
Ewing Sarcoma—Diagnosis, Treatment, Clinical Challenges and Future Perspectives

 , , , , , , , , and
, , , , , , , , and 
Abstract
1. Introduction
2. Diagnosis
2.1. Imaging (by V. Vieth)
2.1.1. Diagnostic Workup—The Timeless Value of Plain Radiographs for Deciphering Bone Lesions
2.1.2. Local Tumor Assessment and Staging—“Trust in T1”
2.1.3. Therapeutic Assessment and Follow-Up
2.2. Biopsy (by E. de Álava, W. Hartmann, and V. Vieth)
2.2.1. How to Biopsy in EwS?
2.2.2. The Risk of Tumor Seeding along the Access Path of Biopsy
2.2.3. Biopsy—The Holy Tissue Grail
2.3. Pathological Diagnosis (by E. de Álava, T. G. Grünewald, and W. Hartmann)
2.3.1. How to Diagnose EwS?
2.3.2. Historical Evolution of EwS and EwS-Related Entities
2.3.3. Round Cell Sarcoma with Non-ETS-Fusions and CIC/BCOR-Rearranged Sarcoma
3. Local Therapy
3.1. Operative Local Therapy (by S. Collaud, J. Hardes, and A. Streitbürger)
3.1.1. The Matter of Local Therapy—Scientifically Hard to Resolve, but Clinically Guided by Interdisciplinary Tumor Board Recommendations
3.1.2. Surgical Strategies—Both Form and Function Follow Local Control
3.1.3. Surgical Margins and Histopathological Response to Systemic Treatment—Implications for Additional Local Therapy
3.1.4. Initial Versus Chemotherapy Responsive Tumor—Operate to What Extent?
3.1.5. Pathological Fracture in EwS
3.1.6. EwS of the Extremities and the Role of Limb Perfusion
3.1.7. Pelvic and Sacral EwS—When and How to Operate?
3.1.8. Primary Thoracic EwS
3.1.9. Patients May Benefit from Early Referral—Even Prior Diagnostic Biopsy—But Do Not Benefit from Re-Resection or Debulking Strategies
3.1.10. Disseminated and Relapsed EwS
3.2. Radiotherapy (by B. Timmermann)
3.2.1. Role of RT and Timing
3.2.2. Modern RT Strategies and Techniques
3.2.3. Primary Tumor Site, Prescription Dose, and Target Volume Definition
3.2.4. RT for Relapse, Metastases and Whole-Lung Irradiation
3.2.5. Radiation Toxicity with High-Dose Treatment
3.2.6. Irradiation for Palliation
4. Systemic Therapy
4.1. Evolution of the Current Systemic Backbone for Classical EwS (by U. Dirksen, S. G. DuBois, and D. S. Shulman)
4.1.1. Development of VACA-Based Regimens—Multi-Agent Systemic Therapy Improves Outcomes
4.1.2. Addition of Ifosfamide and Etoposide Further Improves Outcomes
4.1.3. Intensified Therapies—Time but Not Dose or Duration Matters
4.1.4. Adding Conventional Agents to Existing Backbone Regimens Has Not Thus Far Improved Outcomes
4.2. Systemic Treatment of Relapsed Classical EwS Including Combination Therapies (by S. G. DuBois and D. S. Shulman)
4.2.1. Approach Relapse Therapy
4.2.2. Systemic Therapies for Relapsed Disease—Time to Relapse Dictates Novel Agents Versus Re-Challenge with Frontline Drugs
4.2.3. Maintenance Therapy in EwS
4.3. EwS-Targeted Therapy—Low-Hanging Fruit or Unfair Rumor? (by S. Bauer, U. Dirksen, S. G. DuBois, J. A. Toretsky, and D. S. Shulman)
4.3.1. Targeted Agents Will Be Necessary to Overcome the Limitations of Conventional Chemotherapy and to Reduce the Burden of Late Effects in EwS
4.3.2. Adding Targeted Therapies to Existing Backbone Regimens
4.3.3. Established and Emerging Targeted Therapies for Relapsed Disease
4.3.4. The FET-ETS Translocations—A Clear Target, with Both Direct and Indirect Strategies
4.3.5. Where Is Targeted Therapy Heading?
4.3.6. Challenges to Treat EwS in Low- and Middle-Income Countries
5. Scientific Perspectives on Clinical Enigmas of Disseminated EwS Disease
5.1. How Similar Are Primary Tumors with Metastatic Lesions? (by J. F. Amatruda and H. Kovar)
5.1.1. Clonal Evolution of Metastases Seeds in Intratumor Heterogeneity and Correlates with Mutational Burden
5.1.2. Lessons from Bulk Gene Expression Analyses Including Immune Contextures of EwS Tumors and in Peripheral Blood
5.2. How Immunogenic Are EwS Tumors and What Clinical Value Lies within? (by J. F. Amatruda and H. Kovar)
5.2.1. Prognostic Immune Contextures of EwS Tumors and in Peripheral Blood
5.2.2. Immunotargeting of EwS Tumors
5.3. Oncogene Plasticity—Myth or Tumor Strategy with Clinical Impact and Potential Therapeutic Consequence? (by J. F. Amatruda and H. Kovar)
EWSR1-FLI1 Oncogene Fluctuations as Metastatic Drivers in EwS
5.4. Why Does Outcome Differ between Lung Metastases and Metastases at Other Locations? (by J. F. Amatruda and H. Kovar)
5.4.1. Therapeutic Accessibility
5.4.2. Biological Concepts for Organotropism of EwS Metastasis
5.5. Are We on Track with Preclinical Models? (by J. F. Amatruda and H. Kovar)
5.5.1. Patient-Derived EwS Models
5.5.2. Non-Patient Derived EwS Models
5.5.3. “The” EwS Mouse Model—Chronically Unavailable (So Far)
6. Biomarkers
6.1. Status Report on Biomarkers in EwS (by E. de Álava, M. Metzler, and V. Vieth)
6.1.1. Diagnostic, Prognostic, and Therapeutic Markers in EwS
6.1.2. Limitations and Future Perspectives for EwS Biomarkers
7. Concluding Remarks (by Y. Uhlenbruch)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, C.C.; Schulz, M.D. Ewing’s sarcoma; a study of fifty cases treated at the Massachusetts General Hospital, 1930-1952 inclusive. N. Engl. J. Med. 1953, 248, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, D.C.; Coventry, M.B.; Scanlon, P.W. Ewing’s sarcoma. A critical analysis of 165 cases. J. Bone Joint. Surg. Am. 1961, 43-A, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Ewing, J. The Classic: Diffuse endothelioma of bone. Proceedings of the New York Pathological Society. 1921;12:17. Clin. Orthop. Relat. Res. 2006, 450, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Anderton, J.; Moroz, V.; Marec-Berard, P.; Gaspar, N.; Laurence, V.; Martin-Broto, J.; Sastre, A.; Gelderblom, H.; Owens, C.; Kaiser, S.; et al. International randomised controlled trial for the treatment of newly diagnosed EWING sarcoma family of tumours-EURO EWING 2012 Protocol. Trials 2020, 21, 96. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol 2015, 33, 3036–3046. [Google Scholar] [CrossRef]
- Wheatley, K.M.V.; Marec-Berard, P. First results of the EURO EWING 2012 trial comparing two chemotherapy regimens in newly diagnosed Ewing sarcoma. In Proceedings of the Connective Tissue Oncology Society (CTOS)-Oral Presentation/Abstract, Annual Meeting 2019. Tokyo, Japan, 13–16 November 2019. [Google Scholar]
- Whelan, J.; Le Deley, M.C.; Dirksen, U.; Le Teuff, G.; Brennan, B.; Gaspar, N.; Hawkins, D.S.; Amler, S.; Bauer, S.; Bielack, S.; et al. High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J. Clin. Oncol 2018, 36, JCO2018782516. [Google Scholar] [CrossRef]
- Haeusler, J.; Ranft, A.; Boelling, T.; Gosheger, G.; Braun-Munzinger, G.; Vieth, V.; Burdach, S.; van den Berg, H.; Juergens, H.; Dirksen, U. The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 2010, 116, 443–450. [Google Scholar] [CrossRef]
- Ladenstein, R.; Potschger, U.; Le Deley, M.C.; Whelan, J.; Paulussen, M.; Oberlin, O.; van den Berg, H.; Dirksen, U.; Hjorth, L.; Michon, J.; et al. Primary disseminated multifocal Ewing sarcoma: Results of the Euro-EWING 99 trial. J. Clin. Oncol 2010, 28, 3284–3291. [Google Scholar] [CrossRef]
- Barker, L.M.; Pendergrass, T.W.; Sanders, J.E.; Hawkins, D.S. Survival after recurrence of Ewing’s sarcoma family of tumors. J. Clin. Oncol 2005, 23, 4354–4362. [Google Scholar] [CrossRef]
- Rodriguez-Galindo, C.; Billups, C.A.; Kun, L.E.; Rao, B.N.; Pratt, C.B.; Merchant, T.E.; Santana, V.M.; Pappo, A.S. Survival after recurrence of Ewing tumors: The St Jude Children’s Research Hospital experience, 1979–1999. Cancer 2002, 94, 561–569. [Google Scholar] [CrossRef]
- Stahl, M.; Ranft, A.; Paulussen, M.; Bolling, T.; Vieth, V.; Bielack, S.; Gortitz, I.; Braun-Munzinger, G.; Hardes, J.; Jurgens, H.; et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr. Blood Cancer 2011, 57, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Kovar, H.; Aryee, D.N.; Jug, G.; Henockl, C.; Schemper, M.; Delattre, O.; Thomas, G.; Gadner, H. EWS/FLI-1 antagonists induce growth inhibition of Ewing tumor cells in vitro. Cell Growth Differ. 1996, 7, 429–437. [Google Scholar] [PubMed]
- Lambert, G.; Bertrand, J.R.; Fattal, E.; Subra, F.; Pinto-Alphandary, H.; Malvy, C.; Auclair, C.; Couvreur, P. EWS fli-1 antisense nanocapsules inhibits ewing sarcoma-related tumor in mice. Biochem. Biophys. Res. Commun. 2000, 279, 401–406. [Google Scholar] [CrossRef]
- Anderson, W.J.; Doyle, L.A. Updates from the 2020 World Health Organization Classification of Soft Tissue and Bone Tumours. Histopathology 2021. [Google Scholar] [CrossRef]
- Gerrand, C.; Bate, J.; Seddon, B.; Dirksen, U.; Randall, R.L.; van de Sande, M.; O’Donnell, P.; Tuckett, J.; Peake, D.; Jeys, L.; et al. Seeking international consensus on approaches to primary tumour treatment in Ewing sarcoma. Clin. Sarcoma Res. 2020, 10, 21. [Google Scholar] [CrossRef]
- Ulaner, G.; Hwang, S.; Landa, J.; Lefkowitz, R.A.; Panicek, D.M. Musculoskeletal tumours and tumour-like conditions: Common and avoidable pitfalls at imaging in patients with known or suspected cancer: Part B: Malignant mimics of benign tumours. Int. Orthop. 2013, 37, 877–882. [Google Scholar] [CrossRef]
- Ulaner, G.; Hwang, S.; Lefkowitz, R.A.; Landa, J.; Panicek, D.M. Musculoskeletal tumors and tumor-like conditions: Common and avoidable pitfalls at imaging in patients with known or suspected cancer: Part A: Benign conditions that may mimic malignancy. Int. Orthop. 2013, 37, 871–876. [Google Scholar] [CrossRef]
- Daniel, A., Jr.; Ullah, E.; Wahab, S.; Kumar, V., Jr. Relevance of MRI in prediction of malignancy of musculoskeletal system--a prospective evaluation. BMC Musculoskelet. Disord. 2009, 10, 125. [Google Scholar] [CrossRef]
- Lodwick, G.S. Radiographic diagnosis and grading of bone tumors, with comments on computer evaluation. Proc. Natl. Cancer Conf. 1964, 5, 369–380. [Google Scholar]
- Caracciolo, J.T.; Temple, H.T.; Letson, G.D.; Kransdorf, M.J. A Modified Lodwick-Madewell Grading System for the Evaluation of Lytic Bone Lesions. AJR Am. J. Roentgenol. 2016, 207, 150–156. [Google Scholar] [CrossRef]
- Rana, R.S.; Wu, J.S.; Eisenberg, R.L. Periosteal reaction. AJR Am. J. Roentgenol. 2009, 193, W259–W272. [Google Scholar] [CrossRef]
- Wenaden, A.E.; Szyszko, T.A.; Saifuddin, A. Imaging of periosteal reactions associated with focal lesions of bone. Clin. Radiol. 2005, 60, 439–456. [Google Scholar] [CrossRef]
