapy Targeting Bcma and CD38 for Relapsed/Refractory Multiple Myeloma: Updated Results from a Phase 1 Dose-Climbing Trial. Blood 2019, 134, 930. [Google Scholar] [CrossRef]
- Kergueris, M.F.; Milpied, N.; Moreau, P.; Harousseau, J.L.; Larousse, C. Pharmacokinetics of high-dose melphalan in adults: Influence of renal function. Anticancer Res. 1994, 14, 2379–2382. [Google Scholar]
- Bosanquet, A.G.; Gilby, E.D. Pharmacokinetics of oral and intravenous melphalan during routine treatment of multiple myeloma. Eur. J. Cancer Clin. Oncol. 1982, 18, 355–362. [Google Scholar] [CrossRef]
- Pinguet, F.; Martel, P.; Fabbro, M.; Petit, I.; Canal, P.; Culine, S.; Astre, C.; Bressolle, F. Pharmacokinetics of high-dose intravenous melphalan in patients undergoing peripheral blood hematopoietic progenitor-cell transplantation. Anticancer Res. 1997, 17, 605–611. [Google Scholar]
- Moreau, P.; Kergueris, M.-F.; Milpied, N.; Le Tortorec, S.; Mahé, B.; Bulabois, C.-E.; Rapp, M.-J.; Larousse, C.; Bataille, R.; Harousseau, J.-L. A pilot study of 220 mg/m2 melphalan followed by autologous stem cell transplantation in patients with advanced haematological malignancies: Pharmacokinetics and toxicity. Br. J. Haematol. 1996, 95, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Reece, P.; Hill, H.; Green, R.; Morris, R.; Dale, B.; Kotasek, D.; Sage, R. Renal clearance and protein binding of melphalan in patients with cancer. Cancer Chemother. Pharmacol. 1988, 22, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Mougenot, P.; Fabbro, M.; Poujol, S.; Pinguet, F.; Culine, S.; Astre, C.; Bressolle, F. Population pharmacokinetics of melphalan, infused over a 24-hour period, in patients with advanced malignancies. Cancer Chemother. Pharmacol. 2004, 53, 503–512. [Google Scholar] [CrossRef]
- Pinguet, F.; Culine, S.; Bressolle, F.; Astre, C.; Serre, M.P.; Chevillard, C.; Fabbro, M. A phase I and pharmacokinetic study of melphalan using a 24-hour continuous infusion in patients with advanced malignancies. Clin. Cancer Res. 2000, 6, 57–63. [Google Scholar] [PubMed]
- Hersh, M.R.; Ludden, T.M.; Kuhn, J.G.; Knight, W.A. Pharmacokinetics of high dose melphalan. Investig. New Drugs 1983, 1, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Ehrsson, H.; Eksborg, S.; Osterborg, A.; Mellstedt, H.; Lindfors, A. Oral melphalan pharmacokinetics--relation to dose in patients with multiple myeloma. Cancer Immunol. Immunother. 1989, 6, 151–154. [Google Scholar]
- Alberts, D.S.; Chang, S.Y.; Chen, H.-S.G.; Evans, T.L.; Moon, T.E. Oral melphalan kinetics. Clin Pharmacol Ther. 1979, 26, 737–745. [Google Scholar] [CrossRef]
- Reece, P.A.; Kotasek, D.; Morris, R.G.; Dale, B.M.; Sage, R.E. The effect of food on oral melphalan absorption. Cancer Chemother. Pharmacol. 1986, 16, 194–197. [Google Scholar] [CrossRef]
- Kuczma, M.; Ding, Z.-C.; Zhou, G. Immunostimulatory Effects of Melphalan and Usefulness in Adoptive Cell Therapy with Antitumor CD4+ T Cells. Crit. Rev. Immunol. 2016, 36, 179–191. [Google Scholar] [CrossRef]
- Haines, D.R.; Fuller, R.W.; Ahmad, S.; Vistica, D.T.; Marquez, V.E. Selective cytotoxicity of a system L specific amino acid nitrogen mustard. J. Med. Chem. 1987, 30, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Vistica, D. Cytotoxicity as an indicator for transport mechanism: Evidence that murine bone marrow progenitor cells lack a high-affinity leucine carrier that transports melphalan in murine L1210 leukemia cells. Blood 1980, 56, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Begleiter, A.; Lam, H.Y.; Grover, J.; Froese, E.; Goldenberg, G.J. Evidence for active transport of melphalan by two amino acid carriers in L5178Y lymphoblasts in vitro. Cancer Res. 1979, 39, 353–359. [Google Scholar]
- Goldenberg, G.; Lam, H.; Begleiter, A. Active carrier-mediated transport of melphalan by two separate amino acid transport systems in LPC-1 plasmacytoma cells in vitro. J. Biol. Chem. 1979, 254, 1057–1064. [Google Scholar] [CrossRef]
