
Role of Proteasomes in Inflammation


{kind=link}
{kind=link}
{kind=link}
Abstract
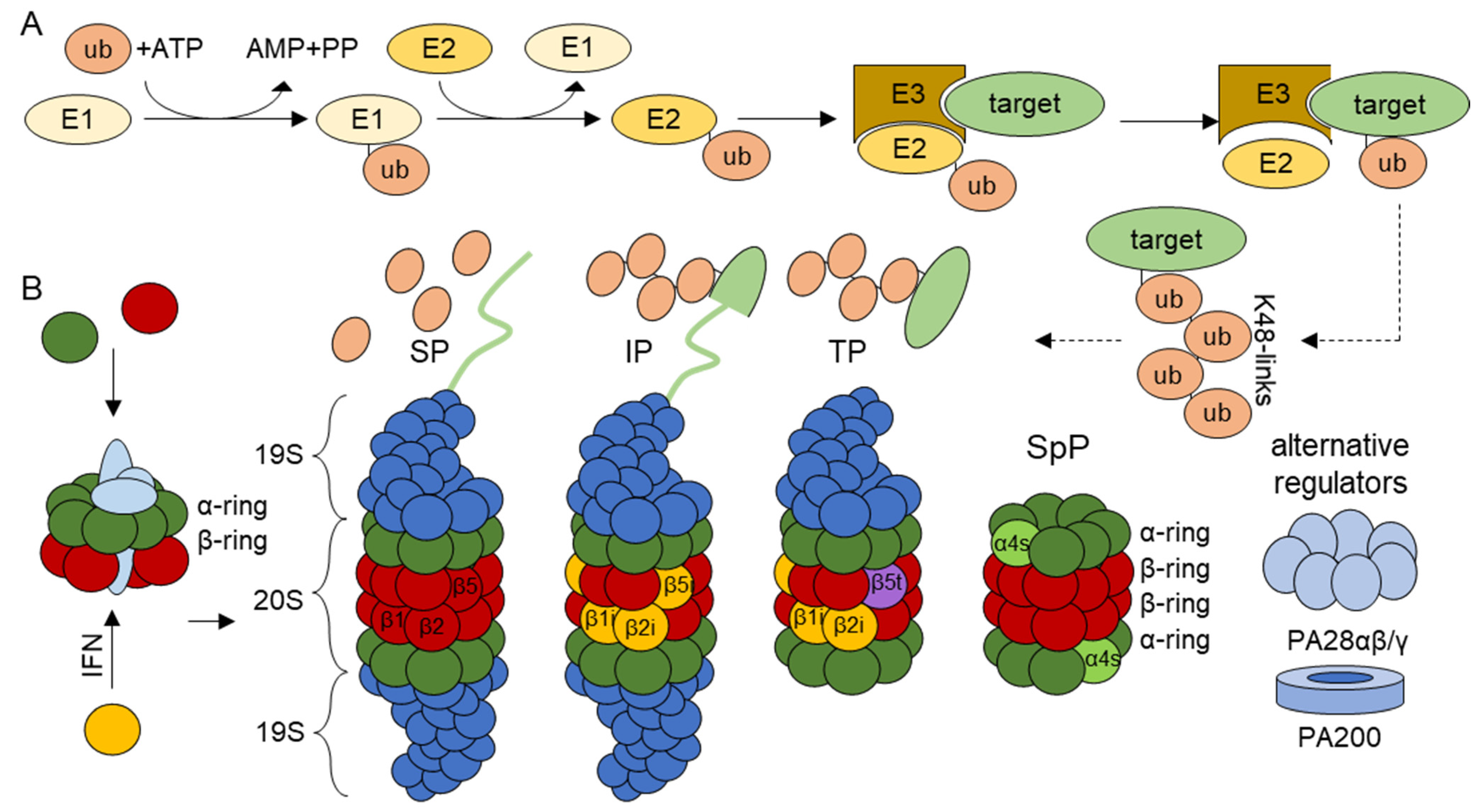
1. The Ubiquitin–Proteasome System
1.1. Proteasome Isoforms
1.2. Proteasomes and Cellular Pro-Inflammatory-Pathways
2. Impaired Proteasomal Function—(Mono)genetic Defects in the Ubiquitin Proteasome-System in Autoinflammatory Disorders
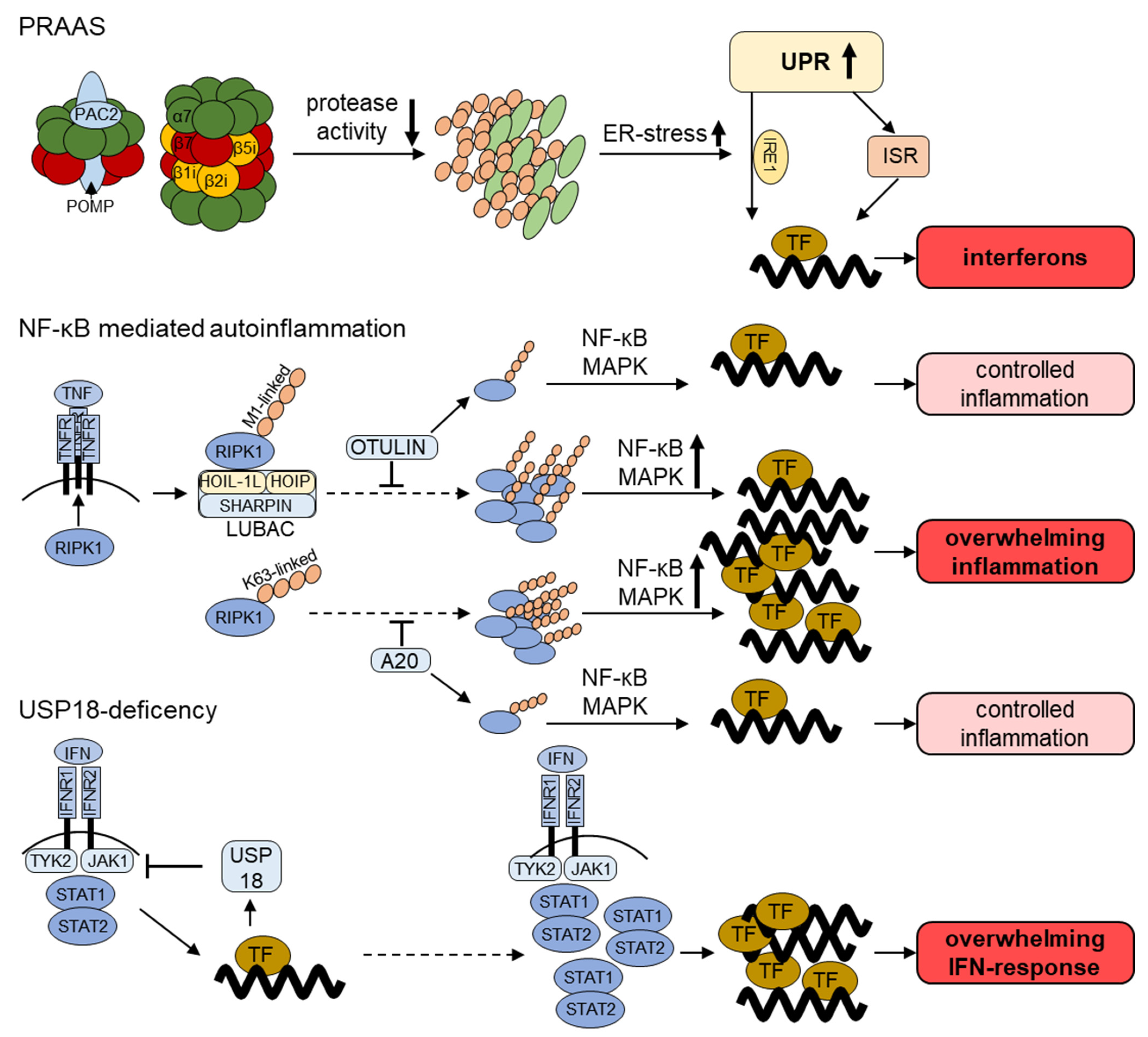
2.1. Proteasome-Associated-Autoinflammatory-Syndrome (PRAAS)
2.2. Further Genetic Inflammatory Diseases with a Link to the UPS
3. Inflammation and Increased Proteasomal Activity
3.1. Rheumatoid Arthritis
3.2. Systemic Lupus Erythematosus
3.3. Sjögren Syndrome
3.4. Inflammatory Bowel Disease
3.5. Multiple Sclerosis
3.6. Further Inflammatory Diseases with a Link to the UPS
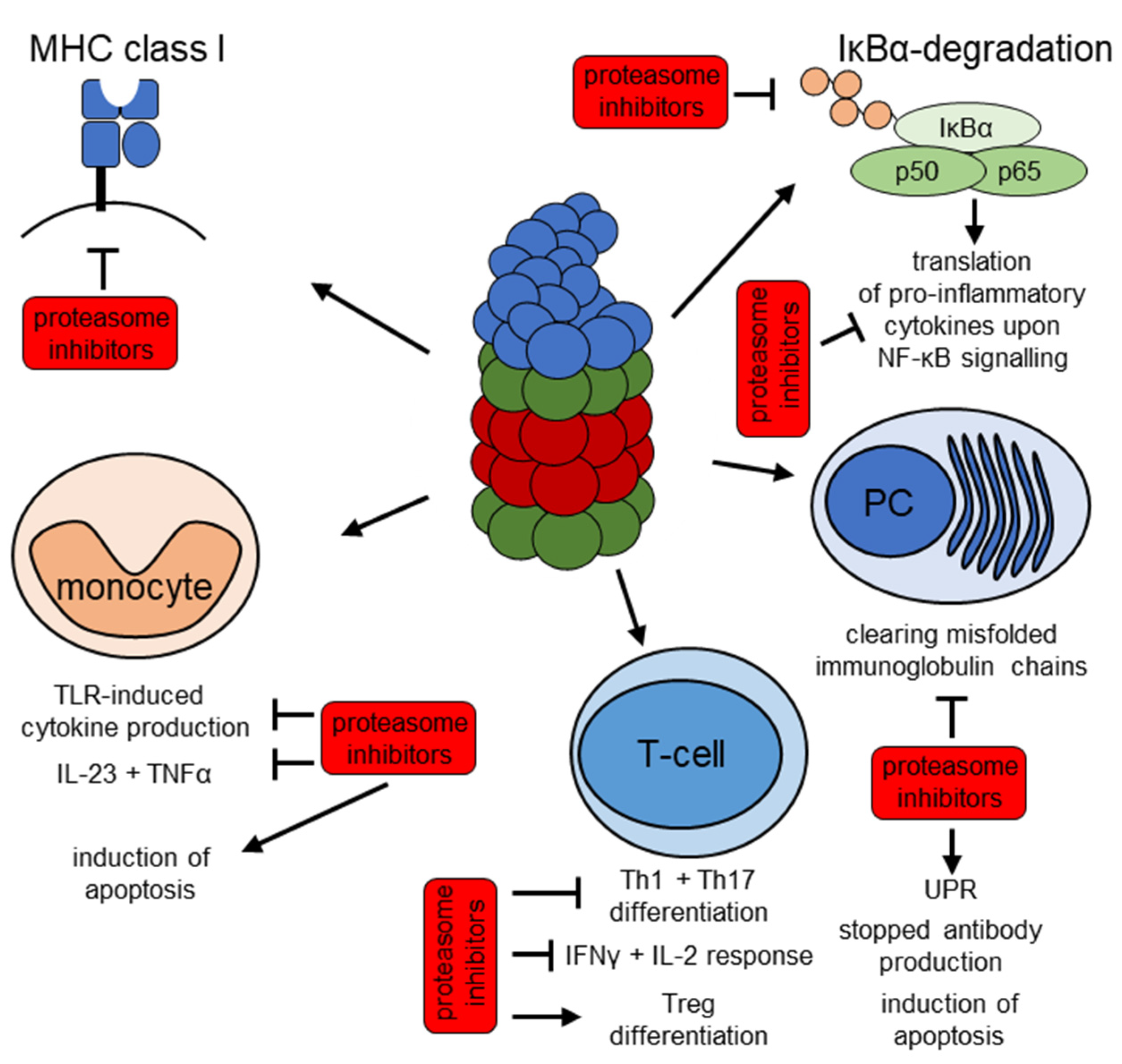
4. The UPS as a Therapeutic Target
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005, 12, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin–proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Bergann, T.; Krüger, E. Oxidation matters: The ubiquitin proteasome system connects innate immune mechanisms with MHC class I antigen presentation. Mol. Immunol. 2013, 55, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Geetha, T.; Babu, J.R. Failure of Ubiquitin Proteasome System: Risk for Neurodegenerative Diseases. Neurodegener. Dis. 2014, 14, 161–175. [Google Scholar] [CrossRef]
- Çetin, G.; Klafack, S.; Studencka-Turski, M.; Krüger, E.; Ebstein, F. The Ubiquitin–Proteasome System in Immune Cells. Biomolecules 2021, 11, 60. [Google Scholar] [CrossRef]
- Brehm, A.; Krüger, E. Dysfunction in protein clearance by the proteasome: Impact on autoinflammatory diseases. Semin. Immunopathol. 2015, 37, 323–333. [Google Scholar] [CrossRef]
- Haas, A.L.; Siepmann, T.J. Pathways of ubiquitin conjugation. FASEB J. 1997, 11, 1257–1268. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Back to the Future with Ubiquitin. Cell 2004, 116, 181–190. [Google Scholar] [CrossRef]
- Berndsen, C.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef]
- McDowell, G.S.; Kucerova, R.; Philpott, A. Non-canonical ubiquitylation of the proneural protein Ngn2 occurs in both Xenopus embryos and mammalian cells. Biochem. Biophys. Res. Commun. 2010, 400, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K. Ubiquitination on Nonlysine Residues by a Viral E3 Ubiquitin Ligase. Science 2005, 309, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Vosper, J.M.; McDowell, G.S.; Hindley, C.J.; Fiore-Heriche, C.S.; Kucerova, R.; Horan, I.; Philpott, A. Ubiquitylation on Canonical and Non-canonical Sites Targets the Transcription Factor Neurogenin for Ubiquitin-mediated Proteolysis. J. Biol. Chem. 2009, 284, 15458–15468. [Google Scholar] [CrossRef] [PubMed]
- Mallette, F.A.; Richard, S. K48-linked ubiquitination and protein degradation regulate 53BP1 recruitment at DNA damage sites. Cell Res. 2012, 22, 1221–1223. [Google Scholar] [CrossRef]
- Pickart, C.M.; Fushman, D. Polyubiquitin chains: Polymeric protein signals. Curr. Opin. Chem. Biol. 2004, 8, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Sun, L.; Chen, Z.J. The novel functions of ubiquitination in signaling. Curr. Opin. Cell Biol. 2004, 16, 119–126. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef]
- Eletr, Z.M.; Wilkinson, K.D. Regulation of proteolysis by human deubiquitinating enzymes. Biochim. Biophys. Acta 2014, 1843, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B. Proteasomes. Essays Biochem. 2005, 41, 31–48. [Google Scholar] [CrossRef]
- Tanaka, K.; Mizushima, T.; Saeki, Y. The proteasome: Molecular machinery and pathophysiological roles. Biol. Chem. 2012, 393, 217–234. [Google Scholar] [CrossRef]
- Bard, J.A.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Orlowski, M.; Wilk, S. Catalytic Activities of the 20 S Proteasome, a Multicatalytic Proteinase Complex. Arch. Biochem. Biophys. 2000, 383, 1–16. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Songyang, Z.; Goldberg, A.L. Why Does Threonine, and Not Serine, Function as the Active Site Nucleophile in Proteasomes? J. Biol. Chem. 2000, 275, 14831–14837. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Bedford, L.; Paine, S.; Sheppard, P.W.; Mayer, R.J.; Roelofs, J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010, 20, 391–401. [Google Scholar] [CrossRef]
- Liu, C.-W.; Jacobson, A.D. Functions of the 19S complex in proteasomal degradation. Trends Biochem. Sci. 2013, 38, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.; van Nocker, S.; Mahaffey, D.; Vierstra, R.; Rechsteiner, M. Inhibition of Ubiquitin-mediated Proteolysis by the Arabidopsis 26 S Protease Subunit S5a. J. Biol. Chem. 1995, 270, 29660–29663. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nat. Cell Biol. 2008, 453, 481–488. [Google Scholar] [CrossRef]