- Erlemann, R. [MRI morphology of bone tumors and tumor-like lesions]. Radiologe 2010, 50, 61–80. [Google Scholar] [CrossRef]
- Kasalak, O.; Overbosch, J.; Adams, H.J.; Dammann, A.; Dierckx, R.A.; Jutte, P.C.; Kwee, T.C. Diagnostic value of MRI signs in differentiating Ewing sarcoma from osteomyelitis. Acta Radiol. 2019, 60, 204–212. [Google Scholar] [CrossRef]
- Brady, E.J.; Hameed, M.; Tap, W.D.; Hwang, S. Imaging features and clinical course of undifferentiated round cell sarcomas with CIC-DUX4 and BCOR-CCNB3 translocations. Skeletal Radiol. 2021, 50, 521–529. [Google Scholar] [CrossRef]
- Meyer, J.S.; Nadel, H.R.; Marina, N.; Womer, R.B.; Brown, K.L.; Eary, J.F.; Gorlick, R.; Grier, H.E.; Randall, R.L.; Lawlor, E.R.; et al. Imaging guidelines for children with Ewing sarcoma and osteosarcoma: A report from the Children’s Oncology Group Bone Tumor Committee. Pediatr. Blood Cancer 2008, 51, 163–170. [Google Scholar] [CrossRef]
- Thompson, M.J.; Shapton, J.C.; Punt, S.E.; Johnson, C.N.; Conrad, E.U., 3rd. MRI Identification of the Osseous Extent of Pediatric Bone Sarcomas. Clin. Orthop. Relat. Res. 2018, 476, 559–564. [Google Scholar] [CrossRef]
- Shahid, M.; Albergo, N.; Purvis, T.; Heron, K.; Gaston, L.; Carter, S.; Grimer, R.; Jeys, L. Management of sarcomas possibly involving the knee joint when to perform extra-articular resection of the knee joint and is it safe? Eur. J. Surg. Oncol. 2017, 43, 175–180. [Google Scholar] [CrossRef]
- van Trommel, M.F.; Kroon, H.M.; Bloem, J.L.; Hogendoorn, P.C.; Taminiau, A.H. MR imaging based strategies in limb salvage surgery for osteosarcoma of the distal femur. Skeletal Radiol. 1997, 26, 636–641. [Google Scholar] [CrossRef]
- Enneking, W.F.; Kagan, A. “Skip” metastases in osteosarcoma. Cancer 1975, 36, 2192–2205. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.J.; Parisi, M.T.; Shulkin, B.L. The Role of (18)F-FDG-PET/CT in Pediatric Sarcoma. Semin Nucl. Med. 2017, 47, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.; Mosci, C.; von Eyben, R.; Mittra, E.; Ganjoo, K.; Biswal, S.; Gambhir, S.S.; Iagaru, A. Combined 18F-NaF and 18F-FDG PET/CT in the Evaluation of Sarcoma Patients. Clin. Nucl. Med. 2015, 40, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.P.; Schoenberg, S.O.; Schmid, R.; Stahl, R.; Tiling, R.; Becker, C.R.; Reiser, M.F.; Baur-Melnyk, A. Screening for bone metastases: Whole-body MRI using a 32-channel system versus dual-modality PET-CT. Eur. Radiol. 2007, 17, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Daldrup-Link, H. How PET/MR Can Add Value For Children With Cancer. Curr. Radiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Heinemann, M.; Ranft, A.; Langer, T.; Jurgens, H.; Kreyer, J.; Vieth, V.; Schafers, M.; Weckesser, M.; Simon, T.; Hassenpflug, W.; et al. Recurrence of Ewing sarcoma: Is detection by imaging follow-up protocol associated with survival advantage? Pediatr Blood Cancer 2018, 65, e27011. [Google Scholar] [CrossRef]
- Kaste, S.C. Imaging pediatric bone sarcomas. Radiol. Clin. N. Am. 2011, 49, 749–765. [Google Scholar] [CrossRef]
- Exner, G.U.; Kurrer, M.O.; Mamisch-Saupe, N.; Cannon, S.R. The tactics and technique of musculoskeletal biopsy. EFORT Open Rev. 2017, 2, 51–57. [Google Scholar] [CrossRef]
- Traina, F.; Errani, C.; Toscano, A.; Pungetti, C.; Fabbri, D.; Mazzotti, A.; Donati, D.; Faldini, C. Current concepts in the biopsy of musculoskeletal tumors: AAOS exhibit selection. J. Bone Joint. Surg. Am. 2015, 97, e7. [Google Scholar] [CrossRef]
- Khoo, M.M.; Saifuddin, A. The role of MRI in image-guided needle biopsy of focal bone and soft tissue neoplasms. Skeletal Radiol. 2013, 42, 905–915. [Google Scholar] [CrossRef]
- Singh, H.K.; Kilpatrick, S.E.; Silverman, J.F. Fine needle aspiration biopsy of soft tissue sarcomas: Utility and diagnostic challenges. Adv. Anat. Pathol. 2004, 11, 24–37. [Google Scholar] [CrossRef]
- Pohlig, F.; Kirchhoff, C.; Lenze, U.; Schauwecker, J.; Burgkart, R.; Rechl, H.; von Eisenhart-Rothe, R. Percutaneous core needle biopsy versus open biopsy in diagnostics of bone and soft tissue sarcoma: A retrospective study. Eur. J. Med. Res. 2012, 17, 29. [Google Scholar] [CrossRef]
- Mitsuyoshi, G.; Naito, N.; Kawai, A.; Kunisada, T.; Yoshida, A.; Yanai, H.; Dendo, S.; Yoshino, T.; Kanazawa, S.; Ozaki, T. Accurate diagnosis of musculoskeletal lesions by core needle biopsy. J. Surg. Oncol. 2006, 94, 21–27. [Google Scholar] [CrossRef]
- Altuntas, A.O.; Slavin, J.; Smith, P.J.; Schlict, S.M.; Powell, G.J.; Ngan, S.; Toner, G.; Choong, P.F. Accuracy of computed tomography guided core needle biopsy of musculoskeletal tumours. ANZ J. Surg. 2005, 75, 187–191. [Google Scholar] [CrossRef]
- Didolkar, M.M.; Anderson, M.E.; Hochman, M.G.; Rissmiller, J.G.; Goldsmith, J.D.; Gebhardt, M.G.; Wu, J.S. Image guided core needle biopsy of musculoskeletal lesions: Are nondiagnostic results clinically useful? Clin. Orthop. Relat. Res. 2013, 471, 3601–3609. [Google Scholar] [CrossRef]
- Dupuy, D.E.; Rosenberg, A.E.; Punyaratabandhu, T.; Tan, M.H.; Mankin, H.J. Accuracy of CT-guided needle biopsy of musculoskeletal neoplasms. AJR Am. J. Roentgenol. 1998, 171, 759–762. [Google Scholar] [CrossRef]
- Interiano, R.B.; Malkan, A.D.; Loh, A.H.; Hinkle, N.; Wahid, F.N.; Bahrami, A.; Mao, S.; Wu, J.; Bishop, M.W.; Neel, M.D.; et al. Initial diagnostic management of pediatric bone tumors. J. Pediatr. Surg. 2016, 51, 981–985. [Google Scholar] [CrossRef][Green Version]
- Jelinek, J.S.; Murphey, M.D.; Welker, J.A.; Henshaw, R.M.; Kransdorf, M.J.; Shmookler, B.M.; Malawer, M.M. Diagnosis of primary bone tumors with image-guided percutaneous biopsy: Experience with 110 tumors. Radiology 2002, 223, 731–737. [Google Scholar] [CrossRef]
- Kalus, S.; Vidoni, A.; Oliveira, I.; Saifuddin, A. Image-guided core needle biopsy for Ewing sarcoma of bone: A 10-year single-institution review. Eur. Radiol. 2020, 30, 5308–5314. [Google Scholar] [CrossRef]
- McCarville, M.B.; Chen, J.Y.; Coleman, J.L.; Li, Y.; Li, X.; Adderson, E.E.; Neel, M.D.; Gold, R.E.; Kaufman, R.A. Distinguishing Osteomyelitis From Ewing Sarcoma on Radiography and MRI. AJR Am. J. Roentgenol. 2015, 205, 640–650. [Google Scholar] [CrossRef]
- Shimose, S.; Sugita, T.; Kubo, T.; Matsuo, T.; Nobuto, H.; Ochi, M. Differential diagnosis between osteomyelitis and bone tumors. Acta Radiol. 2008, 49, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Oudjhane, K.; Azouz, E.M. Imaging of osteomyelitis in children. Radiol Clin. N. Am. 2001, 39, 251–266. [Google Scholar] [CrossRef]
- Mankin, H.J.; Mankin, C.J.; Simon, M.A. The hazards of the biopsy, revisited. Members of the Musculoskeletal Tumor Society. J. Bone Joint. Surg. Am. 1996, 78, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Jalgaonkar, A.; Dawson-Bowling, S.J.; Mohan, A.T.; Spiegelberg, B.; Saifuddin, A.; Pollock, R.; Skinner, J.A.; Briggs, T.W.; Aston, W. Identification of the biopsy track in musculoskeletal tumour surgery: A novel technique using India ink. Bone Joint. J. 2013, 95-B, 250–253. [Google Scholar] [CrossRef]
- Cannon, S.R.; Dyson, P.H.P. Relationship of the site of open biopsy of malignant bone tumours to local recurrence following resection and prosthetic replacement. J. Bone Joint. Surg. Br. 1987, 69, 492. [Google Scholar]
- Grohs, J.G.; Zoubek, A.; Jugovic, D.; Kovar, H.; Windhager, R. Intraoperative dissemination of tumour cells in patients with Ewing tumours detected by RT-PCR. Int. Orthop. 2004, 28, 222–225. [Google Scholar] [CrossRef][Green Version]
- Zoubek, A.; Kovar, H.; Kronberger, M.; Amann, G.; Windhager, R.; Gruber, B.; Gadner, H. Mobilization of tumour cells during biopsy in an infant with Ewing sarcoma. Eur. J. Pediatr. 1996, 155, 373–376. [Google Scholar] [CrossRef]
- Kiatisevi, P.; Thanakit, V.; Sukunthanak, B.; Boonthatip, M.; Bumrungchart, S.; Witoonchart, K. Computed tomography-guided core needle biopsy versus incisional biopsy in diagnosing musculoskeletal lesions. J. Orthop. Surg. 2013, 21, 204–208. [Google Scholar] [CrossRef]
- Berger-Richardson, D.; Swallow, C.J. Needle tract seeding after percutaneous biopsy of sarcoma: Risk/benefit considerations. Cancer 2017, 123, 560–567. [Google Scholar] [CrossRef]
- Mankin, H.J.; Lange, T.A.; Spanier, S.S. The hazards of biopsy in patients with malignant primary bone and soft-tissue tumors. J. Bone Joint. Surg. Am. 1982, 64, 1121–1127. [Google Scholar] [CrossRef]
- Liu, P.T.; Valadez, S.D.; Chivers, F.S.; Roberts, C.C.; Beauchamp, C.P. Anatomically based guidelines for core needle biopsy of bone tumors: Implications for limb-sparing surgery. Radiographics 2007, 27, 189–205. [Google Scholar] [CrossRef]
- Saghieh, S.; Masrouha, K.Z.; Musallam, K.M.; Mahfouz, R.; Abboud, M.; Khoury, N.J.; Haidar, R. The risk of local recurrence along the core-needle biopsy tract in patients with bone sarcomas. Iowa Orthop. J. 2010, 30, 80–83. [Google Scholar]
- Oliveira, M.P.; Lima, P.M.; da Silva, H.J.; de Mello, R.J. Neoplasm seeding in biopsy tract of the musculoskeletal system. A systematic review. Acta Ortop. Bras. 2014, 22, 106–110. [Google Scholar] [CrossRef]
- Barrientos-Ruiz, I.; Ortiz-Cruz, E.J.; Serrano-Montilla, J.; Bernabeu-Taboada, D.; Pozo-Kreilinger, J.J. Are Biopsy Tracts a Concern for Seeding and Local Recurrence in Sarcomas? Clin. Orthop. Relat. Res. 2017, 475, 511–518. [Google Scholar] [CrossRef]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients-The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef]
- Casali, P.G.; Bielack, S.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brennan, B.; et al. Bone sarcomas: ESMO-PaedCan-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv79–iv95. [Google Scholar] [CrossRef] [PubMed]
- Marcilla, D.; Machado, I.; Grunewald, T.G.P.; Llombart-Bosch, A.; de Alava, E. (Immuno)histological Analysis of Ewing Sarcoma. Methods Mol. Biol. 2021, 2226, 49–64. [Google Scholar] [CrossRef]
- Nascimento, A.G.; Unii, K.K.; Pritchard, D.J.; Cooper, K.L.; Dahlin, D.C. A clinicopathologic study of 20 cases of large-cell (atypical) Ewing’s sarcoma of bone. Am. J. Surg. Pathol. 1980, 4, 29–36. [Google Scholar] [CrossRef]