- Singh, N.; Ecker, G.F. Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1. Int. J. Mol. Sci. 2018, 19, 1278. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Nath, C.E.; Lazarus, H.M. Not too little, not too much—Just right! (Better ways to give high dose melphalan). Bone Marrow Transpl. 2014, 49, 1457–1465. [Google Scholar] [CrossRef]
- Maybury, B.; Cook, G.; Pratt, G.; Yong, K.; Ramasamy, K. Augmenting Autologous Stem Cell Transplantation to Improve Outcomes in Myeloma. Biol. Blood Marrow Transpl. 2016, 22, 1926–1937. [Google Scholar] [CrossRef]
- Diamond, B.; MacLachlan, K.; Chung, D.J.; Lesokhin, A.M.; Landgren, C.O. Maintenance therapy and need for cessation studies in multiple myeloma: Focus on the future. Best Pract. Res. Clin. Haematol. 2020, 33, 101140. [Google Scholar] [CrossRef] [PubMed]
- McElwain, T.J.; Powles, R.L. High-dose intravenous melphalan for plasma-cell leukaemia and myeloma. Lancet 1983, 322, 822–824. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Miguel, J.F.S. Management of multiple myeloma in the newly diagnosed patient. Hematology 2017, 2017, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Dew, A.; Hill, E. The changing role of high dose melphalan with stem cell rescue in the treatment of newly diagnosed multiple myeloma in the era of modern therapies-back to the future! Best Pract. Res. Clin. Haematol. 2020, 33, 101150. [Google Scholar] [CrossRef]
- Cavo, M.; Rajkumar, S.V.; Palumbo, A.; Moreau, P.; Orlowski, R.; Bladé, J.; Sezer, O.; Ludwig, H.; Dimopoulos, M.A.; Attal, M.; et al. International Myeloma Working Group consensus approach to the treatment of multiple myeloma patients who are candidates for autologous stem cell transplantation. Blood 2011, 117, 6063–6073. [Google Scholar] [CrossRef]
- Merz, M.; Neben, K.; Raab, M.S.; Sauer, S.; Egerer, G.; Hundemer, M.; Hose, D.; Kunz, C.; Heiß, C.; Ho, A.D.; et al. Autologous stem cell transplantation for elderly patients with newly diagnosed multiple myeloma in the era of novel agents. Ann. Oncol. 2014, 25, 189–195. [Google Scholar] [CrossRef]
- Attal, M.; Harousseau, J.-L.; Stoppa, A.-M.; Sotto, J.-J.; Fuzibet, J.-G.; Rossi, J.-F.; Casassus, P.; Maisonneuve, H.; Facon, T.; Ifrah, N.; et al. A Prospective, Randomized Trial of Autologous Bone Marrow Transplantation and Chemotherapy in Multiple Myeloma. N. Engl. J. Med. 1996, 335, 91–97. [Google Scholar] [CrossRef]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef]
- Cavo, M.; Gay, F.; Beksac, M.; Pantani, L.; Petrucci, M.T.; Dimopoulos, M.A.; Dozza, L.; van der Holt, B.; Zweegman, S.; Oliva, S.; et al. Autologous haematopoietic stem-cell transplantation versus bortezomib–melphalan–prednisone, with or without bortezomib–lenalidomide–dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): A multicentre, randomised, open-label, phase 3 study. Lancet Haematol. 2020, 7, e456–e468. [Google Scholar] [CrossRef] [PubMed]
- Gay, F.; Oliva, S.; Petrucci, M.T.; Conticello, C.; Catalano, L.; Corradini, P.; Siniscalchi, A.; Magarotto, V.; Pour, L.; Carella, A.; et al. Chemotherapy plus lenalidomide versus autologous transplantation, followed by lenalidomide plus prednisone versus lenalidomide maintenance, in patients with multiple myeloma: A randomised, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 1617–1629. [Google Scholar] [CrossRef]
- Palumbo, A.; Cavallo, F.; Gay, F.; Di Raimondo, F.; Ben Yehuda, D.; Petrucci, M.T.; Pezzatti, S.; Caravita, T.; Cerrato, C.; Ribakovsky, E.; et al. Autologous Transplantation and Maintenance Therapy in Multiple Myeloma. N. Engl. J. Med. 2014, 371, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Kastritis, E.; Terpos, E.; Dimopoulos, M.A. Multiple myeloma: Role of autologous transplantation. Cancer Treat. Rev. 2020, 82, 101929. [Google Scholar] [CrossRef]