- Smith, D.M.; Chang, S.-C.; Park, S.; Finley, D.; Cheng, Y.; Goldberg, A.L. Docking of the Proteasomal ATPases’ Carboxyl Termini in the 20S Proteasome’s α Ring Opens the Gate for Substrate Entry. Mol. Cell 2007, 27, 731–744. [Google Scholar] [CrossRef]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Shin, J.Y.; Muniyappan, S.; Tran, N.-N.; Park, H.; Lee, S.B.; Lee, B.-H. Deubiquitination Reactions on the Proteasome for Proteasome Versatility. Int. J. Mol. Sci. 2020, 21, 5312. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B. Mammalian proteasome subtypes: Their diversity in structure and function. Arch. Biochem. Biophys. 2016, 591, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Kloetzel, P.-M. The proteasome and MHC class I antigen processing. Biochim. Biophys. Acta 2004, 1695, 225–233. [Google Scholar] [CrossRef]
- Seyffer, F.; Tampé, R. ABC transporters in adaptive immunity. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Townsend, A.; Trowsdale, J. The transporters associated with antigen presentation. Semin. Cell Biol. 1993, 4, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. Role of proteasomes modified by interferon-γ in antigen processing. J. Leukoc. Biol. 1994, 56, 571–575. [Google Scholar] [CrossRef]
- Ebstein, F.; Kloetzel, P.-M.; Krüger, E.; Seifert, U.; Krueger, E. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell. Mol. Life Sci. 2012, 69, 2543–2558. [Google Scholar] [CrossRef]
- Strehl, B.; Seifert, U.; Kruger, E.; Heink, S.; Kuckelkorn, U.; Kloetzel, P.-M.; Krueger, E. Interferon-gamma, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol. Rev. 2005, 207, 19–30. [Google Scholar] [CrossRef]
- Aki, M.; Shimbara, N.; Takashina, M.; Akiyama, K.; Kagawa, S.; Tamura, T.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Interferon-Gamma Induces Different Subunit Organizations and Functional Diversity of Proteasomes. J. Biochem. 1994, 115, 257–269. [Google Scholar] [CrossRef]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.S.; Kim, K.H.; Tschida, B.; Sachs, Z.; Noble-Orcutt, K.E.; Moriarity, B.S.; Ai, T.; Ding, R.; Williams, J.; Chen, L.; et al. mTORC1 Coordinates Protein Synthesis and Immunoproteasome Formation via PRAS40 to Prevent Accumulation of Protein Stress. Mol. Cell 2016, 61, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Niewerth, D.; Kaspers, G.J.; Assaraf, Y.G.; Van Meerloo, J.; Kirk, C.J.; Anderl, J.; Blank, J.L.; Van De Ven, P.M.; Zweegman, S.; Jansen, G.; et al. Interferon-γ-induced upregulation of immunoproteasome subunit assembly overcomes bortezomib resistance in human hematological cell lines. J. Hematol. Oncol. 2014, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Opitz, E.; Koch, A.; Klingel, K.; Schmidt, F.; Prokop, S.; Rahnefeld, A.; Sauter, M.; Heppner, F.L.; Völker, U.; Kandolf, R.; et al. Impairment of Immunoproteasome Function by β5i/LMP7 Subunit Deficiency Results in Severe Enterovirus Myocarditis. PLoS Pathog. 2011, 7, e1002233. [Google Scholar] [CrossRef] [PubMed]
- Krüger, E.; Kloetzel, P.-M. Immunoproteasomes at the interface of innate and adaptive immune responses: Two faces of one enzyme. Curr. Opin. Immunol. 2012, 24, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Mishto, M.; Liepe, J.; Textoris-Taube, K.; Keller, C.; Henklein, P.; Weberruß, M.; Dahlmann, B.; Enenkel, C.; Voigt, A.; Kuckelkorn, U.; et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur. J. Immunol. 2014, 44, 3508–3521. [Google Scholar] [CrossRef]
- Murata, S.; Sasaki, K.; Kishimoto, T.; Niwa, S.-I.; Hayashi, H.; Takahama, Y.; Tanaka, K. Regulation of CD8+ T Cell Development by Thymus-Specific Proteasomes. Science 2007, 316, 1349–1353. [Google Scholar] [CrossRef]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef]
- Kniepert, A.; Groettrup, M. The unique functions of tissue-specific proteasomes. Trends Biochem. Sci. 2014, 39, 17–24. [Google Scholar] [CrossRef]
- Iwai, K.; Fujita, H.; Sasaki, Y. Linear ubiquitin chains: NF-κB signalling, cell death and beyond. Nat. Rev. Mol. Cell Biol. 2014, 15, 503–508. [Google Scholar] [CrossRef]
- Alkalay, I.; Yaron, A.; Hatzubai, A.; Orian, A.; Ciechanover, A.; Ben-Neriah, Y. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1995, 92, 10599–10603. [Google Scholar] [CrossRef]
- Fiil, B.K.; Gyrd-Hansen, M. OTULIN deficiency causes auto-inflammatory syndrome. Cell Res. 2016, 26, 1176–1177. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.T.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nat. Cell Biol. 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Althof, N.; Goetzke, C.C.; Kespohl, M.; Voss, K.; Heuser, A.; Pinkert, S.; Kaya, Z.; Klingel, K.; Beling, A. The immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis. EMBO Mol. Med. 2018, 10, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Bockstahler, M.; Fischer, A.; Goetzke, C.C.; Neumaier, H.L.; Sauter, M.; Kespohl, M.; Müller, A.-M.; Meckes, C.; Salbach, C.; Schenk, M.; et al. Heart-Specific Immune Responses in an Animal Model of Autoimmune-Related Myocarditis Mitigated by an Immunoproteasome Inhibitor and Genetic Ablation. Circulation 2020, 141, 1885–1902. [Google Scholar] [CrossRef]