- Ambros, I.M.; Ambros, P.F.; Strehl, S.; Kovar, H.; Gadner, H.; Salzer-Kuntschik, M. MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing’s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer 1991, 67, 1886–1893. [Google Scholar] [CrossRef]
- Baldauf, M.C.; Orth, M.F.; Dallmayer, M.; Marchetto, A.; Gerke, J.S.; Rubio, R.A.; Kiran, M.M.; Musa, J.; Knott, M.M.L.; Ohmura, S.; et al. Robust diagnosis of Ewing sarcoma by immunohistochemical detection of super-enhancer-driven EWSR1-ETS targets. Oncotarget 2018, 9, 1587–1601. [Google Scholar] [CrossRef]
- Hornick, J.L. Novel uses of immunohistochemistry in the diagnosis and classification of soft tissue tumors. Mod. Pathol. 2014, 27 (Suppl. 1), S47–S63. [Google Scholar] [CrossRef]
- Llombart-Bosch, A.; Machado, I.; Navarro, S.; Bertoni, F.; Bacchini, P.; Alberghini, M.; Karzeladze, A.; Savelov, N.; Petrov, S.; Alvarado-Cabrero, I.; et al. Histological heterogeneity of Ewing’s sarcoma/PNET: An immunohistochemical analysis of 415 genetically confirmed cases with clinical support. Virchows Arch. 2009, 455, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Machado, I.; Yoshida, A.; Morales, M.G.N.; Abrahao-Machado, L.F.; Navarro, S.; Cruz, J.; Lavernia, J.; Parafioriti, A.; Picci, P.; Llombart-Bosch, A. Review with novel markers facilitates precise categorization of 41 cases of diagnostically challenging, “undifferentiated small round cell tumors”. A clinicopathologic, immunophenotypic and molecular analysis. Ann. Diagn. Pathol. 2018, 34, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Sorensen, P.H.; Liu, X.F.; Delattre, O.; Rowland, J.M.; Biggs, C.A.; Thomas, G.; Triche, T.J. Reverse transcriptase PCR amplification of EWS/FLI-1 fusion transcripts as a diagnostic test for peripheral primitive neuroectodermal tumors of childhood. Diagn. Mol. Pathol. 1993, 2, 147–157. [Google Scholar] [CrossRef]
- Huang, S.C.; Zhang, L.; Sung, Y.S.; Chen, C.L.; Kao, Y.C.; Agaram, N.P.; Antonescu, C.R. Secondary EWSR1 gene abnormalities in SMARCB1-deficient tumors with 22q11-12 regional deletions: Potential pitfalls in interpreting EWSR1 FISH results. Genes Chromosomes Cancer 2016, 55, 767–776. [Google Scholar] [CrossRef]
- Chen, S.; Deniz, K.; Sung, Y.S.; Zhang, L.; Dry, S.; Antonescu, C.R. Ewing sarcoma with ERG gene rearrangements: A molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 2016, 55, 340–349. [Google Scholar] [CrossRef]
- Italiano, A.; Di Mauro, I.; Rapp, J.; Pierron, G.; Auger, N.; Alberti, L.; Chibon, F.; Escande, F.; Voegeli, A.C.; Ghnassia, J.P.; et al. Clinical effect of molecular methods in sarcoma diagnosis (GENSARC): A prospective, multicentre, observational study. Lancet Oncol. 2016, 17, 532–538. [Google Scholar] [CrossRef]
- Askin, F.B.; Rosai, J.; Sibley, R.K.; Dehner, L.P.; McAlister, W.H. Malignant small cell tumor of the thoracopulmonary region in childhood: A distinctive clinicopathologic entity of uncertain histogenesis. Cancer 1979, 43, 2438–2451. [Google Scholar] [CrossRef]
- Schmidt, D.; Herrmann, C.; Jurgens, H.; Harms, D. Malignant peripheral neuroectodermal tumor and its necessary distinction from Ewing’s sarcoma. A report from the Kiel Pediatric Tumor Registry. Cancer 1991, 68, 2251–2259. [Google Scholar] [CrossRef]
- Tsokos, M. Peripheral primitive neuroectodermal tumors. Diagnosis, classification, and prognosis. Perspect Pediatr. Pathol. 1992, 16, 27–98. [Google Scholar]
- Sankar, S.; Lessnick, S.L. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011, 204, 351–365. [Google Scholar] [CrossRef]
- Watson, S.; Perrin, V.; Guillemot, D.; Reynaud, S.; Coindre, J.M.; Karanian, M.; Guinebretiere, J.M.; Freneaux, P.; Le Loarer, F.; Bouvet, M.; et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J. Pathol. 2018, 245, 29–40. [Google Scholar] [CrossRef]
- Doyle, L.A.; Wong, K.K.; Bueno, R.; Dal Cin, P.; Fletcher, J.A.; Sholl, L.M.; Kuo, F. Ewing sarcoma mimicking atypical carcinoid tumor: Detection of unexpected genomic alterations demonstrates the use of next generation sequencing as a diagnostic tool. Cancer Genet. 2014, 207, 335–339. [Google Scholar] [CrossRef]
- Jo, V.Y.; Doyle, L.A. Refinements in Sarcoma Classification in the Current 2013 World Health Organization Classification of Tumours of Soft Tissue and Bone. Surg. Oncol. Clin. N. Am. 2016, 25, 621–643. [Google Scholar] [CrossRef]
- Koelsche, C.; Kriegsmann, M.; Kommoss, F.K.F.; Stichel, D.; Kriegsmann, K.; Vokuhl, C.; Grunewald, T.G.P.; Romero-Perez, L.; Kirchner, T.; de Alava, E.; et al. DNA methylation profiling distinguishes Ewing-like sarcoma with EWSR1-NFATc2 fusion from Ewing sarcoma. J. Cancer Res. Clin. Oncol. 2019, 145, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Owosho, A.A.; Zhang, L.; Chen, S.; Deniz, K.; Huryn, J.M.; Kao, Y.C.; Huang, S.C.; Singer, S.; Tap, W.; et al. Sarcomas With CIC-rearrangements Are a Distinct Pathologic Entity With Aggressive Outcome: A Clinicopathologic and Molecular Study of 115 Cases. Am. J. Surg. Pathol. 2017, 41, 941–949. [Google Scholar] [CrossRef]
- Gambarotti, M.; Benini, S.; Gamberi, G.; Cocchi, S.; Palmerini, E.; Sbaraglia, M.; Donati, D.; Picci, P.; Vanel, D.; Ferrari, S.; et al. CIC-DUX4 fusion-positive round-cell sarcomas of soft tissue and bone: A single-institution morphological and molecular analysis of seven cases. Histopathology 2016, 69, 624–634. [Google Scholar] [CrossRef]
- Le Guellec, S.; Velasco, V.; Perot, G.; Watson, S.; Tirode, F.; Coindre, J.M. ETV4 is a useful marker for the diagnosis of CIC-rearranged undifferentiated round-cell sarcomas: A study of 127 cases including mimicking lesions. Mod. Pathol. 2016, 29, 1523–1531. [Google Scholar] [CrossRef]
- Kao, Y.C.; Owosho, A.A.; Sung, Y.S.; Zhang, L.; Fujisawa, Y.; Lee, J.C.; Wexler, L.; Argani, P.; Swanson, D.; Dickson, B.C.; et al. BCOR-CCNB3 Fusion Positive Sarcomas: A Clinicopathologic and Molecular Analysis of 36 Cases With Comparison to Morphologic Spectrum and Clinical Behavior of Other Round Cell Sarcomas. Am. J. Surg. Pathol. 2018, 42, 604–615. [Google Scholar] [CrossRef]
- Pierron, G.; Tirode, F.; Lucchesi, C.; Reynaud, S.; Ballet, S.; Cohen-Gogo, S.; Perrin, V.; Coindre, J.M.; Delattre, O. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat. Genet. 2012, 44, 461–466. [Google Scholar] [CrossRef]
- Szuhai, K.; Ijszenga, M.; de Jong, D.; Karseladze, A.; Tanke, H.J.; Hogendoorn, P.C. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin. Cancer Res. 2009, 15, 2259–2268. [Google Scholar] [CrossRef]
- Renzi, S.; Anderson, N.D.; Light, N.; Gupta, A. Ewing-like sarcoma: An emerging family of round cell sarcomas. J. Cell Physiol. 2019, 234, 7999–8007. [Google Scholar] [CrossRef]
- Bridge, J.A.; Sumegi, J.; Druta, M.; Bui, M.M.; Henderson-Jackson, E.; Linos, K.; Baker, M.; Walko, C.M.; Millis, S.; Brohl, A.S. Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod. Pathol. 2019, 32, 1593–1604. [Google Scholar] [CrossRef]
- Sumegi, J.; Nishio, J.; Nelson, M.; Frayer, R.W.; Perry, D.; Bridge, J.A. A novel t(4;22)(q31;q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod. Pathol. 2011, 24, 333–342. [Google Scholar] [CrossRef]
- Wang, L.; Bhargava, R.; Zheng, T.; Wexler, L.; Collins, M.H.; Roulston, D.; Ladanyi, M. Undifferentiated small round cell sarcomas with rare EWS gene fusions: Identification of a novel EWS-SP3 fusion and of additional cases with the EWS-ETV1 and EWS-FEV fusions. J. Mol. Diagn. 2007, 9, 498–509. [Google Scholar] [CrossRef]
- Siegfried, A.; Rousseau, A.; Maurage, C.A.; Pericart, S.; Nicaise, Y.; Escudie, F.; Grand, D.; Delrieu, A.; Gomez-Brouchet, A.; Le Guellec, S.; et al. EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol. 2019, 29, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Kreyer, J.; Ranft, A.; Timmermann, B.; Juergens, H.; Jung, S.; Wiebe, K.; Boelling, T.; Schuck, A.; Vieth, V.; Streitbuerger, A.; et al. Impact of the Interdisciplinary Tumor Board of the Cooperative Ewing Sarcoma Study Group on local therapy and overall survival of Ewing sarcoma patients after induction therapy. Pediatr. Blood Cancer 2018, 65, e27384. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.S.; Torrey, M.; Link, M.P.; Glicksman, A.; Gilula, L.; Laurie, F.; Manning, J.; Neff, J.; Reinus, W.; Thompson, E.; et al. A multidisciplinary study investigating radiotherapy in Ewing’s sarcoma: End results of POG #8346. Pediatric Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, M.; Ahrens, S.; Dunst, J.; Winkelmann, W.; Exner, G.U.; Kotz, R.; Amann, G.; Dockhorn-Dworniczak, B.; Harms, D.; Muller-Weihrich, S.; et al. Localized Ewing tumor of bone: Final results of the cooperative Ewing’s Sarcoma Study CESS 86. J. Clin. Oncol. 2001, 19, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Forni, C.; Longhi, A.; Ferrari, S.; Donati, D.; De Paolis, M.; Barbieri, E.; Pignotti, E.; Rosito, P.; Versari, M. Long-term outcome for patients with non-metastatic Ewing’s sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur. J. Cancer 2004, 40, 73–83. [Google Scholar] [CrossRef]
- Craft, A.W.; Cotterill, S.J.; Bullimore, J.A.; Pearson, D. Long-term results from the first UKCCSG Ewing’s Tumour Study (ET-1). United Kingdom Children’s Cancer Study Group (UKCCSG) and the Medical Research Council Bone Sarcoma Working Party. Eur. J. Cancer 1997, 33, 1061–1069. [Google Scholar] [CrossRef]
- DuBois, S.G.; Krailo, M.D.; Gebhardt, M.C.; Donaldson, S.S.; Marcus, K.J.; Dormans, J.; Shamberger, R.C.; Sailer, S.; Nicholas, R.W.; Healey, J.H.; et al. Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: A report from the Children’s Oncology Group. Cancer 2015, 121, 467–475. [Google Scholar] [CrossRef]
- Granowetter, L.; Womer, R.; Devidas, M.; Krailo, M.; Wang, C.; Bernstein, M.; Marina, N.; Leavey, P.; Gebhardt, M.; Healey, J.; et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2009, 27, 2536–2541. [Google Scholar] [CrossRef]
- Krasin, M.J.; Rodriguez-Galindo, C.; Davidoff, A.M.; Billups, C.A.; Fuller, C.E.; Neel, M.D.; Kun, L.E.; Merchant, T.E. Efficacy of combined surgery and irradiation for localized Ewings sarcoma family of tumors. Pediatr. Blood Cancer 2004, 43, 229–236. [Google Scholar] [CrossRef]
- Schuck, A.; Ahrens, S.; Paulussen, M.; Kuhlen, M.; Konemann, S.; Rube, C.; Winkelmann, W.; Kotz, R.; Dunst, J.; Willich, N.; et al. Local therapy in localized Ewing tumors: Results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int. J. Radiat. Oncol. Biol. Phys. 2003, 55, 168–177. [Google Scholar] [CrossRef]