- Al Hamed, R.; Bazarbachi, A.H.; Malard, F.; Harousseau, J.-L.; Mohty, M. Current status of autologous stem cell transplantation for multiple myeloma. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Cook, G.; Ashcroft, A.J.; Cairns, D.A.; Williams, C.D.; Brown, J.M.; Cavenagh, J.D.; Snowden, J.A.; Parrish, C.; Yong, K.; Cavet, J.; et al. The effect of salvage autologous stem-cell transplantation on overall survival in patients with relapsed multiple myeloma (final results from BSBMT/UKMF Myeloma X Relapse [Intensive]): A randomised, open-label, phase 3 trial. Lancet Haematol. 2016, 3, e340–e351. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Pasquini, M.C.; Blackwell, B.; Hari, P.; Bashey, A.; Devine, S.; Efebera, Y.; Ganguly, S.; Gasparetto, C.; Geller, N.; et al. Autologous Transplantation, Consolidation, and Maintenance Therapy in Multiple Myeloma: Results of the BMT CTN 0702 Trial. J. Clin. Oncol. 2019, 37, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Stojanoski, Z.; Georgievski, B.; Cevreska, L.; Stojanovic, A.; Pivkova, A.; Genadieva-Stavric, S.; Stankovic, S.; Karadzova-Stojanoska, A. Autologous stem-cell trans-plantation in patients with multiple myeloma. Prilozi 2008, 29, 265–279. [Google Scholar]
- Garderet, L.; Morris, C.; Beksac, M.; Gahrton, G.; Schönland, S.; Yakoub-Agha, I.; Hayden, P.J. Are Autologous Stem Cell Transplants Still Required to Treat Myeloma in the Era of Novel Therapies? A Review from the Chronic Malignancies Working Party of the EBMT. Biol. Blood Marrow Transpl. 2020, 26, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Soekojo, C.Y.; Kumar, S.K. Stem-cell transplantation in multiple myeloma: How far have we come? Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef]
- Cook, G.; Royle, K.; O’Connor, S.; Cairns, D.A.; Ashcroft, A.J.; Williams, C.D.; Hockaday, A.; Cavenagh, J.D.; Snowden, J.A.; Ademokun, D.; et al. The impact of cytogenetics on duration of response and overall survival in patients with relapsed multiple myeloma (long-term follow-up results from BSBMT / UKMF Myeloma X Relapse [Intensive]): A randomised, open-label, phase 3 trial. Br. J. Haematol. 2019, 185, 450–467. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Gay, F.; Schjesvold, F.; Beksac, M.; Hajek, R.; Weisel, K.C.; Goldschmidt, H.; Maisnar, V.; Moreau, P.; Min, C.K.; et al. Oral ixazomib maintenance following autologous stem cell transplantation (TOURMALINE-MM3): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2019, 393, 253–264. [Google Scholar] [CrossRef]
- Rosiñol, L.; Oriol, A.; Rios, R.; Sureda, A.; Blanchard, M.J.; Hernández, M.T.; Martínez-Martínez, R.; Moraleda, J.M.; Jarque, I.; Bargay, J.; et al. Bortezomib, lenalidomide, and dexamethasone as induction therapy prior to autologous transplant in multiple myeloma. Blood 2019, 134, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Lahuerta, J.J.; Mateos, M.V.; Martínez-López, J.; Grande, C.; de La Rubia, J.; Rosinol, L.; Sureda, A.; García-Laraña, J.; Díaz-Mediavilla, J.; Hernández-García, M.T.; et al. Busulfan 12 mg/kg plus melphalan 140 mg/m2 versus melphalan 200 mg/m2 as conditioning regimens for autologous transplantation in newly diagnosed multiple myeloma patients included in the PETHEMA/GEM2000 study. Haematologica 2010, 95, 1913–1920. [Google Scholar] [CrossRef]
- Bashir, Q.; Thall, P.F.; Milton, D.R.; Fox, P.S.; Kawedia, J.D.; Kebriaei, P.; Shah, N.; Patel, K.; Andersson, B.S.; Nieto, Y.L.; et al. Conditioning with busulfan plus melphalan versus melphalan alone before autologous haemopoietic cell transplantation for multiple myeloma: An open-label, randomised, phase 3 trial. Lancet Haematol. 2019, 6, e266–e275. [Google Scholar] [CrossRef]
- Garderet, L.; Kuhnowski, F.; Berge, B.; Roussel, M.; Escoffre-Barbe, M.; Lafon, I.; Facon, T.; Leleu, X.; Karlin, L.; Perrot, A.; et al. Pomalidomide, cyclophosphamide, and dexamethasone for relapsed multiple myeloma. Blood 2018, 132, 2555–2563. [Google Scholar] [CrossRef]
- Martino, M.; Gori, M.; Tripepi, G.; Recchia, A.G.; Cimminiello, M.; Provenzano, P.F.; Naso, V.; Ferreri, A.; Moscato, T.; Console, G.; et al. A comparative effectiveness study of lipegfilgrastim in multiple myeloma patients after high dose melphalan and autologous stem cell transplant. Ann. Hematol. 2020, 99, 331–341. [Google Scholar] [CrossRef]
- Alexanian, R. Treatment for multiple myeloma. Combination chemotherapy with different melphalan dose regimens. JAMA 1969, 208, 1680–1685. [Google Scholar] [CrossRef]