- Schmidt, C.; Berger, T.; Groettrup, M.; Basler, M. Immunoproteasome Inhibition Impairs T and B Cell Activation by Restraining ERK Signaling and Proteostasis. Front. Immunol. 2018, 9, 2386. [Google Scholar] [CrossRef]
- Kalim, K.W.; Basler, M.; Kirk, C.J.; Groettrup, M. Immunoproteasome Subunit LMP7 Deficiency and Inhibition Suppresses Th1 and Th17 but Enhances Regulatory T Cell Differentiation. J. Immunol. 2012, 189, 4182–4193. [Google Scholar] [CrossRef]
- Goetzke, C.C.; Althof, N.; Neumaier, H.L.; Heuser, A.; Kaya, Z.; Kespohl, M.; Klingel, K.; Beling, A. Mitigated viral myocarditis in A/J mice by the immunoproteasome inhibitor ONX 0914 depends on inhibition of systemic inflammatory responses in CoxsackievirusB3 infection. Basic Res. Cardiol. 2021, 116, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Alexander, T.; Sarfert, R.; Klotsche, J.; Kühl, A.; Rubbert-Roth, A.; Lorenz, H.-M.; Rech, J.; Hoyer, B.F.; Cheng, Q.; Waka, A.; et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann. Rheum. Dis. 2015, 74, 1474–1478. [Google Scholar] [CrossRef]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A Phase 2 Study of Bortezomib in Relapsed, Refractory Myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Koerner, J.; Basler, M.; Brunner, T.; Kirk, C.J.; Groettrup, M. Immunoproteasome inhibition induces plasma cell apoptosis and preserves kidney allografts by activating the unfolded protein response and suppressing plasma cell survival factors. Kidney Int. 2019, 95, 611–623. [Google Scholar] [CrossRef]
- Ebstein, F.; Harlowe, M.C.P.; Studencka-Turski, M.; Krüger, E. Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS). Front. Immunol. 2019, 10, 2756. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Xing, C.; DeMartino, G.N.; Mizrachi, D.; Hernandez, M.D.; Sousa, A.B.; De Villarreal, L.M.; Dos Santos, H.G.; Garg, A. PSMB8 Encoding the β5i Proteasome Subunit Is Mutated in Joint Contractures, Muscle Atrophy, Microcytic Anemia, and Panniculitis-Induced Lipodystrophy Syndrome. Am. J. Hum. Genet. 2010, 87, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Arima, K.; Kinoshita, A.; Mishima, H.; Kanazawa, N.; Kaneko, T.; Mizushima, T.; Ichinose, K.; Nakamura, H.; Tsujino, A.; Kawakami, A.; et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 14914–14919. [Google Scholar] [CrossRef]
- Kitamura, A.; Maekawa, Y.; Uehara, H.; Izumi, K.; Kawachi, I.; Nishizawa, M.; Toyoshima, Y.; Takahashi, H.; Standley, D.M.; Tanaka, K.; et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Investig. 2011, 121, 4150–4160. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ramot, Y.; Torrelo, A.; Paller, A.S.; Si, N.; Babay, S.; Kim, P.W.; Sheikh, A.; Lee, C.-C.R.; Chen, Y.; et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2011, 64, 895–907. [Google Scholar] [CrossRef]
- McDermott, A.; Jacks, J.; Kessler, M.; Emanuel, P.D.; Gao, L. Proteasome-associated autoinflammatory syndromes: Advances in pathogeneses, clinical presentations, diagnosis, and management. Int. J. Dermatol. 2014, 54, 121–129. [Google Scholar] [CrossRef] [PubMed]
- McDermott, A.; Jesus, A.A.; Liu, Y.; Kim, P.; Jacks, J.; Sanchez, G.A.M.; Chen, Y.; Kannan, A.; Schnebelen, A.; Emanuel, P.D.; et al. A case of proteasome-associated auto-inflammatory syndrome with compound heterozygous mutations. J. Am. Acad. Dermatol. 2013, 69, e29–e32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Torrelo, A.; Patel, S.; Colmenero, I.; Gurbindo, D.; Lendínez, F.; Hernández, A.; López-Robledillo, J.C.; Dadban, A.; Requena, L.; Paller, A.S. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J. Am. Acad. Dermatol. 2010, 62, 489–495. [Google Scholar] [CrossRef]
- Feist, E.; Brehm, A.; Kallinich, T.; Krüger, E. Klinik und Genetik bei Proteasomen-assoziierten autoinflammatorischen Syndromen (PRAAS) Clinical aspects and genetics of proteasome-associated autoinflammatory syndromes (PRAAS). Z. Rheumatol. 2017, 76, 328–334. [Google Scholar] [CrossRef]
- Brehm, A.; Liu, Y.; Sheikh, A.; Marrero, B.; Omoyinmi, E.; Zhou, Q.; Montealegre, G.; Biancotto, A.; Reinhardt, A.; De Jesus, A.A.; et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J. Clin. Investig. 2015, 125, 4196–4211. [Google Scholar] [CrossRef] [PubMed]
- Goldbach-Mansky, R. Immunology in clinic review series; focus on autoinflammatory diseases: Update on monogenic autoinflammatory diseases: The role of interleukin (IL)-1 and an emerging role for cytokines beyond IL-1. Clin. Exp. Immunol. 2012, 167, 391–404. [Google Scholar] [CrossRef]
- Rice, G.I.; Melki, I.; Frémond, M.-L.; Briggs, T.A.; Rodero, M.P.; Kitabayashi, N.; Oojageer, A.; Bader-Meunier, B.; Belot, A.; Bodemer, C.; et al. Assessment of Type I Interferon Signaling in Pediatric Inflammatory Disease. J. Clin. Immunol. 2017, 37, 123–132. [Google Scholar] [CrossRef]