- Schuck, A.; Rube, C.; Konemann, S.; Rube, C.E.; Ahrens, S.; Paulussen, M.; Dunst, J.; Jurgens, H.; Willich, N. Postoperative radiotherapy in the treatment of Ewing tumors: Influence of the interval between surgery and radiotherapy. Strahlenther. Onkol. 2002, 178, 25–31. [Google Scholar] [CrossRef]
- Dunst, J.; Ahrens, S.; Paulussen, M.; Rube, C.; Winkelmann, W.; Zoubek, A.; Harms, D.; Jurgens, H. Second malignancies after treatment for Ewing’s sarcoma: A report of the CESS-studies. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 379–384. [Google Scholar] [CrossRef]
- Hardes, J.; von Eiff, C.; Streitbuerger, A.; Balke, M.; Budny, T.; Henrichs, M.P.; Hauschild, G.; Ahrens, H. Reduction of periprosthetic infection with silver-coated megaprostheses in patients with bone sarcoma. J. Surg. Oncol. 2010, 101, 389–395. [Google Scholar] [CrossRef]
- Zahlten-Hinguranage, A.; Bernd, L.; Ewerbeck, V.; Sabo, D. Equal quality of life after limb-sparing or ablative surgery for lower extremity sarcomas. Br. J. Cancer 2004, 91, 1012–1014. [Google Scholar] [CrossRef]
- Gupta, A.A.; Pappo, A.; Saunders, N.; Hopyan, S.; Ferguson, P.; Wunder, J.; O’Sullivan, B.; Catton, C.; Greenberg, M.; Blackstein, M. Clinical outcome of children and adults with localized Ewing sarcoma: Impact of chemotherapy dose and timing of local therapy. Cancer 2010, 116, 3189–3194. [Google Scholar] [CrossRef]
- Lin, T.A.; Ludmir, E.B.; Liao, K.P.; McAleer, M.F.; Grosshans, D.R.; McGovern, S.L.; Bishop, A.J.; Woodhouse, K.D.; Paulino, A.C.; Yeboa, D.N. Timing of Local Therapy Affects Survival in Ewing Sarcoma. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 127–136. [Google Scholar] [CrossRef]
- Enneking, W. Musculoskeletal Tumour Surgery; Churchill Livingstone: Edinburgh, UK, 1983. [Google Scholar]
- Gilg, M.M.; Gaston, C.L.; Parry, M.C.; Jeys, L.; Abudu, A.; Tillman, R.M.; Carter, S.R.; Grimer, R.J. What is the morbidity of a non-invasive growing prosthesis? Bone Joint. J. 2016, 98-B, 1697–1703. [Google Scholar] [CrossRef]
- Jeys, L.M.; Grimer, R.J.; Carter, S.R.; Tillman, R.M. Risk of amputation following limb salvage surgery with endoprosthetic replacement, in a consecutive series of 1261 patients. Int. Orthop. 2003, 27, 160–163. [Google Scholar] [CrossRef]
- Jeys, L.M.; Luscombe, J.S.; Grimer, R.J.; Abudu, A.; Tillman, R.M.; Carter, S.R. The risks and benefits of radiotherapy with massive endoprosthetic replacement. J. Bone Joint. Surg. Br. 2007, 89, 1352–1355. [Google Scholar] [CrossRef]
- Nagarajan, R.; Clohisy, D.R.; Neglia, J.P.; Yasui, Y.; Mitby, P.A.; Sklar, C.; Finklestein, J.Z.; Greenberg, M.; Reaman, G.H.; Zeltzer, L.; et al. Function and quality-of-life of survivors of pelvic and lower extremity osteosarcoma and Ewing’s sarcoma: The Childhood Cancer Survivor Study. Br. J. Cancer 2004, 91, 1858–1865. [Google Scholar] [CrossRef]
- Hardes, J.; Gosheger, G.; Vachtsevanos, L.; Hoffmann, C.; Ahrens, H.; Winkelmann, W. Rotationplasty type BI versus type BIIIa in children under the age of ten years. Should the knee be preserved? J. Bone Joint. Surg. Br. 2005, 87, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Winkelmann, W.W. Rotationplasty. Orthop. Clin. N. Am. 1996, 27, 503–523. [Google Scholar] [CrossRef]
- Fernandez-Pineda, I.; Hudson, M.M.; Pappo, A.S.; Bishop, M.W.; Klosky, J.L.; Brinkman, T.M.; Srivastava, D.K.; Neel, M.D.; Rao, B.N.; Davidoff, A.M.; et al. Long-term functional outcomes and quality of life in adult survivors of childhood extremity sarcomas: A report from the St. Jude Lifetime Cohort Study. J. Cancer Surviv. 2017, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Hillmann, A.; Hoffmann, C.; Rube, C.; Blasius, S.; Dunst, J.; Jurgens, H.; Winkelmann, W. Significance of surgical margin on the prognosis of patients with Ewing’s sarcoma. A report from the Cooperative Ewing’s Sarcoma Study. Cancer 1996, 78, 892–900. [Google Scholar] [CrossRef]
- Sluga, M.; Windhager, R.; Lang, S.; Heinzl, H.; Krepler, P.; Mittermayer, F.; Dominkus, M.; Zoubek, A.; Kotz, R. The role of surgery and resection margins in the treatment of Ewing’s sarcoma. Clin. Orthop. Relat. Res. 2001, 394–399. [Google Scholar] [CrossRef]
- Durr, H.R.; Bakhshai, Y.; Rechl, H.; Tunn, P.U. Resection margins in bone tumors: What is adequate? Unfallchirurg 2014, 117, 593–599. [Google Scholar] [CrossRef]
- Gomez-Brouchet, A.; Mascard, E.; Siegfried, A.; de Pinieux, G.; Gaspar, N.; Bouvier, C.; Aubert, S.; Marec-Berard, P.; Piperno-Neumann, S.; Marie, B.; et al. Assessment of resection margins in bone sarcoma treated by neoadjuvant chemotherapy: Literature review and guidelines of the bone group (GROUPOS) of the French sarcoma group and bone tumor study group (GSF-GETO/RESOS). Orthop. Traumatol. Surg. Res. 2019, 105, 773–780. [Google Scholar] [CrossRef]
- Lin, P.P.; Jaffe, N.; Herzog, C.E.; Costelloe, C.M.; Deavers, M.T.; Kelly, J.S.; Patel, S.R.; Madewell, J.E.; Lewis, V.O.; Cannon, C.P.; et al. Chemotherapy response is an important predictor of local recurrence in Ewing sarcoma. Cancer 2007, 109, 603–611. [Google Scholar] [CrossRef]
- Andreou, D.; Ranft, A.; Gosheger, G.; Timmermann, B.; Ladenstein, R.; Hartmann, W.; Bauer, S.; Baumhoer, D.; van den Berg, H.; Dijkstra, P.D.S.; et al. Which Factors Are Associated with Local Control and Survival of Patients with Localized Pelvic Ewing’s Sarcoma? A Retrospective Analysis of Data from the Euro-EWING99 Trial. Clin. Orthop. Relat. Res. 2020, 478, 290–302. [Google Scholar] [CrossRef]
- Foulon, S.; Brennan, B.; Gaspar, N.; Dirksen, U.; Jeys, L.; Cassoni, A.; Claude, L.; Seddon, B.; Marec-Berard, P.; Whelan, J.; et al. Can postoperative radiotherapy be omitted in localised standard-risk Ewing sarcoma? An observational study of the Euro-E.W.I.N.G group. Eur. J. Cancer 2016, 61, 128–136. [Google Scholar] [CrossRef]
- Bosma, S.E.; Rueten-Budde, A.J.; Lancia, C.; Ranft, A.; Dirksen, U.; Krol, A.D.; Gelderblom, H.; van de Sande, M.A.J.; Dijkstra, P.D.S.; Fiocco, M. Individual risk evaluation for local recurrence and distant metastasis in Ewing sarcoma: A multistate model: A multistate model for Ewing sarcoma. Pediatr. Blood Cancer 2019, 66, e27943. [Google Scholar] [CrossRef]
- Schultheiss, M.; Traub, S.E.; v Baer, A. Pathological fractures due to malignant bone tumors. Unfallchirurg 2014, 117, 583–592. [Google Scholar] [CrossRef]
- Hoffmann, C.; Jabar, S.; Ahrens, S.; Rodl, R.; Rube, C.; Winkelmann, W.; Dunst, J.; Jurgens, H. Prognosis in Ewing sarcoma patients with initial pathological fractures of the primary tumor site. Klin. Padiatr. 1995, 207, 151–157. [Google Scholar] [CrossRef]
- Wagner, L.M.; Neel, M.D.; Pappo, A.S.; Merchant, T.E.; Poquette, C.A.; Rao, B.N.; Rodriguez-Galindo, C. Fractures in pediatric Ewing sarcoma. J. Pediatr. Hematol. Oncol. 2001, 23, 568–571. [Google Scholar] [CrossRef]
- Schlegel, M.; Zeumer, M.; Prodinger, P.M.; Woertler, K.; Steinborn, M.; von Eisenhart-Rothe, R.; Burdach, S.; Rechl, H.; von Luettichau, I. Impact of Pathological Fractures on the Prognosis of Primary Malignant Bone Sarcoma in Children and Adults: A Single-Center Retrospective Study of 205 Patients. Oncology 2018, 94, 354–362. [Google Scholar] [CrossRef]
- Hardes, J.; Gosheger, G.; Budny, T. Knochensarkome. Z Orthop. Unfall 2018, 156, 105–124. [Google Scholar] [CrossRef]
- Bacci, G.; Picci, P.; Gherlinzoni, F.; Capanna, R.; Calderoni, P.; Putti, C.; Mancini, A.; Campanacci, M. Localized Ewing’s sarcoma of bone: Ten years’ experience at the Istituto Ortopedico Rizzoli in 124 cases treated with multimodal therapy. Eur. J. Cancer Clin. Oncol. 1985, 21, 163–173. [Google Scholar] [CrossRef]
- Sinkovics, J.G.; Plager, C.; Ayala, A.G.; Lindberg, R.D.; Samuels, M.L. Ewing sarcoma: Its course and treatment in 50 adult patients. Oncology 1980, 37, 114–119. [Google Scholar] [CrossRef]
- Hesla, A.C.; Tsagozis, P.; Jebsen, N.; Zaikova, O.; Bauer, H.; Brosjo, O. Improved Prognosis for Patients with Ewing Sarcoma in the Sacrum Compared with the Innominate Bones: The Scandinavian Sarcoma Group Experience. J. Bone Joint. Surg. Am. 2016, 98, 199–210. [Google Scholar] [CrossRef]
- Hoffmann, C.; Ahrens, S.; Dunst, J.; Hillmann, A.; Winkelmann, W.; Craft, A.; Gobel, U.; Rube, C.; Voute, P.A.; Harms, D.; et al. Pelvic Ewing sarcoma: A retrospective analysis of 241 cases. Cancer 1999, 85, 869–877. [Google Scholar] [CrossRef]
- Guder, W.K.; Hardes, J.; Nottrott, M.; Steffen, A.J.; Dirksen, U.; Streitbürger, A. Pelvic Ewing sarcoma: A retrospective outcome analysis of 104 patients who underwent pelvic tumor resection at a single supra-regional center. J. Orthop. Surg. Res. 2020, 15, 534. [Google Scholar] [CrossRef]
- Uezono, H.; Indelicato, D.J.; Rotondo, R.L.; Mailhot Vega, R.B.; Bradfield, S.M.; Morris, C.G.; Bradley, J.A. Treatment Outcomes After Proton Therapy for Ewing Sarcoma of the Pelvis. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 974–981. [Google Scholar] [CrossRef]
- Paulussen, M.; Ahrens, S.; Braun-Munzinger, G.; Craft, A.W.; Dockhorn-Dworniczak, B.; Dorffel, W.; Dunst, J.; Frohlich, B.; Gobel, U.; Haussler, M.; et al. EICESS 92 (European Intergroup Cooperative Ewing’s Sarcoma Study)—Preliminary results. Klin. Padiatr. 1999, 211, 276–283. [Google Scholar] [CrossRef]
- Rodl, R.W.; Hoffmann, C.; Gosheger, G.; Leidinger, B.; Jurgens, H.; Winkelmann, W. Ewing’s sarcoma of the pelvis: Combined surgery and radiotherapy treatment. J. Surg. Oncol. 2003, 83, 154–160. [Google Scholar] [CrossRef]
- Gebert, C.; Gosheger, G.; Winkelmann, W. Hip transposition as a universal surgical procedure for periacetabular tumors of the pelvis. J. Surg. Oncol. 2009, 99, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Gebert, C.; Wessling, M.; Hoffmann, C.; Roedl, R.; Winkelmann, W.; Gosheger, G.; Hardes, J. Hip transposition as a limb salvage procedure following the resection of periacetabular tumors. J. Surg. Oncol. 2011, 103, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Lex, J.R.; Stevenson, J.D.; Tsuda, Y.; Clark, R.; Parry, M.C.; Grimer, R.J.; Jeys, L.M. Surgical treatment for pelvic Ewing sarcoma: What is a safe and functional acetabular reconstruction when combined with modern multidisciplinary treatments? J. Surg. Oncol. 2019, 120, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Shamberger, R.C.; Grier, H.E. Ewing’s sarcoma/primitive neuroectodermal tumor of the chest wall. Semin Pediatr. Surg. 2001, 10, 153–160. [Google Scholar] [CrossRef]