- Kazandjian, D.; Landgren, O. Delaying the use of high-dose melphalan with stem cell rescue in multiple myeloma is ready for prime time. Clin. Adv. Hematol. Oncol. 2019, 17, 559–568. [Google Scholar]
- Terpos, E.; Ntanasis-Stathopoulos, I. Multiple Myeloma: Clinical Updates from the American Society of Hematology Annual Meeting 2018. Clin. Lymphoma Myeloma Leuk. 2019, 19, e324–e336. [Google Scholar] [CrossRef]
- Sekine, L.; Ziegelmann, P.K.; Manica, D.; Pithan, C.D.F.; Sosnoski, M.; Morais, V.D.; Falcetta, F.S.; Ribeiro, M.R.; Salazar, A.P.; Ribeiro, R.A. Frontline treatment for transplant-eligible multiple myeloma: A 6474 patients network meta-analysis. Hematol. Oncol. 2019, 37, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Durie, B.G.M.; Hoering, A.; Abidi, M.H.; Rajkumar, S.V.; Epstein, J.; Kahanic, S.P.; Thakuri, M.; Reu, F.; Reynolds, C.M.; Sexton, R.; et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): A randomised, open-label, phase 3 trial. Lancet 2017, 389, 519–527. [Google Scholar] [CrossRef]
- Uttervall, K.; Bruchfeld, J.B.; Gran, C.; Wålinder, G.; Månsson, R.; Lund, J.; Gahrton, G.; Alici, E.; Nahi, H. Upfront bortezomib, lenalidomide, and dexamethasone compared to bortezomib, cyclophosphamide, and dexamethasone in multiple myeloma. Eur. J. Haematol. 2019, 103, 247–254. [Google Scholar] [CrossRef]
- Goldschmidt, H.; Ashcroft, J.; Szabo, Z.; Garderet, L. Navigating the treatment landscape in multiple myeloma: Which combinations to use and when? Ann. Hematol. 2019, 98, 1–18. [Google Scholar] [CrossRef]
- Syed, Y.Y. Daratumumab: A Review in Combination Therapy for Transplant-Ineligible Newly Diagnosed Multiple Myeloma. Drugs 2019, 79, 447–454. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef] [PubMed]
- Nooka, A.K.; Kaufman, J.L.; Hofmeister, C.C.; Joseph, N.S.; Heffner, T.L.; Gupta, V.A.; Sullivan, H.C.; Neish, A.S.; Dhodapkar, M.V.; Lonial, S. Daratumumab in multiple myeloma. Cancer 2019, 125, 2364–2382. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lúcio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Bonello, F.; Grasso, M.; D’Agostino, M.; Celeghini, I.; Castellino, A.; Boccadoro, M.; Bringhen, S. The Role of Monoclonal Antibodies in the First-Line Treatment of Transplant-Ineligible Patients with Newly Diagnosed Multiple Myeloma. Pharmaceuticals 2020, 14, 20. [Google Scholar] [CrossRef]
- Leleu, X.; Fouquet, G.; Richez, V.; Guidez, S.; Duhamel, A.; Machuron, F.; Karlin, L.; Kolb, B.; Tiab, M.; Araujo, C.; et al. Carfilzomib Weekly plus Melphalan and Prednisone in Newly Diagnosed Transplant-Ineligible Multiple Myeloma (IFM 2012-03): A Phase I Trial. Clin. Cancer Res. 2019, 25, 4224–4230. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Goldschmidt, H.; Niesvizky, R.; Joshua, D.; Chng, W.-J.; Oriol, A.; Orlowski, R.Z.; Ludwig, H.; Facon, T.; Hajek, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327–1337. [Google Scholar] [CrossRef]
- Kazandjian, D.; Mo, C.C.; Landgren, O.; Richardson, P.G. The role of high-dose melphalan with autologous stem-cell transplant in multiple myeloma: Is it time for a paradigm shift? Br. J. Haematol. 2020, 191, 692–703. [Google Scholar] [CrossRef]
- Kühne, A.; Sezer, O.; Heider, U.; Meineke, I.; Muhlke, S.; Niere, W.; Overbeck, T.; Hohloch, K.; Trümper, L.; Brockmöller, J.; et al. Population Pharmacokinetics of Melphalan and Glutathione S-transferase Polymorphisms in Relation to Side Effects. Clin. Pharmacol. Ther. 2008, 83, 749–757. [Google Scholar] [CrossRef]
- Isoda, A.; Saito, R.; Komatsu, F.; Negishi, Y.; Oosawa, N.; Ishikawa, T.; Miyazawa, Y.; Matsumoto, M.; Sawamura, M.; Manaka, A. Palonosetron, aprepitant, and dexamethasone for prevention of nausea and vomiting after high-dose melphalan in autologous transplantation for multiple myeloma: A phase II study. Int. J. Hematol. 2016, 105, 478–484. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012–2020. Melphalan. [Updated 2020 Jan 15]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548280/ (accessed on 18 April 2021).