- Kim, H.; De Jesus, A.A.; Brooks, S.R.; Liu, Y.; Huang, Y.; VanTries, R.; Sanchez, G.A.M.; Rotman, Y.; Gadina, M.; Goldbach-Mansky, R. Development of a Validated Interferon Score Using NanoString Technology. J. Interf. Cytokine Res. 2018, 38, 171–185. [Google Scholar] [CrossRef]
- Orak, B.; Ngoumou, G.; Ebstein, F.; Zieba, B.; Goetzke, C.C.; Knierim, E.; Kaindl, A.M.; Panzer, A.; Theophil, M.; Berns, M.; et al. SIGLEC1 (CD169) as a potential diagnostical screening marker for monogenic interferonopathies. Pediatr. Allergy Immunol. 2021, 32, 621–625. [Google Scholar] [CrossRef]
- Poli, M.C.; Ebstein, F.; Nicholas, S.K.; De Guzman, M.M.; Forbes, L.R.; Chinn, I.K.; Mace, E.M.; Vogel, T.P.; Carisey, A.F.; Benavides, F.; et al. Heterozygous Truncating Variants in POMP Escape Nonsense-Mediated Decay and Cause a Unique Immune Dysregulatory Syndrome. Am. J. Hum. Genet. 2018, 102, 1126–1142. [Google Scholar] [CrossRef]
- De Jesus, A.A.; Brehm, A.; VanTries, R.; Pillet, P.; Parentelli, A.-S.; Sanchez, G.A.M.; Deng, Z.; Paut, I.K.; Goldbach-Mansky, R.; Krüger, E. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J. Allergy Clin. Immunol. 2019, 143, 1939–1943.e8. [Google Scholar] [CrossRef] [PubMed]
- Sarrabay, G.; Méchin, D.; Salhi, A.; Boursier, G.; Rittore, C.; Crow, Y.; Rice, G.; Tran, T.-A.; Cezar, R.; Duffy, D.; et al. PSMB10, the last immunoproteasome gene missing for PRAAS. J. Allergy Clin. Immunol. 2020, 145, 1015–1017.e6. [Google Scholar] [CrossRef]
- Ansar, M.; Ebstein, F.; Özkoç, H.; Paracha, S.; Iwaszkiewicz, J.; Gesemann, M.; Zoete, V.; Ranza, E.; Santoni, F.; Sarwar, M.T.; et al. Biallelic variants in PSMB1 encoding the proteasome subunit β6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum. Mol. Genet. 2020, 29, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Küry, S.; Besnard, T.; Ebstein, F.; Khan, T.N.; Gambin, T.; Douglas, J.; Bacino, C.A.; Sanders, S.J.; Lehmann, A.; Latypova, X.; et al. De Novo Disruption of the Proteasome Regulatory Subunit PSMD12 Causes a Syndromic Neurodevelopmental Disorder. Am. J. Hum. Genet. 2017, 100, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Kröll-Hermi, A.; Ebstein, F.; Stoetzel, C.; Geoffroy, V.; Schaefer, E.; Scheidecker, S.; Bär, S.; Takamiya, M.; Kawakami, K.; Zieba, B.; et al. Proteasome subunit PSMC3 variants cause neurosensory syndrome combining deafness and cataract due to proteotoxic stress. EMBO Mol. Med. 2020, 12, 11861. [Google Scholar] [CrossRef]
- Nikesitch, N.; Tao, C.; Lai, K.; Killingsworth, M.; Bae, S.; Wang, M.; Harrison, S.; Roberts, T.L.; Ling, S.C.W. Predicting the response of multiple myeloma to the proteasome inhibitor Bortezomib by evaluation of the unfolded protein response. Blood Cancer J. 2016, 6, e432. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Janssens, S.; Pulendran, B.; Lambrecht, B.N. Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 2014, 15, 910–919. [Google Scholar] [CrossRef]
- Studencka-Turski, M.; Çetin, G.; Junker, H.; Ebstein, F.; Krüger, E. Molecular Insight into the IRE1α-Mediated Type I Interferon Response Induced by Proteasome Impairment in Myeloid Cells of the Brain. Front. Immunol. 2019, 10, 2900. [Google Scholar] [CrossRef] [PubMed]
- Mazor, K.M.; Stipanuk, M.H. GCN2- and eIF2α-phosphorylation-independent, but ATF4-dependent, induction of CARE-containing genes in methionine-deficient cells. Amino Acids 2016, 48, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Palm, W.; Peng, M.; King, B.; Lindsten, T.; Li, M.O.; Koumenis, C.; Thompson, C.B. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev. 2015, 29, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4–CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Kimball, S.R.; Jefferson, L.S. Induction of REDD1 gene expression in the liver in response to endoplasmic reticulum stress is mediated through a PERK, eIF2α phosphorylation, ATF4-dependent cascade. Biochem. Biophys. Res. Commun. 2012, 427, 485–489. [Google Scholar] [CrossRef]
- York, A.G.; Williams, K.J.; Argus, J.P.; Zhou, Q.D.; Brar, G.; Vergnes, L.; Gray, E.E.; Zhen, A.; Wu, N.C.; Yamada, D.H.; et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 2015, 163, 1716–1729. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.A.M.; Reinhardt, A.; Ramsey, S.; Wittkowski, H.; Hashkes, P.J.; Berkun, Y.; Schalm, S.; Murias, S.; Dare, J.A.; Brown, D.; et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J. Clin. Investig. 2018, 128, 3041–3052. [Google Scholar] [CrossRef]
- Patel, P.N.; Hunt, R.; Pettigrew, Z.J.; Shirley, J.B.; Vogel, T.P.; de Guzman, M.M. Successful treatment of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome with tofacitinib. Pediatr. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Steiner, A.; Harapas, C.R.; Masters, S.L.; Davidson, S. An Update on Autoinflammatory Diseases: Relopathies. Curr. Rheumatol. Rep. 2018, 20, 39. [Google Scholar] [CrossRef]