- Silver, J.M.; Losken, A.; Young, A.N.; Mansour, K.A. Ewing’s sarcoma presenting as a posterior mediastinal mass: A lesson learned. Ann. Thorac. Surg. 1999, 67, 845–847. [Google Scholar] [CrossRef]
- Sabanathan, S.; Salama, F.D.; Morgan, W.E.; Harvey, J.A. Primary chest wall tumors. Ann. Thorac. Surg. 1985, 39, 4–15. [Google Scholar] [CrossRef]
- Saenz, N.C.; Hass, D.J.; Meyers, P.; Wollner, N.; Gollamudi, S.; Bains, M.; LaQuaglia, M.P. Pediatric chest wall Ewing’s sarcoma. J. Pediatr. Surg. 2000, 35, 550–555. [Google Scholar] [CrossRef]
- Hayes, F.A.; Thompson, E.I.; Meyer, W.H.; Kun, L.; Parham, D.; Rao, B.; Kumar, M.; Hancock, M.; Parvey, L.; Magill, L.; et al. Therapy for localized Ewing’s sarcoma of bone. J. Clin. Oncol. 1989, 7, 208–213. [Google Scholar] [CrossRef]
- Meys, K.M.; Heinen, R.C.; van den Berg, H.; Aronson, D.C. Recurrence of Ewing sarcomas of the chest wall. Pediatr. Blood Cancer 2008, 51, 765–767. [Google Scholar] [CrossRef]
- Rao, B.N.; Hayes, F.A.; Thompson, E.I.; Kumar, A.P.; Fleming, I.D.; Green, A.A.; Austin, B.A.; Pate, J.W.; Hustu, H.O. Chest wall resection for Ewing’s sarcoma of the rib: An unnecessary procedure. 1988. Updated in 1995. Ann. Thorac. Surg. 1995, 60, 1454–1455. [Google Scholar] [CrossRef]
- Bedetti, B.; Wiebe, K.; Ranft, A.; Aebert, H.; Schmidt, J.; Jurgens, H.; Dirksen, U. Local control in Ewing sarcoma of the chest wall: Results of the EURO-EWING 99 trial. Ann. Surg. Oncol. 2015, 22, 2853–2859. [Google Scholar] [CrossRef] [PubMed]
- Collaud, S.; Stork, T.; Dirksen, U.; Pottgen, C.; Hegedus, B.; Schildhaus, H.U.; Bauer, S.; Aigner, C. Surgical Treatment for Primary Chest Wall Sarcoma: A Single-Institution Study. J. Surg. Res. 2020, 260, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Saltsman, J.A.; Danzer, E.; Hammond, W.J.; Rhee, D.; Berhe, S.; Monteagudo, J.; Price, A.P.; Heaton, T.E.; Jones, D.R.; LaQuaglia, M.P. Survival and Scoliosis Following Resection of Chest Wall Tumors in Children and Adolescents: A Single-center Retrospective Analysis. Ann. Surg. 2019. [Google Scholar] [CrossRef] [PubMed]
- Scalabre, A.; Parot, R.; Hameury, F.; Cunin, V.; Jouve, J.L.; Chotel, F. Prognostic risk factors for the development of scoliosis after chest wall resection for malignant tumors in children. J. Bone Joint. Surg. Am. 2014, 96, e10. [Google Scholar] [CrossRef]
- Collaud, S.; Stork, T.; Schildhaus, H.U.; Pottgen, C.; Plones, T.; Valdivia, D.; Zaatar, M.; Dirksen, U.; Bauer, S.; Aigner, C. Multimodality treatment including surgery for primary pulmonary sarcoma: Size does matter. J. Surg. Oncol. 2020, 122, 506–514. [Google Scholar] [CrossRef]
- Halliday, J.; Soon, S.Y.; Monaghan, H.; Walker, W.S.; Zamvar, V. Extraskeletal Ewing’s sarcoma presenting as a mediastinal mass. Ann. Thorac. Surg. 2010, 90, 1016–1017. [Google Scholar] [CrossRef]
- Shamberger, R.C.; LaQuaglia, M.P.; Gebhardt, M.C.; Neff, J.R.; Tarbell, N.J.; Marcus, K.C.; Sailer, S.L.; Womer, R.B.; Miser, J.S.; Dickman, P.S.; et al. Ewing sarcoma/primitive neuroectodermal tumor of the chest wall: Impact of initial versus delayed resection on tumor margins, survival, and use of radiation therapy. Ann. Surg. 2003, 238, 563–567. [Google Scholar] [CrossRef]
- Brasme, J.F.; Chalumeau, M.; Oberlin, O.; Valteau-Couanet, D.; Gaspar, N. Time to diagnosis of Ewing tumors in children and adolescents is not associated with metastasis or survival: A prospective multicenter study of 436 patients. J. Clin. Oncol. 2014, 32, 1935–1940. [Google Scholar] [CrossRef]
- Xue, R.; Lewis, V.O.; Moon, B.S.; Lin, P.P. Local recurrence of Ewing sarcoma: Is wide excision an acceptable treatment? J. Surg. Oncol. 2019, 120, 746–752. [Google Scholar] [CrossRef]
- Rombi, B.; Ares, C.; Hug, E.B.; Schneider, R.; Goitein, G.; Staab, A.; Albertini, F.; Bolsi, A.; Lomax, A.J.; Timmermann, B. Spot-scanning proton radiation therapy for pediatric chordoma and chondrosarcoma: Clinical outcome of 26 patients treated at paul scherrer institute. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 578–584. [Google Scholar] [CrossRef]
- Bacci, G.; Briccoli, A.; Picci, P.; Ferrari, S. Metachronous pulmonary metastases resection in patients with Ewing’s sarcoma initially treated with adjuvant or neoadjuvant chemotherapy. Eur. J. Cancer 1995, 31A, 999–1001. [Google Scholar] [CrossRef]
- Briccoli, A.; Rocca, M.; Ferrari, S.; Mercuri, M.; Ferrari, C.; Bacci, G. Surgery for lung metastases in Ewing’s sarcoma of bone. Eur. J. Surg. Oncol. 2004, 30, 63–67. [Google Scholar] [CrossRef]
- Lanza, L.A.; Miser, J.S.; Pass, H.I.; Roth, J.A. The role of resection in the treatment of pulmonary metastases from Ewing’s sarcoma. J. Thorac. Cardiovasc. Surg. 1987, 94, 181–187. [Google Scholar] [CrossRef]
- Ewing, J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA A Cancer J. Clin. 1972, 22, 95–98. [Google Scholar] [CrossRef]
- Rao, A.; Chen, Q.; Ermoian, R.; Alcorn, S.; Figueiredo, M.; Chen, M.; Dieckmann, K.; Macdonald, S.; Ladra, M.; Kobyzeva, D.; et al. Practice patterns of palliative radiation therapy in pediatric oncology patients in an international pediatric research consortium: Rao et al. Pediatric Blood Cancer 2017, 64, e26589. [Google Scholar] [CrossRef]
- Oberlin, O.; Deley, M.C.; Bui, B.N.; Gentet, J.C.; Philip, T.; Terrier, P.; Carrie, C.; Mechinaud, F.; Schmitt, C.; Babin-Boillettot, A.; et al. Prognostic factors in localized Ewing’s tumours and peripheral neuroectodermal tumours: The third study of the French Society of Paediatric Oncology (EW88 study). Br. J. Cancer 2001, 85, 1646–1654. [Google Scholar] [CrossRef]
- Talleur, A.C.; Navid, F.; Spunt, S.L.; McCarville, M.B.; Wu, J.; Mao, S.; Davidoff, A.M.; Neel, M.D.; Krasin, M.J. Limited Margin Radiation Therapy for Children and Young Adults With Ewing Sarcoma Achieves High Rates of Local Tumor Control. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.; Timmermann, B. The Evolving Role of Proton Beam Therapy for Sarcomas. Clin. Oncol. (R Coll. Radiol.) 2017, 29, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Mounessi, F.S.; Lehrich, P.; Haverkamp, U.; Willich, N.; Bolling, T.; Eich, H.T. Pelvic Ewing sarcomas. Three-dimensional conformal vs. intensity-modulated radiotherapy. Strahlenther. Onkol. Organ. Dtsch. Rontgenges. 2013, 189, 308–314. [Google Scholar] [CrossRef]
- DeLaney, T.F.; Trofimov, A.V.; Engelsman, M.; Suit, H.D. Advanced-technology radiation therapy in the management of bone and soft tissue sarcomas. Cancer Control J. Moffitt Cancer Center 2005, 12, 27–35. [Google Scholar] [CrossRef]
- Keole, S.; Ashman, J.B.; Daniels, T.B. Proton therapy for sarcomas. Cancer J. 2014, 20, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Indelicato, D.J.; Mailhot, R.B.; Bradley, J.A. Impact of different treatment techniques for pediatric Ewing sarcoma of the chest wall: IMRT, 3DCPT, and IMPT with/without beam aperture. J. Appl. Clin. Med Phys./Am. Coll. Med Phys. 2020, 21, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Grevener, K.; Haveman, L.M.; Ranft, A.; van den Berg, H.; Jung, S.; Ladenstein, R.; Klco-Brosius, S.; Juergens, H.; Merks, J.H.; Dirksen, U. Management and Outcome of Ewing Sarcoma of the Head and Neck. Pediatr. Blood Cancer 2016, 63, 604–610. [Google Scholar] [CrossRef]
- Rombi, B.; DeLaney, T.F.; MacDonald, S.M.; Huang, M.S.; Ebb, D.H.; Liebsch, N.J.; Raskin, K.A.; Yeap, B.Y.; Marcus, K.J.; Tarbell, N.J.; et al. Proton radiotherapy for pediatric Ewing’s sarcoma: Initial clinical outcomes. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 1142–1148. [Google Scholar] [CrossRef]
- Ladra, M.M.; Szymonifka, J.D.; Mahajan, A.; Friedmann, A.M.; Yong Yeap, B.; Goebel, C.P.; MacDonald, S.M.; Grosshans, D.R.; Rodriguez-Galindo, C.; Marcus, K.J.; et al. Preliminary results of a phase II trial of proton radiotherapy for pediatric rhabdomyosarcoma. J. Clin. Oncol. 2014, 32, 3762–3770. [Google Scholar] [CrossRef]
- Rieber, J.G.; Kessel, K.A.; Witt, O.; Behnisch, W.; Kulozik, A.E.; Debus, J.; Combs, S.E. Treatment tolerance of particle therapy in pediatric patients. Acta Oncol. 2015, 54, 1049–1055. [Google Scholar] [CrossRef]
- Leroy, R.; Benahmed, N.; Hulstaert, F.; Van Damme, N.; De Ruysscher, D. Proton Therapy in Children: A Systematic Review of Clinical Effectiveness in 15 Pediatric Cancers. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 267–278. [Google Scholar] [CrossRef]
- Bolek, T.W.; Marcus, R.B., Jr.; Mendenhall, N.P.; Scarborough, M.T.; Graham-Pole, J. Local control and functional results after twice-daily radiotherapy for Ewing’s sarcoma of the extremities. Int. J. Radiat. Oncol. Biol. Phys. 1996, 35, 687–692. [Google Scholar] [CrossRef]
- Indelicato, D.J.; Keole, S.R.; Shahlaee, A.H.; Shi, W.; Morris, C.G.; Marcus, R.B., Jr. Definitive radiotherapy for ewing tumors of extremities and pelvis: Long-term disease control, limb function, and treatment toxicity. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 871–877. [Google Scholar] [CrossRef]
- Chuba, P.J. Radiation therapy strategies and clinical trials in pediatric Ewing’s sarcoma. J. Radiat. Oncol. 2013, 2, 149–158. [Google Scholar] [CrossRef]
- Bolling, T.; Braun-Munzinger, G.; Burdach, S.; Calaminus, G.; Craft, A.; Delattre, O.; Deley, M.C.; Dirksen, U.; Dockhorn-Dworniczak, B.; Dunst, J.; et al. Development of curative therapies for Ewing sarcomas by interdisciplinary cooperative groups in Europe. Klin. Padiatr. 2015, 227, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Longhi, A.; Briccoli, A.; Bertoni, F.; Versari, M.; Picci, P. The role of surgical margins in treatment of Ewing’s sarcoma family tumors: Experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.S. Ewing sarcoma: Radiation dose and target volume. Pediatr. Blood Cancer 2004, 42, 471–476. [Google Scholar] [CrossRef]
- Lopez, J.L.; Cabrera, P.; Ordonez, R.; Marquez, C.; Ramirez, G.L.; Praena-Fernandez, J.M.; Ortiz, M.J. Role of radiation therapy in the multidisciplinary management of Ewing’s Sarcoma of bone in pediatric patients: An effective treatment for local control. Rep. Pract. Oncol. Radiother. 2011, 16, 103–109. [Google Scholar] [CrossRef]