- Spencer, A.; Horvath, N.; Gibson, J.; Prince, H.M.; Herrmann, R.; Bashford, J.; Joske, D.; Grigg, A.; McKendrick, J.; Deveridge, S.; et al. Prospective randomised trial of amifostine cytoprotection in myeloma patients undergoing high-dose melphalan conditioned autologous stem cell transplantation. Bone Marrow Transpl. 2005, 35, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.; Tobinai, K.; Uike, N.; Asakawa, T.; Saito, I.; Fukuda, H.; Mizoroki, F.; Ando, K.; Iida, S.; Ueda, R.; et al. Melphalan-Prednisolone and Vincristine-Doxorubicin-Dexamethasone Chemotherapy followed by Prednisolone/Interferon Maintenance Therapy for Multiple Myeloma: Japan Clinical Oncology Group Study, JCOG0112. Jpn. J. Clin. Oncol. 2011, 41, 586–589. [Google Scholar] [CrossRef][Green Version]
- Ma, L.-L.; Liu, Y.; Jia, S.-X.; Lv, H.-C.; Fang, M.-Y.; Xia, Y.-L. A case report: High dose melphalan as a conditioning regimen for multiple myeloma induces sinus arrest. Cardio-Oncology 2020, 6, 4. [Google Scholar] [CrossRef]
- Tuzovic, M.; Mead, M.; Young, P.A.; Schiller, G.; Yang, E.H. Cardiac Complications in the Adult Bone Marrow Transplant Patient. Curr. Oncol. Rep. 2019, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.; Sylvester, M.; Parrondo, R.; Mariotti, V.; Eloy, J.A.; Chang, V.T. In-Hospital Mortality and Post-Transplantation Complications in Elderly Multiple Myeloma Patients Undergoing Autologous Hematopoietic Stem Cell Transplantation: A Population-Based Study. Biol. Blood Marrow Transpl. 2017, 23, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Savani, B.N.; Mukherjee, A.; Savani, G.T.; Ankit, C. Utilization Trend and in-Hospital Complications of Autologous Hematopoietic Stem Cell Transplantation in Multiple Myeloma in the United States: 13 Years Perspective. Blood 2014, 124, 3978. [Google Scholar] [CrossRef]
- Bachier-Rodriguez, L.; Shah, G.L.; Knezevic, A.; Devlin, S.M.; Maloy, M.; Koehne, G.; Chung, D.J.; Landau, H.; Giralt, S.A. Engraftment Kinetics after High-Dose Melphalan Autologous Stem Cell Transplant in Patients with Multiple Myeloma. Biol. Blood Marrow Transpl. 2018, 24, S144. [Google Scholar] [CrossRef]
- Bakalov, V.; Tang, A.; Shaikh, H.G.; Shah, S.; Chahine, Z.; Kaplan, R.B.; Lister, J.; Sadashiv, S. In-Hospital Complications and Outcomes of Autologous Stem Cell Transplantation in Multiple Myeloma Patients Older Than 65-Years Old in the United States: National Inpatient Sample Analysis, 2011–2015. Blood 2019, 134, 4764. [Google Scholar] [CrossRef]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-H.; Shin, H.-J.; Jung, K.S.; Kim, D.Y. Dose Adjustment Helps Obtain Better Outcomes in Multiple Myeloma Patients with Bortezomib, Melphalan, and Prednisolone (VMP) Treatment. Turk. J. Hematol. 2019, 36, 106–111. [Google Scholar] [CrossRef]
- Hungria, V.; Martínez-Baños, D.M.; Mateos, M.-V.; Dimopoulos, M.A.; Cavo, M.; Heeg, B.; Garcia, A.; Lam, A.; Machnicki, G.; He, J.; et al. Daratumumab Plus Bortezomib, Melphalan, and Prednisone Versus Standard of Care in Latin America for Transplant-Ineligible Newly Diagnosed Multiple Myeloma: Propensity Score Matching Analysis. Adv. Ther. 2020, 37, 4996–5009. [Google Scholar] [CrossRef]
- Moreau, P.; Milpied, N.; Mahé, B.; Juge-Morineau, N.; Rapp, M.-J.; Bataille, R.; Harousseau, J.-L. Melphalan 220 mg/m2 followed by peripheral blood stem cell transplantation in 27 patients with advanced multiple myeloma. Bone Marrow Transpl. 1999, 23, 1003–1006. [Google Scholar] [CrossRef]
- Auner, H.W.; Iacobelli, S.; Sbianchi, G.; Knol-Bout, C.; Blaise, D.; Russell, N.H.; Apperley, J.F.; Pohlreich, D.; Browne, P.V.; Kobbe, G.; et al. Melphalan 140 mg/m 2 or 200 mg/m 2 for autologous transplantation in myeloma: Results from the Collaboration to Collect Autologous Transplant Outcomes in Lymphoma and Myeloma (CALM) study. A report by the EBMT Chronic Malignancies Working Party. Haematologica 2018, 103, 514–521. [Google Scholar] [CrossRef]