- Kallinich, T.; Hinze, C.; Wittkowski, H. Klassifikation autoinflammatorischer Erkrankungen anhand pathophysiologischer Mechanismen. Z. Rheumatol. 2020, 79, 624–638. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israël, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Laplantine, E.; Dobbs, K.; Cobat, A.; Tarantino, N.; Hazen, M.; Lidov, H.G.; Hopkins, G.W.; Du, L.; Belkadi, A.; et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 2015, 212, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yu, X.; Demirkaya, E.; Deuitch, N.; Stone, D.; Tsai, W.L.; Kuehn, H.S.; Wang, H.; Yang, D.; Park, Y.H.; et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA 2016, 113, 10127–10132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef]
- Takagi, M.; Ogata, S.; Ueno, H.; Yoshida, K.; Yeh, T.; Hoshino, A.; Piao, J.; Yamashita, M.; Nanya, M.; Okano, T.; et al. Haploinsufficiency of TNFAIP3 (A20) by germline mutation is involved in autoimmune lymphoproliferative syndrome. J. Allergy Clin. Immunol. 2017, 139, 1914–1922. [Google Scholar] [CrossRef]
- Meuwissen, M.E.; Schot, R.; Buta, S.; Oudesluijs, G.; Tinschert, S.; Speer, S.D.; Li, Z.; Van Unen, L.; Heijsman, D.; Goldmann, T.; et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 2016, 213, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Basters, A.; Knobeloch, K.-P.; Fritz, G. USP18—A multifunctional component in the interferon response. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Gruber, C.; Martin-Fernandez, M.; Ailal, F.; Qiu, X.; Taft, J.; Altman, J.; Rosain, J.; Buta, S.; Bousfiha, A.; Casanova, J.-L.; et al. Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Alsohime, F.; Martin-Fernandez, M.; Temsah, M.-H.; Alabdulhafid, M.; Le Voyer, T.; Alghamdi, M.; Qiu, X.; Alotaibi, N.; Alkahtani, A.; Buta, S.; et al. JAK Inhibitor Therapy in a Child with Inherited USP18 Deficiency. N. Engl. J. Med. 2020, 382, 256–265. [Google Scholar] [CrossRef]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Cardona, D.O.; Wu, Z.; et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N. Engl. J. Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef]
- Poulter, J.; Collins, J.C.; Cargo, C.; de Tute, R.M.; Evans, P.; Cardona, D.O.; Bowen, D.T.; Cunnington, J.R.; Baguley, E.; Quinn, M.; et al. Novel somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood 2021. [Google Scholar] [CrossRef] [PubMed]
- Egerer, T.; Martínez-Gamboa, L.; Dankof, A.; Stuhlmüller, B.; Dorner, T.; Krenn, V.; Egerer, K.; Rudolph, P.E.; Burmester, G.-R.; Feist, E. Tissue-specific up-regulation of the proteasome subunit β5i (LMP7) in Sjögren’s syndrome. Arthritis Rheum. 2006, 54, 1501–1508. [Google Scholar] [CrossRef]
- Krause, S.; Kuckelkorn, U.; Dörner, T.; Burmester, G.-R.; Feist, E.; Kloetzel, P.-M. Immunoproteasome subunit LMP2 expression is deregulated in Sjogren’s syndrome but not in other autoimmune disorders. Ann. Rheum. Dis. 2006, 65, 1021–1027. [Google Scholar] [CrossRef]
- Feist, E.; Dörner, T.; Kuckelkorn, U.; Schmidtke, G.; Micheel, B.; Hiepe, F.; Burmester, G.R.; Kloetzel, P.M. Proteasome alpha-type subunit C9 is a primary target of autoantibodies in sera of patients with myositis and systemic lupus erythematosus. J. Exp. Med. 1996, 184, 1313–1318. [Google Scholar] [CrossRef]
- Feist, E.; Kuckelkorn, U.; Dörner, T.; Dönitz, H.; Scheffler, S.; Hiepe, F.; Kloetzel, P.M.; Burmester, G.R. Autoantibodies in primary Sjögren’s syndrome are directed against proteasomal subunits of the alpha and beta type. Arthritis Rheum. 1999, 42, 697–702. [Google Scholar] [CrossRef]
- Mayo, I.; Arribas, J.; Villoslada, P.; Doforno, R.A.; Rodríguez-Vilariño, S.; Montalban, X.; de Sagarra, M.R.; Castaño, J.G. The proteasome is a major autoantigen in multiple sclerosis. Brain 2002, 125, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Brychcy, M.; Kuckelkorn, U.; Hausdorf, G.; Egerer, K.; Kloetzel, P.-M.; Burmester, G.-R.; Feist, E. Anti-20S proteasome autoantibodies inhibit proteasome stimulation by proteasome activator PA28. Arthritis Rheum. 2006, 54, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Egerer, K.; Kuckelkorn, U.; Rudolph, P.; Rückert, J.C.; Dörner, T.; Burmester, G.-R.; Kloetzel, P.-M.; Feist, E. Circulating proteasomes are markers of cell damage and immunologic activity in autoimmune diseases. J. Rheumatol. 2002, 29, 2045–2052. [Google Scholar]
- Maruyama, H.; Hirayama, K.; Yamashita, M.; Ohgi, K.; Tsujimoto, R.; Takayasu, M.; Shimohata, H.; Kobayashi, M. Serum 20S proteasome levels are associated with disease activity in MPO-ANCA-associated microscopic polyangiitis. BMC Rheumatol. 2020, 4, 1–7. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef]
- Basler, M.; Dajee, M.; Moll, C.; Groettrup, M.; Kirk, C.J. Prevention of Experimental Colitis by a Selective Inhibitor of the Immunoproteasome. J. Immunol. 2010, 185, 634–641. [Google Scholar] [CrossRef]