- Wortman, J.R.; Tirumani, S.H.; Jagannathan, J.P.; Rosenthal, M.H.; Shinagare, A.B.; Hornick, J.L.; Baldini, E.H.; Ramaiya, N.H. Radiation Therapy for Soft-Tissue Sarcomas: A Primer for Radiologists. Radiographics 2016, 36, 554–572. [Google Scholar] [CrossRef]
- Casey, D.L.; Wexler, L.H.; Meyers, P.A.; Magnan, H.; Chou, A.J.; Wolden, S.L. Radiation for bone metastases in Ewing sarcoma and rhabdomyosarcoma. Pediatr. Blood Cancer 2015, 62, 445–449. [Google Scholar] [CrossRef]
- Bolling, T.; Schuck, A.; Paulussen, M.; Dirksen, U.; Ranft, A.; Konemann, S.; Dunst, J.; Willich, N.; Jurgens, H. Whole lung irradiation in patients with exclusively pulmonary metastases of Ewing tumors. Toxicity analysis and treatment results of the EICESS-92 trial. Strahlenther. Onkol. 2008, 184, 193–197. [Google Scholar] [CrossRef]
- Scobioala, S.; Ranft, A.; Wolters, H.; Jabar, S.; Paulussen, M.; Timmermann, B.; Juergens, H.; Hassenpflug, W.; Klingebiel, T.; Elsayad, K.; et al. Impact of Whole Lung Irradiation on Survival Outcome in Patients With Lung Relapsed Ewing Sarcoma. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 584–592. [Google Scholar] [CrossRef]
- Bacci, G.; Ferrari, S.; Longhi, A.; Donati, D.; De Paolis, M.; Forni, C.; Versari, M.; Setola, E.; Briccoli, A.; Barbieri, E. Therapy and survival after recurrence of Ewing’s tumors: The Rizzoli experience in 195 patients treated with adjuvant and neoadjuvant chemotherapy from 1979 to 1997. Ann. Oncol. 2003, 14, 1654–1659. [Google Scholar] [CrossRef]
- Luksch, R.; Tienghi, A.; Hall, K.S.; Fagioli, F.; Picci, P.; Barbieri, E.; Gandola, L.; Eriksson, M.; Ruggieri, P.; Daolio, P.; et al. Primary metastatic Ewing’s family tumors: Results of the Italian Sarcoma Group and Scandinavian Sarcoma Group ISG/SSG IV Study including myeloablative chemotherapy and total-lung irradiation. Ann. Oncol. 2012, 23, 2970–2976. [Google Scholar] [CrossRef]
- Scobioala, S.; Eich, H.T. Risk stratification of pulmonary toxicities in the combination of whole lung irradiation and high-dose chemotherapy for Ewing sarcoma patients with lung metastases: A review. Strahlenther. Onkol. 2020, 196, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Bolling, T.; Dirksen, U.; Ranft, A.; Ernst, I.; Jurgens, H.; Willich, N. Radiation toxicity following busulfan/melphalan high-dose chemotherapy in the EURO-EWING-99-trial: Review of GPOH data. Strahlenther. Onkol. 2009, 185 (Suppl. 2), 21–22. [Google Scholar] [CrossRef]
- Seddon, B.M.; Cassoni, A.M.; Galloway, M.J.; Rees, J.H.; Whelan, J.S. Fatal radiation myelopathy after high-dose busulfan and melphalan chemotherapy and radiotherapy for Ewing’s sarcoma: A review of the literature and implications for practice. Clin. Oncol. (R Coll. Radiol.) 2005, 17, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Baeza, M.R.; Barkley, H.T., Jr.; Fernandez, C.H. Total-lung irradiation in the treatment of pulmonary metastases. Radiology 1975, 116, 151–154. [Google Scholar] [CrossRef]
- D’Angio, G.J.; Farber, S.; Maddock, C.L. Potentiation of x-ray effects by actinomycin D. Radiology 1959, 73, 175–177. [Google Scholar] [CrossRef]
- Dritschilo, A.; Piro, A.J.; Belli, J.A. Interaction between radiation and drug damage in mammalian cells. III. The effect of adriamycin and actinomycin-D on the repair of potentially lethal radiation damage. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1979, 35, 549–560. [Google Scholar] [CrossRef]
- Hill, S.A.; Travis, E.L.; Denekamp, J. Actinomycin D and radiation: Effects on mouse lung. Eur. J. Cancer Clin. Oncol. 1986, 22, 577–582. [Google Scholar] [CrossRef]
- Cohen, I.J.; Loven, D.; Schoenfeld, T.; Sandbank, J.; Kaplinsky, C.; Yaniv, Y.; Jaber, L.; Zaizov, R. Dactinomycin potentiation of radiation pneumonitis: A forgotten interaction. Pediatr. Hematol. Oncol. 1991, 8, 187–192. [Google Scholar] [CrossRef]
- Phillips, T.L.; Fu, K.K. Quantification of combined radiation therapy and chemotherapy effects on critical normal tissues. Cancer 1976, 37, 1186–1200. [Google Scholar] [CrossRef]
- Phillips, T.L.; Wharam, M.D.; Margolis, L.W. Modification of radiation injury to normal tissues by chemotherapeutic agents. Cancer 1975, 35, 1678–1684. [Google Scholar] [CrossRef]
- Koontz, B.F.; Clough, R.W.; Halperin, E.C. Palliative radiation therapy for metastatic Ewing sarcoma. Cancer 2006, 106, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.C.; Sutow, W.W.; Lindberg, R.D.; Samuels, M.L.; Murray, J.A.; Johnston, D.A. Management and results of localized Ewing’s sarcoma. Cancer 1979, 43, 1001–1006. [Google Scholar] [CrossRef]
- Rosen, G.; Caparros, B.; Nirenberg, A.; Marcove, R.C.; Huvos, A.G.; Kosloff, C.; Lane, J.; Murphy, M.L. Ewing’s sarcoma: Ten-year experience with adjuvant chemotherapy. Cancer 1981, 47, 2204–2213. [Google Scholar] [CrossRef]
- Zucker, J.M.; Henry-Amar, M.; Sarrazin, D.; Blache, R.; Patte, C.; Schweisguth, O. Intensive systemic chemotherapy in localized Ewing’s sarcoma in childhood. A historical trial. Cancer 1983, 52, 415–423. [Google Scholar] [CrossRef]
- Nesbit, M.E., Jr.; Gehan, E.A.; Burgert, E.O., Jr.; Vietti, T.J.; Cangir, A.; Tefft, M.; Evans, R.; Thomas, P.; Askin, F.B.; Kissane, J.M.; et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: A long-term follow-up of the First Intergroup study. J. Clin. Oncol. 1990, 8, 1664–1674. [Google Scholar] [CrossRef]
- Burgert, E.O., Jr.; Nesbit, M.E.; Garnsey, L.A.; Gehan, E.A.; Herrmann, J.; Vietti, T.J.; Cangir, A.; Tefft, M.; Evans, R.; Thomas, P.; et al. Multimodal therapy for the management of nonpelvic, localized Ewing’s sarcoma of bone: Intergroup study IESS-II. J. Clin. Oncol. 1990, 8, 1514–1524. [Google Scholar] [CrossRef]
- Evans, R.G.; Nesbit, M.E.; Gehan, E.A.; Garnsey, L.A.; Burgert, O., Jr.; Vietti, T.J.; Cangir, A.; Tefft, M.; Thomas, P.; Askin, F.B.; et al. Multimodal therapy for the management of localized Ewing’s sarcoma of pelvic and sacral bones: A report from the second intergroup study. J. Clin. Oncol. 1991, 9, 1173–1180. [Google Scholar] [CrossRef]
- Kung, F.H.; Pratt, C.B.; Vega, R.A.; Jaffe, N.; Strother, D.; Schwenn, M.; Nitschke, R.; Homans, A.C.; Holbrook, C.T.; Golembe, B.; et al. Ifosfamide/etoposide combination in the treatment of recurrent malignant solid tumors of childhood. A Pediatric Oncology Group Phase II study. Cancer 1993, 71, 1898–1903. [Google Scholar] [CrossRef]
- Magrath, I.; Sandlund, J.; Raynor, A.; Rosenberg, S.; Arasi, V.; Miser, J. A phase II study of ifosfamide in the treatment of recurrent sarcomas in young people. Cancer Chemother. Pharmacol. 1986, 18 (Suppl. 2), S25–S28. [Google Scholar] [CrossRef]
- Miser, J.S.; Kinsella, T.J.; Triche, T.J.; Tsokos, M.; Jarosinski, P.; Forquer, R.; Wesley, R.; Magrath, I. Ifosfamide with mesna uroprotection and etoposide: An effective regimen in the treatment of recurrent sarcomas and other tumors of children and young adults. J. Clin. Oncol. 1987, 5, 1191–1198. [Google Scholar] [CrossRef]
- Meyer, W.H.; Kun, L.; Marina, N.; Roberson, P.; Parham, D.; Rao, B.; Fletcher, B.; Pratt, C.B. Ifosfamide plus etoposide in newly diagnosed Ewing’s sarcoma of bone. J. Clin. Oncol. 1992, 10, 1737–1742. [Google Scholar] [CrossRef]
- Wexler, L.H.; DeLaney, T.F.; Tsokos, M.; Avila, N.; Steinberg, S.M.; Weaver-McClure, L.; Jacobson, J.; Jarosinski, P.; Hijazi, Y.M.; Balis, F.M.; et al. Ifosfamide and etoposide plus vincristine, doxorubicin, and cyclophosphamide for newly diagnosed Ewing’s sarcoma family of tumors. Cancer 1996, 78, 901–911. [Google Scholar] [CrossRef]
- Bacci, G.; Mercuri, M.; Longhi, A.; Bertoni, F.; Barbieri, E.; Donati, D.; Giacomini, S.; Bacchini, P.; Pignotti, E.; Forni, C.; et al. Neoadjuvant chemotherapy for Ewing’s tumour of bone: Recent experience at the Rizzoli Orthopaedic Institute. Eur. J. Cancer 2002, 38, 2243–2251. [Google Scholar] [CrossRef]
- Craft, A.; Cotterill, S.; Malcolm, A.; Spooner, D.; Grimer, R.; Souhami, R.; Imeson, J.; Lewis, I. Ifosfamide-containing chemotherapy in Ewing’s sarcoma: The Second United Kingdom Children’s Cancer Study Group and the Medical Research Council Ewing’s Tumor Study. J. Clin. Oncol. 1998, 16, 3628–3633. [Google Scholar] [CrossRef]
- Elomaa, I.; Blomqvist, C.P.; Saeter, G.; Akerman, M.; Stenwig, E.; Wiebe, T.; Bjork, O.; Alvegard, T.A. Five-year results in Ewing’s sarcoma. The Scandinavian Sarcoma Group experience with the SSG IX protocol. Eur. J. Cancer 2000, 36, 875–880. [Google Scholar] [CrossRef]
- Rosito, P.; Mancini, A.F.; Rondelli, R.; Abate, M.E.; Pession, A.; Bedei, L.; Bacci, G.; Picci, P.; Mercuri, M.; Ruggieri, P.; et al. Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: A preliminary report of 6 years of experience. Cancer 1999, 86, 421–428. [Google Scholar] [CrossRef]
- Grier, H.E.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; Perlman, E.J.; Meyers, P.A.; et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N. Engl. J. Med. 2003, 348, 694–701. [Google Scholar] [CrossRef]
- Paulussen, M.; Craft, A.W.; Lewis, I.; Hackshaw, A.; Douglas, C.; Dunst, J.; Schuck, A.; Winkelmann, W.; Kohler, G.; Poremba, C.; et al. Results of the EICESS-92 Study: Two randomized trials of Ewing’s sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J. Clin. Oncol. 2008, 26, 4385–4393. [Google Scholar] [CrossRef]
- Le Deley, M.C.; Paulussen, M.; Lewis, I.; Brennan, B.; Ranft, A.; Whelan, J.; Le Teuff, G.; Michon, J.; Ladenstein, R.; Marec-Berard, P.; et al. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: Results of the randomized noninferiority Euro-EWING99-R1 trial. J. Clin. Oncol. 2014, 32, 2440–2448. [Google Scholar] [CrossRef]
- Kolb, E.A.; Kushner, B.H.; Gorlick, R.; Laverdiere, C.; Healey, J.H.; LaQuaglia, M.P.; Huvos, A.G.; Qin, J.; Vu, H.T.; Wexler, L.; et al. Long-term event-free survival after intensive chemotherapy for Ewing’s family of tumors in children and young adults. J. Clin. Oncol. 2003, 21, 3423–3430. [Google Scholar] [CrossRef]
- Womer, R.B.; West, D.C.; Krailo, M.D.; Dickman, P.S.; Pawel, B.R.; Grier, H.E.; Marcus, K.; Sailer, S.; Healey, J.H.; Dormans, J.P.; et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 4148–4154. [Google Scholar] [CrossRef]