- Kumar, S.K.; Dispenzieri, A.; Fraser, R.; Mingwei, F.; Akpek, G.; Cornell, R.; Kharfan-Dabaja, M.; Freytes, C.; Hashmi, S.; Hildebrandt, G.; et al. Early relapse after autologous hematopoietic cell transplantation remains a poor prognostic factor in multiple myeloma but outcomes have improved over time. Leukemia 2018, 32, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Landi, S.; Reiman, T.; Perry, T.E.; Plesa, A.; Bellini, I.; Barale, R.; Pilarski, L.M.; Troncy, J.; Tavtigian, S.V.; et al. Genetic polymorphisms associated with outcome in multiple myeloma patients receiving high-dose melphalan. Bone Marrow Transpl. 2009, 45, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Vangsted, A.J.; Gimsing, P.; Klausen, T.W.; Nexø, B.A.; Wallin, H.; Andersen, P.; Hokland, P.; Lillevang, S.T.; Vogel, U. Polymorphisms in the genes ERCC2, XRCC3 and CD3EAP influence treatment outcome in multiple myeloma patients undergoing autologous bone marrow transplantation. Int. J. Cancer 2006, 120, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Mouhieddine, T.H.; Sperling, A.S.; Redd, R.; Park, J.; Leventhal, M.; Gibson, C.J.; Manier, S.; Nassar, A.H.; Capelletti, M.; Huynh, D.; et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Komai, M.; Nishinobo, M.; Yamashita, M.; Yanae, M.; Yamazoe, Y.; Nishida, S. Overexpression of MDR1 and survivin, and decreased Bim expression mediate multidrug-resistance in multiple myeloma cells. Leuk. Res. 2012, 36, 1315–1322. [Google Scholar] [CrossRef]
- Sousa, M.M.L.; Zub, K.A.; Aas, P.A.; Hanssen-Bauer, A.; Demirovic, A.; Sarno, A.; Tian, E.; Liabakk, N.B.; Slupphaug, G. An Inverse Switch in DNA Base Excision and Strand Break Repair Contributes to Melphalan Resistance in Multiple Myeloma Cells. PLoS ONE 2013, 8, e55493. [Google Scholar] [CrossRef]
- Lin, J.; Raoof, D.A.; Thomas, D.G.; Greenson, J.K.; Giordano, T.J.; Robinson, G.S.; Bourner, M.J.; Bauer, C.T.; Orringer, M.B.; Beer, D.G. L-Type Amino Acid Transporter-1 Overexpression and Melphalan Sensitivity in Barrett’s Adenocarcinoma. Neoplasia 2004, 6, 74–84. [Google Scholar] [CrossRef]
- Munawar, U.; Roth, M.; Barrio, S.; Wajant, H.; Siegmund, D.; Bargou, R.C.; Kortüm, K.M.; Stühmer, T. Assessment of TP53 lesions for p53 system functionality and drug resistance in multiple myeloma using an isogenic cell line model. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef]
- Jovanović, K.K.; Escure, G.; Demonchy, J.; Willaume, A.; van de Wyngaert, Z.; Farhat, M.; Chauvet, P.; Facon, T.; Quesnel, B.; Manier, S. Deregulation and Targeting of TP53 Pathway in Multiple Myeloma. Front. Oncol. 2019, 8. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic abnormalities and survival in multiple myeloma: The experience of the Intergroupe Francophone du Myélome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef]
- Krejčí, J.; Harničarová, A.; Štreitová, D.; Hajek, R.; Pour, L.; Kozubek, S.; Bártová, E. Epigenetics of multiple myeloma after treatment with cytostatics and gamma radiation. Leuk. Res. 2009, 33, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.; D’Agnano, I.; Vitelli, G.; Vona, R.; Marino, M.; Mottolese, M.; Zuppi, C.; Capoluongo, E.; Ameglio, F. C-Myc Deregulation is Involved in Melphalan Resistance of Multiple Myeloma: Role of PDGF-BB. Int. J. Immunopathol. Pharmacol. 2006, 19, 67–79. [Google Scholar] [CrossRef]
- Abraham, J.; Salama, N.N.; Azab, A.K. The role of P-glycoprotein in drug resistance in multiple myeloma. Leuk. Lymphoma 2014, 56, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Gullà, A.; Di Martino, M.T.; Cantafio, M.E.G.; Morelli, E.; Amodio, N.; Botta, C.; Pitari, M.R.; Lio, S.G.; Britti, D.; Stamato, M.A.; et al. A 13 mer LNA-i-miR-221 Inhibitor Restores Drug Sensitivity in Melphalan-Refractory Multiple Myeloma Cells. Clin. Cancer Res. 2016, 22, 1222–1233. [Google Scholar] [CrossRef]