- Schmidt, N.; Gonzalez, E.; Visekruna, A.; Kühl, A.; Loddenkemper, C.; Mollenkopf, H.; Kaufmann, S.H.; Steinhoff, U.; Joeris, T. Targeting the proteasome: Partial inhibition of the proteasome by bortezomib or deletion of the immunosubunit LMP7 attenuates experimental colitis. Gut 2010, 59, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Mundt, S.; Engelhardt, B.; Kirk, C.J.; Groettrup, M.; Basler, M. Inhibition and deficiency of the immunoproteasome subunit LMP7 attenuates LCMV-induced meningitis. Eur. J. Immunol. 2015, 46, 104–113. [Google Scholar] [CrossRef]
- Basler, M.; Claus, M.; Klawitter, M.; Goebel, H.; Groettrup, M. Immunoproteasome Inhibition Selectively Kills Human CD14+ Monocytes and as a Result Dampens IL-23 Secretion. J. Immunol. 2019, 203, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Mundt, S.; Muchamuel, T.; Moll, C.; Jiang, J.; Groettrup, M.; Kirk, C.J. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol. Med. 2014, 6, 226–238. [Google Scholar] [CrossRef]
- Feist, E.; Burmester, G.-R.; Krüger, E. The proteasome—Victim or culprit in autoimmunity. Clin. Immunol. 2016, 172, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Neubert, K.; Meister, S.; Moser, K.; Weisel, F.; Maseda, D.; Amann, K.; Wiethe, C.; Winkler, T.H.; Kalden, J.R.; Manz, R.; et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med. 2008, 14, 748–755. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Ilchovska, D.; Barrow, M. An Overview of the NF-kB mechanism of pathophysiology in rheumatoid arthritis, investigation of the NF-kB ligand RANKL and related nutritional interventions. Autoimmun. Rev. 2021, 20, 102741. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, J.W.; Oerlemans, R.; Lems, W.F.; Scheper, R.J.; Dijkmans, B.A.; Jansen, G. The proteasome inhibitor bortezomib inhibits the release of NFkappaB-inducible cytokines and induces apoptosis of activated T cells from rheumatoid arthritis patients. Clin. Exp. Rheumatol. 2009, 27, 92–98. [Google Scholar]
- Migita, K.; Tanaka, F.; Yamasaki, S.; Shibatomi, K.; Ida, H.; Kawakami, A.; Aoyagi, T.; Kawabe, Y.; Eguchi, K. Regulation of rheumatoid synoviocyte proliferation by endogenous p53 induction. Clin. Exp. Immunol. 2001, 126, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Dörner, T.; Furie, R. Novel paradigms in systemic lupus erythematosus. Lancet 2019, 393, 2344–2358. [Google Scholar] [CrossRef]
- Ichikawa, H.T.; Conley, T.; Muchamuel, T.; Jiang, J.; Lee, S.; Owen, T.; Barnard, J.; Nevarez, S.; Goldman, B.I.; Kirk, C.J.; et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012, 64, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.; Grützkau, A.; Hirseland, H.; Huscher, D.; Dähnrich, C.; Dzionek, A.; Ozimkowski, T.; Schlumberger, W.; Enghard, P.; Radbruch, A.; et al. IFNα and its response proteins, IP-10 and SIGLEC-1, are biomarkers of disease activity in systemic lupus erythematosus. Ann. Rheum. Dis. 2012, 72, 1639–1645. [Google Scholar] [CrossRef]
- Yao, L.; Zhou, L.; Xuan, Y.; Zhang, P.; Wang, X.; Wang, T.; Meng, T.; Xue, Y.; Ma, X.; Shah, A.S.; et al. The proteasome activator REGγ counteracts immunoproteasome expression and autoimmunity. J. Autoimmun. 2019, 103, 102282. [Google Scholar] [CrossRef] [PubMed]
- Gruner, M.; Moncsek, A.; Rödiger, S.; Kühnhardt, D.; Feist, E.; Stohwasser, R. Increased proteasome activator 28 gamma (PA28γ) levels are unspecific but correlate with disease activity in rheumatoid arthritis. BMC Musculoskelet. Disord. 2014, 15, 1–10. [Google Scholar] [CrossRef][Green Version]
- Witte, T. Sjögren-Syndrom. Z. Rheumatol. 2019, 78, 511–517. [Google Scholar] [CrossRef]
- Feist, E.; Dörner, T.; Kuckelkorn, U.; Scheffler, S.; Burmester, G.-R.; Kloetzel, P.-M. Diagnostic importance of anti-proteasome antibodies. Int. Arch. Allergy Immunol. 2000, 123, 92–97. [Google Scholar] [CrossRef]
- Morawietz, L.; Martinez-Gamboa, L.; Scheffler, S.; Hausdorf, G.; Dankof, A.; Kuckelkorn, U.; Doerner, T.; Egerer, K.; Burmester, G.-R.; Faustman, D.L.; et al. Expression of Proteasomal Immunosubunit ß1i Is Dysregulated in Inflammatory Infiltrates of Minor Salivary Glands in Sjögren’s Syndrome. J. Rheumatol. 2009, 36, 2694–2703. [Google Scholar] [CrossRef]
- Martinez-Gamboa, L.; Lesemann, K.; Kuckelkorn, U.; Scheffler, S.; Ghannam, K.; Hahne, M.; Gaber-Elsner, T.; Egerer, K.; Naumann, L.; Buttgereit, F.; et al. Gene Expression of Catalytic Proteasome Subunits and Resistance Toward Proteasome Inhibition of B Lymphocytes from Patients with Primary Sjögren Syndrome. J. Rheumatol. 2013, 40, 663–673. [Google Scholar] [CrossRef]
- Xiaohui, W.; Lingyun, S.; Tian, J.; Wang, X.; Chen, Q.; Rui, K.; Ma, J.; Wang, S.; Wang, Q.; Wang, X.; et al. Proteasome inhibition suppresses Th17 cell generation and ameliorates autoimmune development in experimental Sjögren’s syndrome. Cell. Mol. Immunol. 2017, 14, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Kochi, Y.; Muro, R.; Tomofuji, Y.; Okamura, T.; Murata, S.; Suzuki, H.; Sumida, T.; Yamamoto, K.; Takayanagi, H. Human thymoproteasome variations influence CD8 T cell selection. Sci. Immunol. 2017, 2, eaan5165. [Google Scholar] [CrossRef]