- Gaspar, N.; Rey, A.; Berard, P.M.; Michon, J.; Gentet, J.C.; Tabone, M.D.; Roche, H.; Defachelles, A.S.; Lejars, O.; Plouvier, E.; et al. Risk adapted chemotherapy for localised Ewing’s sarcoma of bone: The French EW93 study. Eur. J. Cancer 2012, 48, 1376–1385. [Google Scholar] [CrossRef]
- Dirksen, U.; Brennan, B.; Le Deley, M.C.; Cozic, N.; van den Berg, H.; Bhadri, V.; Brichard, B.; Claude, L.; Craft, A.; Amler, S.; et al. High-Dose Chemotherapy Compared With Standard Chemotherapy and Lung Radiation in Ewing Sarcoma With Pulmonary Metastases: Results of the European Ewing Tumour Working Initiative of National Groups, 99 Trial and EWING 2008. J. Clin. Oncol. 2019, 37, 3192–3202. [Google Scholar] [CrossRef]
- Dirksen, U.; Bhadri, V.; Brichard, B.; Butterfass-Bahloul, T.; Cyprova, S.; Faldum, A.; Gelderblom, H.; Hardes, J.; Hauser, P.; Havemann, L.; et al. Efficacy of add-on treosulfan and melphalan high-dose therapy in patients with high-risk metastatic Ewing sarcoma: Report from the International Ewing 2008R3 trial. J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Bisogno, G.; De Salvo, G.L.; Bergeron, C.; Gallego Melcon, S.; Merks, J.H.; Kelsey, A.; Martelli, H.; Minard-Colin, V.; Orbach, D.; Glosli, H.; et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 1566–1575. [Google Scholar] [CrossRef]
- Bernstein, M.L.; Devidas, M.; Lafreniere, D.; Souid, A.K.; Meyers, P.A.; Gebhardt, M.; Stine, K.; Nicholas, R.; Perlman, E.J.; Dubowy, R.; et al. Intensive therapy with growth factor support for patients with Ewing tumor metastatic at diagnosis: Pediatric Oncology Group/Children’s Cancer Group Phase II Study 9457--a report from the Children’s Oncology Group. J. Clin. Oncol. 2006, 24, 152–159. [Google Scholar] [CrossRef]
- Leavey, P.K.M.; DuBois, S. A Phase III randomized trial of adding vincristine-topotecan-cyclophosphamide (VTC) to standard chemotherapy in initial treatment of non-metastatic Ewing sarcoma-A report from the Children’s Oncology Group. In Proceedings of the Connective Tissue Oncology Society (CTOS)-Oral Presentation/Abstract, Annual Meeting 2019. Tokyo, Japan, 13–16 November 2019. [Google Scholar]
- Ferrari, S.; Sundby Hall, K.; Luksch, R.; Tienghi, A.; Wiebe, T.; Fagioli, F.; Alvegard, T.A.; Brach Del Prever, A.; Tamburini, A.; Alberghini, M.; et al. Nonmetastatic Ewing family tumors: High-dose chemotherapy with stem cell rescue in poor responder patients. Results of the Italian Sarcoma Group/Scandinavian Sarcoma Group III protocol. Ann. Oncol. 2011, 22, 1221–1227. [Google Scholar] [CrossRef]
- Brunetto, A.L.; Castillo, L.A.; Petrilli, A.S.; Macedo, C.D.; Boldrini, E.; Costa, C.; Almeida, M.T.; Kirst, D.; Rodriguez-Galindo, C.; Pereira, W.V.; et al. Carboplatin in the treatment of Ewing sarcoma: Results of the first Brazilian collaborative study group for Ewing sarcoma family tumors-EWING1. Pediatr. Blood Cancer 2015, 62, 1747–1753. [Google Scholar] [CrossRef]
- DuBois, S.G.B.J.; Buxton, A. Randomized phase 3 trial of ganitumab added to interval compressed chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma: A report from the Children’s Oncology Group (COG). In Proceedings of the Connective Tissue Oncology Society (CTOS)-Oral Presentation/Abstract, Annual Meeting 2019. Tokyo, Japan, 13–16 November 2019. [Google Scholar]
- Leavey, P.J.; Mascarenhas, L.; Marina, N.; Chen, Z.; Krailo, M.; Miser, J.; Brown, K.; Tarbell, N.; Bernstein, M.L.; Granowetter, L.; et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2008, 51, 334–338. [Google Scholar] [CrossRef]
- Shankar, A.G.; Ashley, S.; Craft, A.W.; Pinkerton, C.R. Outcome after relapse in an unselected cohort of children and adolescents with Ewing sarcoma. Med. Pediatr. Oncol. 2003, 40, 141–147. [Google Scholar] [CrossRef]
- McLean, T.W.; Hertel, C.; Young, M.L.; Marcus, K.; Schizer, M.A.; Gebhardt, M.; Weinstein, H.J.; Perez-Atayde, A.; Grier, H.E. Late events in pediatric patients with Ewing sarcoma/primitive neuroectodermal tumor of bone: The Dana-Farber Cancer Institute/Children’s Hospital experience. J. Pediatr. Hematol. Oncol. 1999, 21, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; del Prever, A.B.; Palmerini, E.; Staals, E.; Berta, M.; Balladelli, A.; Picci, P.; Fagioli, F.; Bacci, G.; Vanel, D. Response to high-dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr. Blood Cancer 2009, 52, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Hayes, F.A.; Thompson, E.I.; Kumar, M.; Hustu, H.O. Long-term survival in patients with Ewing’s sarcoma relapsing after completing therapy. Med. Pediatr. Oncol. 1987, 15, 254–256. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.G.; Kirton, L.; Khan, M.; Fenwick, N.; Dirksen, U.; Gaspar, N.; Kanerva, J.; Kuehne, T.; Longhi, A.; Luksch, R.; et al. Results of the second interim assessment of rEECur, an international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma (RR-ES). J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- McCabe, M.G.; Moroz, V.; Khan, M.; Dirksen, U.; Evans, A.; Fenwick, N.; Gaspar, N.; Kanerva, J.; Kuhne, T.; Longhi, A.; et al. Results of the first interim assessment of rEECur, an international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Bradfield, S.; Whitlock, J.A.; Krailo, M.; Franklin, J.; Blaney, S.M.; Adamson, P.C.; Reaman, G. Topotecan by 21-day continuous infusion in children with relapsed or refractory solid tumors: A Children’s Oncology Group study. Pediatr. Blood Cancer 2006, 47, 790–794. [Google Scholar] [CrossRef]
- Pratt, C.B.; Stewart, C.; Santana, V.M.; Bowman, L.; Furman, W.; Ochs, J.; Marina, N.; Kuttesch, J.F.; Heideman, R.; Sandlund, J.T.; et al. Phase I study of topotecan for pediatric patients with malignant solid tumors. J. Clin. Oncol. 1994, 12, 539–543. [Google Scholar] [CrossRef]
- Hunold, A.; Weddeling, N.; Paulussen, M.; Ranft, A.; Liebscher, C.; Jurgens, H. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr. Blood Cancer 2006, 47, 795–800. [Google Scholar] [CrossRef]
- Kushner, B.H.; Kramer, K.; Meyers, P.A.; Wollner, N.; Cheung, N.K. Pilot study of topotecan and high-dose cyclophosphamide for resistant pediatric solid tumors. Med. Pediatr. Oncol. 2000, 35, 468–474. [Google Scholar] [CrossRef]
- Wagner, L.M.; Crews, K.R.; Iacono, L.C.; Houghton, P.J.; Fuller, C.E.; McCarville, M.B.; Goldsby, R.E.; Albritton, K.; Stewart, C.F.; Santana, V.M. Phase I trial of temozolomide and protracted irinotecan in pediatric patients with refractory solid tumors. Clin. Cancer Res. 2004, 10, 840–848. [Google Scholar] [CrossRef]
- Casey, D.A.; Wexler, L.H.; Merchant, M.S.; Chou, A.J.; Merola, P.R.; Price, A.P.; Meyers, P.A. Irinotecan and temozolomide for Ewing sarcoma: The Memorial Sloan-Kettering experience. Pediatr. Blood Cancer 2009, 53, 1029–1034. [Google Scholar] [CrossRef]
- Fox, E.; Patel, S.; Wathen, J.K.; Schuetze, S.; Chawla, S.; Harmon, D.; Reinke, D.; Chugh, R.; Benjamin, R.S.; Helman, L.J. Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or locally recurrent chondrosarcoma: Results of Sarcoma Alliance for Research Through Collaboration Study 003. Oncologist 2012, 17, 321. [Google Scholar] [CrossRef]
- Mora, J.; Cruz, C.O.; Parareda, A.; de Torres, C. Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. J. Pediatr. Hematol. Oncol. 2009, 31, 723–729. [Google Scholar] [CrossRef]
- Dharmarajan, K.V.; Wexler, L.H.; Wolden, S.L. Concurrent radiation with irinotecan and carboplatin in intermediate- and high-risk rhabdomyosarcoma: A report on toxicity and efficacy from a prospective pilot phase II study. Pediatr. Blood Cancer 2013, 60, 242–247. [Google Scholar] [CrossRef]
- Rasper, M.; Jabar, S.; Ranft, A.; Jurgens, H.; Amler, S.; Dirksen, U. The value of high-dose chemotherapy in patients with first relapsed Ewing sarcoma. Pediatr. Blood Cancer 2014, 61, 1382–1386. [Google Scholar] [CrossRef]
- Attia, S.; Bolejack, V.; Ganjoo, K.N.; George, S.; Agulnik, M.; Rushing, D.A.; Loggers, E.T.; Livingston, M.B.; Wright, J.A.; Chawla, S.P.; et al. A phase II trial of regorafenib (REGO) in patients (pts) with advanced Ewing sarcoma and related tumors (EWS) of soft tissue and bone: SARCO24 trial results. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Italiano, A.; Mir, O.; Mathoulin-Pelissier, S.; Penel, N.; Piperno-Neumann, S.; Bompas, E.; Chevreau, C.; Duffaud, F.; Entz-Werle, N.; Saada, E.; et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 446–455. [Google Scholar] [CrossRef]
- Olmos, D.; Postel-Vinay, S.; Molife, L.R.; Okuno, S.H.; Schuetze, S.M.; Paccagnella, M.L.; Batzel, G.N.; Yin, D.; Pritchard-Jones, K.; Judson, I.; et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: A phase 1 expansion cohort study. Lancet Oncol. 2010, 11, 129–135. [Google Scholar] [CrossRef]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.P.; Chawla, S.P.; Toner, G.C.; Maki, R.G.; Meyers, P.A.; Chugh, R.; et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: Results of a phase II Sarcoma Alliance for Research through Collaboration study. J. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef]
- Tap, W.D.; Demetri, G.; Barnette, P.; Desai, J.; Kavan, P.; Tozer, R.; Benedetto, P.W.; Friberg, G.; Deng, H.; McCaffery, I.; et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J. Clin. Oncol. 2012, 30, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.; Butrynski, J.E.; Harmon, D.C.; Morgan, J.A.; George, S.; Wagner, A.J.; D’Adamo, D.; Cote, G.M.; Flamand, Y.; Benes, C.H.; et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. Bmc Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.S.; Rau, R.E.; Berg, S.L.; Liu, X.W.; Minard, C.G.; Bishop, A.J.R.; Romero, J.C.; Hicks, M.J.; Nelson, M.D.; Voss, S.; et al. Phase 1/2 trial of talazoparib in combination with temozolomide in children and adolescents with refractory/recurrent solid tumors including Ewing sarcoma: A Children’s Oncology Group Phase 1 Consortium study (ADVL1411). Pediatric Blood Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Federico, S.M.; Pappo, A.S.; Sahr, N.; Sykes, A.; Campagne, O.; Stewart, C.F.; Clay, M.R.; Bahrami, A.; McCarville, M.B.; Kaste, S.C.; et al. A phase I trial of talazoparib and irinotecan with and without temozolomide in children and young adults with recurrent or refractory solid malignancies. Eur. J. Cancer 2020, 137, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.A.F.N.C.; Anderson, P.; Macy, M.E.; Riedel, R.F.; Davis, L.; Daw, N.C.; Muscal, J.A.; Kim, A.; Ratan, R.; Ianopulos, X.; et al. TK216 Phase 1 Study in Metastatic, Relapsed/Refractory Ewing Sarcoma. In Proceedings of the Connective Tissue Oncology Society (CTOS)-Oral Presentation/Abstract, Virtual Annual Meeting 2020. Vancouver, BC, Canada, 18–21 November 2020. [Google Scholar]