- Lu, D.; Yang, C.; Zhang, Z.; Cong, Y.; Xiao, M. Knockdown of Linc00515 Inhibits Multiple Myeloma Autophagy and Chemoresistance by Upregulating miR-140-5p and Downregulating ATG14. Cell Physiol Biochem. 2018, 48, 2517–2527. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.-F.; Fegueux, N.; Lu, Z.Y.; Legouffe, E.; Exbrayat, C.; Bozonnat, M.-C.; Navarro, R.; Lopez, E.; Quittet, P.; Daures, J.-P.; et al. Optimizing the use of anti-interleukin-6 monoclonal antibody with dexamethasone and 140 mg/m2 of melphalan in multiple myeloma: Results of a pilot study including biological aspects. Bone Marrow Transpl. 2005, 36, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Hunsucker, S.A.; Magarotto, V.; Kuhn, D.J.; Kornblau, S.M.; Wang, M.; Weber, D.M.; Thomas, S.K.; Shah, J.J.; Voorhees, P.M.; Xie, H.; et al. Blockade of interleukin-6 signalling with siltuximab enhances melphalan cytotoxicity in preclinical models of multiple myeloma. Br. J. Haematol. 2011, 152, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of in-tegrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Azab, A.K.; Brentnall, M.; Gratton, K.; Klimowicz, A.C.; Lin, C.; Duggan, P.; Tassone, P.; Mansoor, A.; et al. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood 2011, 117, 6202–6213. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Koh, Y.; Park, H.; Kim, D.Y.; Kim, D.C.; Byun, J.M.; Lee, H.J.; Yoon, A.S.-S. Highly Expressed Integrin-α8 Induces Epithelial to Mesenchymal Transition-Like Features in Multiple Myeloma with Early Relapse. Mol. Cells 2016, 39, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Viziteu, E.; Klein, B.; Basbous, J.; Lin, Y.-L.; Hirtz, C.; Gourzones, C.; Tiers, L.; Bruyer, A.; Vincent, L.; Grandmougin, C.; et al. RECQ1 helicase is involved in replication stress survival and drug resistance in multiple myeloma. Leukemia 2017, 31, 2104–2113. [Google Scholar] [CrossRef]
- Gourzones, C.; Bellanger, C.; Lamure, S.; Gadacha, O.C.; de Paco, E.G.; Vincent, L.; Cartron, G.; Klein, B.; Moreaux, J. Antioxidant Defenses Confer Resistance to High Dose Melphalan in Multiple Myeloma Cells. Cancers 2019, 11, 439. [Google Scholar] [CrossRef]
- Xu, H.; He, J.; Zhang, Y.; Fan, L.; Zhao, Y.; Xu, T.; Nie, Z.; Li, X.; Huang, Z.; Lu, B.; et al. Synthesis and in vitro evaluation of a hyaluronic acid–quantum dots–melphalan conjugate. Carbohydr. Polym. 2015, 121, 132–139. [Google Scholar] [CrossRef]
- Mattheolabakis, G.; Milane, L.; Singh, A.P.; Amiji, M.M. Hyaluronic acid targeting of CD44 for cancer therapy: From receptor biology to nanomedicine. J. Drug Target. 2015, 23, 605–618. [Google Scholar] [CrossRef]
- Lu, B.; Huang, D.; Zheng, H.; Huang, Z.; Xu, P.; Xu, H.; Yin, Y.; Liu, X.; Li, D.; Zhang, X. Preparation, characterization, and in vitro efficacy of O-carboxymethyl chitosan conjugate of melphalan. Carbohydr. Polym. 2013, 98, 36–42. [Google Scholar] [CrossRef]
- Li, D.; Lu, B.; Huang, Z.; Xu, P.; Zheng, H.; Yin, Y.; Xu, H.; Liu, X.; Chen, L.; Lou, Y.; et al. A novel melphalan polymeric prodrug: Preparation and property study. Carbohydr. Polym. 2014, 111, 928–935. [Google Scholar] [CrossRef]
- Ramírez-Arroniz, J.C.; Klimova, E.M.; Pedro-Hernández, L.D.; Organista-Mateos, U.; Cortez-Maya, S.; Ramírez-Ápan, T.; Nieto-Camacho, A.; Calderón-Pardo, J.; Martínez-García, M. Water-soluble porphyrin-PAMAM-conjugates of melphalan and their anticancer activity. Drug Dev. Ind. Pharm. 2018, 44, 1342–1349. [Google Scholar] [CrossRef]
- Wickström, M.; Nygren, P.; Larsson, R.; Harmenberg, J.; Lindberg, J.; Sjöberg, P.; Jerling, M.; Lehmann, F.; Richardson, P.; Anderson, K.; et al. Melflufen—A peptidase-potentiated alkylating agent in clinical trials. Oncotarget 2017, 8, 66641–66655. [Google Scholar] [CrossRef] [PubMed]