- Schreiber, S.; Nikolaus, S.; Hampe, J. Activation of nuclear factor kappa B in inflammatory bowel disease. Gut 1998, 42, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Visekruna, A.; Slavova, N.; Dullat, S.; Gröne, J.; Kroesen, A.-J.; Ritz, J.-P.; Buhr, H.-J.; Steinhoff, U. Expression of catalytic proteasome subunits in the gut of patients with Crohn’s disease. Int. J. Color. Dis. 2009, 24, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Visekruna, A.; Joeris, T.; Schmidt, N.; Lawrenz, M.; Ritz, J.-P.; Buhr, H.J.; Steinhoff, U. Comparative expression analysis and characterization of 20S proteasomes in human intestinal tissues. Inflamm. Bowel Dis. 2009, 15, 526–533. [Google Scholar] [CrossRef]
- Visekruna, A.; Joeris, T.; Seidel, D.; Kroesen, A.; Loddenkemper, C.; Zeitz, M.; Kaufmann, S.H.; Schmidt-Ullrich, R.; Steinhoff, U. Proteasome-mediated degradation of IκBα and processing of p105 in Crohn disease and ulcerative colitis. J. Clin. Investig. 2006, 116, 3195–3203. [Google Scholar] [CrossRef]
- Moallemian, R.; Rehman, A.U.; Zhao, N.; Wang, H.; Chen, H.; Lin, G.; Ma, X.; Yu, J. Immunoproteasome inhibitor DPLG3 attenuates experimental colitis by restraining NF-κB activation. Biochem. Pharmacol. 2020, 177, 113964. [Google Scholar] [CrossRef]
- Vachharajani, N.; Joeris, T.; Luu, M.; Hartmann, S.; Pautz, S.; Jenike, E.; Pantazis, G.; Prinz, I.; Hofer, M.J.; Steinhoff, U.; et al. Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 2017, 8, 50447–50459. [Google Scholar] [CrossRef]
- Basler, M.; Lindstrom, M.M.; LaStant, J.J.; Bradshaw, J.M.; Owens, T.D.; Schmidt, C.; Maurits, E.; Tsu, C.; Overkleeft, H.S.; Kirk, C.J.; et al. Co-inhibition of immunoproteasome subunits LMP2 and LMP7 is required to block autoimmunity. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Mishto, M.; Bellavista, E.; Ligorio, C.; Textoris-Taube, K.; Santoro, A.; Giordano, M.; D’Alfonso, S.; Listì, F.; Nacmias, B.; Cellini, E.; et al. Immunoproteasome LMP2 60HH Variant Alters MBP Epitope Generation and Reduces the Risk to Develop Multiple Sclerosis in Italian Female Population. PLoS ONE 2010, 5, e9287. [Google Scholar] [CrossRef]
- VanderLugt, C.L.; Rahbe, S.M.; Elliott, P.J.; Canto, M.C.D.; Miller, S.D. Treatment of Established Relapsing Experimental Autoimmune Encephalomyelitis with the Proteasome Inhibitor PS-5191. J. Autoimmun. 2000, 14, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, H.; André, P.; Lefevre, N.; Viala, L.; Walzer, T.; Peschanski, M.; Lotteau, V. Protection against experimental autoimmune encephalomyelitis by a proteasome modulator. J. Neuroimmunol. 2001, 118, 233–244. [Google Scholar] [CrossRef]
- Gupta, R.; Debbaneh, M.G.; Liao, W. Genetic Epidemiology of Psoriasis. Curr. Dermatol. Rep. 2014, 3, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guo, W.; Zhang, S.; Wang, G. Ubiquitination-proteasome system: A new player in the pathogenesis of psoriasis and clinical implications. J. Dermatol. Sci. 2018, 89, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.-H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Bakir-Gungor, B.; Remmers, E.F.; Meguro, A.; Mizuki, N.; Kastner, D.L.; Gül, A.; Sezerman, O.U. Identification of possible pathogenic pathways in Behçet’s disease using genome-wide association study data from two different populations. Eur. J. Hum. Genet. 2014, 23, 678–687. [Google Scholar] [CrossRef]
- Sánchez-Serrano, I. Success in translational research: Lessons from the development of bortezomib. Nat. Rev. Drug Discov. 2006, 5, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Richardson, P.; Sonneveld, P.; Schuster, M.; Irwin, D.; Stadtmauer, E.; Facon, T.; Harousseau, J.-L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Ishii, T.; Tanaka, Y.; Kawakami, A.; Saito, K.; Ichinose, K.; Fujii, H.; Shirota, Y.; Shirai, T.; Fujita, Y.; Watanabe, R.; et al. Multicenter double-blind randomized controlled trial to evaluate the effectiveness and safety of bortezomib as a treatment for refractory systemic lupus erythematosus. Mod. Rheumatol. 2018, 28, 986–992. [Google Scholar] [CrossRef]
- Van Dam, L.S.; Osmani, Z.; Kamerling, S.W.; Kraaij, T.; Bakker, J.; Scherer, H.U.; Rabelink, T.J.; Voll, R.; Alexander, T.; Isenberg, D.; et al. A reverse translational study on the effect of rituximab, rituximab plus belimumab, or bortezomib on the humoral autoimmune response in SLE. Rheumatology 2020, 59, 2734–2745. [Google Scholar] [CrossRef] [PubMed]
- Hiepe, F.; Radbruch, F.H.A. Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat. Rev. Nephrol. 2016, 12, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Knops, N.; Emonds, M.; Herman, J.; Levtchenko, E.; Mekahli, D.; Pirenne, J.; Van Geet, C.; Dierickx, D. Bortezomib for autoimmune hemolytic anemia after intestinal transplantation. Pediatr. Transplant. 2020, 24, 13700. [Google Scholar] [CrossRef]
- Beydoun, S.B.; Persaud, Y.; Lafferty, J.; Callaghan, M.U.; Savaşan, S. Bortezomib treatment of steroid-refractory Evans syndrome in children. Pediatr. Blood Cancer 2020, 67, 28725. [Google Scholar] [CrossRef]
- Yates, S.; Matevosyan, K.; Rutherford, C.; Shen, Y.-M.; Sarode, R. Bortezomib for chronic relapsing thrombotic thrombocytopenic purpura: A case report. Transfusion 2014, 54, 2064–2067. [Google Scholar] [CrossRef] [PubMed]
- Jakez-Ocampo, J.; Atisha-Fregoso, Y.; Llorente, L. Refractory Primary Sjögren Syndrome Successfully Treated with Bortezomib. J. Clin. Rheumatol. 2015, 21, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Berges, C.; Haberstock, H.; Fuchs, D.; Miltz, M.; Sadeghi, M.; Opelz, G.; Daniel, V.; Naujokat, C. Proteasome inhibition suppresses essential immune functions of human CD4+T cells. Immunology 2008, 124, 234–246. [Google Scholar] [CrossRef]
- Pletinckx, K.; Vaßen, S.; Schlusche, I.; Nordhoff, S.; Bahrenberg, G.; Dunkern, T.R. Inhibiting the immunoproteasome’s β5i catalytic activity affects human peripheral blood-derived immune cell viability. Pharmacol. Res. Perspect. 2019, 7, e00482. [Google Scholar] [CrossRef]
- Scheibe, F.; Prüss, H.; Mengel, A.M.; Kohler, S.; Nümann, A.; Köhnlein, M.; Ruprecht, K.; Alexander, T.; Hiepe, F.; Meisel, A. Bortezomib for treatment of therapy-refractory anti-NMDA receptor encephalitis. Neurology 2016, 88, 366–370. [Google Scholar] [CrossRef]
- Wickel, J.; Chung, H.-Y.; Platzer, S.; Lehmann, T.; Prüss, H.; Leypoldt, F.; Günther, A.; Scherag, A.; Geis, C.; on behalf of the GENERATE Study Group. Generate-Boost: Study protocol for a prospective, multicenter, randomized controlled, double-blinded phase II trial to evaluate efficacy and safety of bortezomib in patients with severe autoimmune encephalitis. Trials 2020, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Meregalli, C. An Overview of Bortezomib-Induced Neurotoxicity. Toxics 2015, 3, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMINN, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2S,3R)-N-((S)-3-(Cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar] [CrossRef]
- Von Brzezinski, L.; Säring, P.; Landgraf, P.; Cammann, C.; Seifert, U.; Dieterich, D.C. Low neurotoxicity of ONX-0914 supports the idea of specific immunoproteasome inhibition as a side-effect-limiting, therapeutic strategy. Eur. J. Microbiol. Immunol. 2017, 7, 234–245. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, J.; Matos, J.; Dziopa, E.; Dziopa, L.; Yang, Z.; Kirk, C.J.; Assefnia, S.; Korngold, R. Inhibition of the Immunoproteasome Subunit LMP7 with ONX 0914 Ameliorates Graft-versus-Host Disease in an MHC-Matched Minor Histocompatibility Antigen–Disparate Murine Model. Biol. Blood Marrow Transplant. 2015, 21, 1555–1564. [Google Scholar] [CrossRef]
- Liu, H.; Wan, C.; Ding, Y.; Han, R.; He, Y.; Xiao, J.; Hao, J. PR-957, a selective inhibitor of immunoproteasome subunit low-MW polypeptide 7, attenuates experimental autoimmune neuritis by suppressing T h 17-cell differentiation and regulating cytokine production. FASEB J. 2016, 31, 1756–1766. [Google Scholar] [CrossRef]
- Basler, M.; Groettrup, M. Recent insights how combined inhibition of immuno/proteasome subunits enables therapeutic efficacy. Genes Immun. 2020, 21, 273–287. [Google Scholar] [CrossRef]
- Ladi, E.; Everett, C.; Stivala, C.E.; Daniels, B.E.; Durk, M.R.; Harris, S.F.; Huestis, M.P.; Purkey, H.E.; Staben, S.T.; Augustin, M.; et al. Design and Evaluation of Highly Selective Human Immunoproteasome Inhibitors Reveal a Compensatory Process That Preserves Immune Cell Viability. J. Med. Chem. 2019, 62, 7032–7041. [Google Scholar] [CrossRef]
- Leestemaker, Y.; De Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; Van De Kooij, B.; Laufer, S.; Heck, A.J.; et al. Proteasome Activation by Small Molecules. Cell Chem. Biol. 2017, 24, 725–736.e7. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.V.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nat. Cell Biol. 2010, 467, 179–184Bico. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goetzke, C.C.; Ebstein, F.; Kallinich, T. Role of Proteasomes in Inflammation. J. Clin. Med. 2021, 10, 1783. https://doi.org/10.3390/jcm10081783
Goetzke CC, Ebstein F, Kallinich T. Role of Proteasomes in Inflammation. Journal of Clinical Medicine. 2021; 10(8):1783. https://doi.org/10.3390/jcm10081783
Chicago/Turabian StyleGoetzke, Carl Christoph, Frédéric Ebstein, and Tilmann Kallinich. 2021. "Role of Proteasomes in Inflammation" Journal of Clinical Medicine 10, no. 8: 1783. https://doi.org/10.3390/jcm10081783
APA StyleGoetzke, C. C., Ebstein, F., & Kallinich, T. (2021). Role of Proteasomes in Inflammation. Journal of Clinical Medicine, 10(8), 1783. https://doi.org/10.3390/jcm10081783