- Strese, S.; Wickström, M.; Fuchs, P.F.; Fryknäs, M.; Gerwins, P.; Dale, T.; Larsson, R.; Gullbo, J. The novel alkylating prodrug melflufen (J1) inhibits angiogenesis in vitro and in vivo. Biochem. Pharmacol. 2013, 86, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Berglund, Å.; Ullén, A.; Lisyanskaya, A.; Orlov, S.; Hagberg, H.; Tholander, B.; Lewensohn, R.; Nygren, P.; Spira, J.; Harmenberg, J.; et al. First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies. Investig. New Drugs 2015, 33, 1232–1241. [Google Scholar] [CrossRef]
- Delforoush, M.; Strese, S.; Wickström, M.; Larsson, R.; Enblad, G.; Gullbo, J. In vitro and in vivo activity of melflufen (J1) in lymphoma. BMC Cancer 2016, 16, 263. [Google Scholar] [CrossRef]
- Chauhan, D.; Ray, A.; Viktorsson, K.; Spira, J.; Paba-Prada, C.; Munshi, N.; Richardson, P.; Lewensohn, R.; Anderson, K.C. In Vitro and In Vivo Antitumor Activity of a Novel Alkylating Agent, Melphalan-Flufenamide, against Multiple Myeloma Cells. Clin. Cancer Res. 2013, 19, 3019–3031. [Google Scholar] [CrossRef]
- Viktorsson, K.; Shah, C.-H.; Juntti, T.; Hååg, P.; Zielinska-Chomej, K.; Sierakowiak, A.; Holmsten, K.; Tu, J.; Spira, J.; Kanter, L.; et al. Melphalan-flufenamide is cytotoxic and potentiates treatment with chemotherapy and the Src inhibitor dasatinib in urothelial carcinoma. Mol. Oncol. 2016, 10, 719–734. [Google Scholar] [CrossRef]
- Carlier, C.; Strese, S.; Viktorsson, K.; Velander, E.; Nygren, P.; Uustalu, M.; Juntti, T.; Lewensohn, R.; Larsson, R.; Spira, J.; et al. Preclinical activity of melflufen (J1) in ovarian cancer. Oncotarget 2016, 7, 59322–59335. [Google Scholar] [CrossRef]
- Richardson, P.G.; Bringhen, S.; Voorhees, P.; Plesner, T.; Mellqvist, U.-H.; Reeves, B.; Paba-Prada, C.; Zubair, H.; Byrne, C.; Chauhan, D.; et al. Melflufen plus dexamethasone in relapsed and refractory multiple myeloma (O-12-M1): A multicentre, international, open-label, phase 1–2 study. Lancet Haematol. 2020, 7, e395–e407. [Google Scholar] [CrossRef]
- Richardson, P.G.; Oriol, A.; Larocca, A.; Bladé, J.; Cavo, M.; Rodriguez-Otero, P.; Leleu, X.; Nadeem, O.; Hiemenz, J.W.; Hassoun, H.; et al. Melflufen and Dexamethasone in Heavily Pretreated Relapsed and Refractory Multiple Myeloma. J. Clin. Oncol. 2021, 39, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Schjesvold, F.; Robak, P.; Pour, L.; Aschan, J.; Sonneveld, P. OCEAN: A randomized Phase III study of melflufen + dexamethasone to treat relapsed refractory multiple myeloma. Futur. Oncol. 2020, 16, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Gajek, A.; Poczta, A.; Łukawska, M.; Adamczewska, V.-C.; Tobiasz, J.; Marczak, A. Chemical modification of melphalan as a key to improving treatment of haematological malignancies. Sci. Rep. 2020, 10, 4479. [Google Scholar] [CrossRef]
- Antoni, F.; Bernhardt, G. Derivatives of nitrogen mustard anticancer agents with improved cytotoxicity. Arch. Pharm. 2020, e2000366. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poczta, A.; Rogalska, A.; Marczak, A. Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies—Current Research and Clinical Approaches. J. Clin. Med. 2021, 10, 1841. https://doi.org/10.3390/jcm10091841
Poczta A, Rogalska A, Marczak A. Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies—Current Research and Clinical Approaches. Journal of Clinical Medicine. 2021; 10(9):1841. https://doi.org/10.3390/jcm10091841
Chicago/Turabian StylePoczta, Anastazja, Aneta Rogalska, and Agnieszka Marczak. 2021. "Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies—Current Research and Clinical Approaches" Journal of Clinical Medicine 10, no. 9: 1841. https://doi.org/10.3390/jcm10091841
APA StylePoczta, A., Rogalska, A., & Marczak, A. (2021). Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies—Current Research and Clinical Approaches. Journal of Clinical Medicine, 10(9), 1841. https://doi.org/10.3390/jcm10091841