- Reed, D.R.; Mascarenhas, L.; Meyers, P.A.; Chawla, S.P.; Harrison, D.J.; Setty, B.; Metts, J.; Wages, D.S.; Stenehjem, D.D.; Santiesteban, D.Y.; et al. A phase I/II clinical trial of the reversible LSD1 inhibitor, seclidemstat, in patients with relapsed/refractory Ewing sarcoma. J. Clin. Oncol. 2020, 38, TPS11567. [Google Scholar] [CrossRef]
- Dirksen, U.; Koch, R.; Bhadri, V.; Brichard, B.; Butterfass-Bahloul, T.; Cyprova, S.; Gelderblom, H.; Hauser, P.; Havemann, L.; Hjorth, L.; et al. Efficacy of maintenance therapy with zoledronic acid in patients with localized Ewing sarcoma: Report from the international Ewing 2008 trial. J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014, 10, e1004475. [Google Scholar] [CrossRef]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Fox, E.; Teachey, D.T.; Reid, J.M.; Safgren, S.L.; Carol, H.; Lock, R.B.; Houghton, P.J.; Smith, M.A.; Hall, D.; et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children’s Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin. Cancer Res. 2019, 25, 3229–3238. [Google Scholar] [CrossRef]
- Bond, M.; Bernstein, M.L.; Pappo, A.; Schultz, K.R.; Krailo, M.; Blaney, S.M.; Adamson, P.C. A phase II study of imatinib mesylate in children with refractory or relapsed solid tumors: A Children’s Oncology Group study. Pediatr. Blood Cancer 2008, 50, 254–258. [Google Scholar] [CrossRef]
- O’Neill, A.; Shah, N.; Zitomersky, N.; Ladanyi, M.; Shukla, N.; Uren, A.; Loeb, D.; Toretsky, J. Insulin-like growth factor 1 receptor as a therapeutic target in ewing sarcoma: Lack of consistent upregulation or recurrent mutation and a review of the clinical trial literature. Sarcoma 2013, 2013, 450478. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Kalebic, T.; Blakesley, V.; LeRoith, D.; Helman, L.J. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J. Biol. Chem. 1997, 272, 30822–30827. [Google Scholar] [CrossRef]
- Naing, A.; LoRusso, P.; Fu, S.; Hong, D.S.; Anderson, P.; Benjamin, R.S.; Ludwig, J.; Chen, H.X.; Doyle, L.A.; Kurzrock, R. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumors. Clin. Cancer Res. 2012, 18, 2625–2631. [Google Scholar] [CrossRef]
- Guenther, L.M.; Dharia, N.V.; Ross, L.; Conway, A.; Robichaud, A.L.; Catlett, J.L., 2nd; Wechsler, C.S.; Frank, E.S.; Goodale, A.; Church, A.J.; et al. A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 1343–1357. [Google Scholar] [CrossRef]
- de Groot, S.; Rottgering, B.; Gelderblom, H.; Pijl, H.; Szuhai, K.; Kroep, J.R. Unraveling the Resistance of IGF-Pathway Inhibition in Ewing Sarcoma. Cancers 2020, 12, 3568. [Google Scholar] [CrossRef]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef]
- DuBois, S.; Demetri, G. Markers of angiogenesis and clinical features in patients with sarcoma. Cancer 2007, 109, 813–819. [Google Scholar] [CrossRef]
- Fleuren, E.D.G.; Roeffen, M.H.S.; Leenders, W.P.; Flucke, U.E.; Vlenterie, M.; Schreuder, H.W.; Boerman, O.C.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Expression and clinical relevance of MET and ALK in Ewing sarcomas. Int J. Cancer 2013, 133, 427–436. [Google Scholar] [CrossRef]
- Druker, B.J. Taking aim at Ewing’s sarcoma: Is KIT a target and will imatinib work? J. Natl. Cancer I 2002, 94, 1660–1661. [Google Scholar] [CrossRef][Green Version]
- Bauer, S.; Yu, L.K.; Demetri, G.D.; Fletcher, J.A. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006, 66, 9153–9161. [Google Scholar] [CrossRef]
- Chao, J.; Budd, G.T.; Chu, P.G.; Frankel, P.; Garcia, D.; Junqueira, M.; Loera, S.; Somlo, G.; Sato, J.; Chow, W.A. Phase II Clinical Trial of Imatinib Mesylate in Therapy of KIT and/or PDGFR alpha-expressing Ewing Sarcoma Family of Tumors and Desmoplastic Small Round Cell Tumors. Anticancer Res. 2010, 30, 547–552. [Google Scholar]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma (vol 555, pg 387, 2018). Nature 2018, 559. [Google Scholar] [CrossRef]
- Stewart, E.; Goshorn, R.; Bradley, C.; Griffiths, L.M.; Benavente, C.; Twarog, N.R.; Miller, G.M.; Caufield, W.; Freeman, B.B.; Bahrami, A.; et al. Targeting the DNA Repair Pathway in Ewing Sarcoma. Cell Rep. 2014, 9, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Engert, F.; Schneider, C.; Weiss, L.M.; Probst, M.; Fulda, S. PARP Inhibitors Sensitize Ewing Sarcoma Cells to Temozolomide-Induced Apoptosis via the Mitochondrial Pathway. Mol. Cancer Ther. 2015, 14, 2818–2830. [Google Scholar] [CrossRef]
- Smith, M.A.; Reynolds, C.P.; Kang, M.H.; Kolb, E.A.; Gorlick, R.; Carol, H. Synergistic Activity of PARP Inhibition by Talazoparib (BMN 673) with Temozolomide in Pediatric Cancer Models in the Pediatric Preclinical Testing Program (vol 21, pg 819, 2015). Clin. Cancer Res. 2017, 23, 1118–1119. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Heidel, J.D.; Bartlett, D.W.; Davis, M.E.; Triche, T.J. Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res. 2005, 65, 8984–8992. [Google Scholar] [CrossRef]
- Tanaka, K.; Iwakuma, T.; Harimaya, K.; Sato, H.; Iwamoto, Y. EWS-Fli1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing’s sarcoma and primitive neuroectodermal tumor cells. J. Clin. Invest. 1997, 99, 239–247. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Connell, Y.; Neckers, L.; Bhat, N.K. Inhibition of EWS-FLI-1 fusion protein with antisense oligodeoxynucleotides. J. Neurooncol. 1997, 31, 9–16. [Google Scholar] [CrossRef]
- Aynaud, M.M.; Mirabeau, O.; Gruel, N.; Grossetete, S.; Boeva, V.; Durand, S.; Surdez, D.; Saulnier, O.; Zaidi, S.; Gribkova, S.; et al. Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep. 2020, 30, 1767–1779 e1766. [Google Scholar] [CrossRef]
- Franzetti, G.A.; Laud-Duval, K.; van der Ent, W.; Brisac, A.; Irondelle, M.; Aubert, S.; Dirksen, U.; Bouvier, C.; de Pinieux, G.; Snaar-Jagalska, E.; et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017, 36, 3505–3514. [Google Scholar] [CrossRef]
- Pedersen, E.A.; Menon, R.; Bailey, K.M.; Thomas, D.G.; Van Noord, R.A.; Tran, J.; Wang, H.; Qu, P.P.; Hoering, A.; Fearon, E.R.; et al. Activation of Wnt/beta-Catenin in Ewing Sarcoma Cells Antagonizes EWS/ETS Function and Promotes Phenotypic Transition to More Metastatic Cell States. Cancer Res. 2016, 76, 5040–5053. [Google Scholar] [CrossRef] [PubMed]
- Uren, A.; Toretsky, J.A. Ewing’s sarcoma oncoprotein EWS-FLI1: The perfect target without a therapeutic agent. Future Oncol. 2005, 1, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Berg, T. Inhibition of transcription factors with small organic molecules. Curr. Opin. Chem. Biol. 2008, 12, 464–471. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Wright, P.E. Assemblages: Functional units formed by cellular phase separation. J. Cell Biol. 2014, 206, 579–588. [Google Scholar] [CrossRef]
- Chong, S.; Dugast-Darzacq, C.; Liu, Z.; Dong, P.; Dailey, G.M.; Cattoglio, C.; Heckert, A.; Banala, S.; Lavis, L.; Darzacq, X.; et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 2018, 361. [Google Scholar] [CrossRef]
- Selvanathan, S.P.; Moseley, E.; Graham, G.T.; Jessen, K.; Lannutti, B.; Üren, A.; Toretsky, J.A. Abstract 694: TK-216: A novel, first-in-class, small molecule inhibitor of EWS-FLI1 in early clinical development, for the treatment of Ewing Sarcoma. Cancer Res. 2017, 77, 694. [Google Scholar] [CrossRef]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef]
- Erkizan, H.V.; Schneider, J.A.; Sajwan, K.; Graham, G.T.; Griffin, B.; Chasovskikh, S.; Youbi, S.E.; Kallarakal, A.; Chruszcz, M.; Padmanabhan, R.; et al. RNA helicase A activity is inhibited by oncogenic transcription factor EWS-FLI1. Nucleic Acids Res. 2015, 43, 1069–1080. [Google Scholar] [CrossRef]
- Zollner, S.K.; Selvanathan, S.P.; Graham, G.T.; Commins, R.M.T.; Hong, S.H.; Moseley, E.; Parks, S.; Haladyna, J.N.; Erkizan, H.V.; Dirksen, U.; et al. Inhibition of the oncogenic fusion protein EWS-FLI1 causes G2-M cell cycle arrest and enhanced vincristine sensitivity in Ewing’s sarcoma. Sci Signal. 2017, 10. [Google Scholar] [CrossRef]
- Casey, D.L.; Lin, T.Y.; Cheung, N.V. Exploiting Signaling Pathways and Immune Targets Beyond the Standard of Care for Ewing Sarcoma. Front. Oncol. 2019, 9, 537. [Google Scholar] [CrossRef]
- Grohar, P.J.; Glod, J.; Peer, C.J.; Sissung, T.M.; Arnaldez, F.I.; Long, L.; Figg, W.D.; Whitcomb, P.; Helman, L.J.; Widemann, B.C. A phase I/II trial and pharmacokinetic study of mithramycin in children and adults with refractory Ewing sarcoma and EWS-FLI1 fusion transcript. Cancer Chemother. Pharmacol. 2017, 80, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Invest. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hu, H.M.; Zielinska-Kwiatkowska, A.; Chansky, H.A. FOXO1 is a direct target of EWS-Fli1 oncogenic fusion protein in Ewing’s sarcoma cells. Biochem. Biophys. Res. Commun. 2010, 402, 129–134. [Google Scholar] [CrossRef]
- Sakimura, R.; Tanaka, K.; Nakatani, F.; Matsunobu, T.; Li, X.; Hanada, M.; Okada, T.; Nakamura, T.; Matsumoto, Y.; Iwamoto, Y. Antitumor effects of histone deacetylase inhibitor on Ewing’s family tumors. Int. J. Cancer 2005, 116, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Sankar, S.; Theisen, E.R.; Bearss, J.; Mulvihill, T.; Hoffman, L.M.; Sorna, V.; Beckerle, M.C.; Sharma, S.; Lessnick, S.L. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin. Cancer Res. 2014, 20, 4584–4597. [Google Scholar] [CrossRef]
- Harlow, M.L.; Chasse, M.H.; Boguslawski, E.A.; Sorensen, K.M.; Gedminas, J.M.; Kitchen-Goosen, S.M.; Rothbart, S.B.; Taslim, C.; Lessnick, S.L.; Peck, A.S.; et al. Trabectedin Inhibits EWS-FLI1 and Evicts SWI/SNF from Chromatin in a Schedule-dependent Manner. Clin. Cancer Res. 2019, 25, 3417–3429. [Google Scholar] [CrossRef]
- Knott, M.M.L.; Holting, T.L.B.; Ohmura, S.; Kirchner, T.; Cidre-Aranaz, F.; Grunewald, T.G.P. Targeting the undruggable: Exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer Metastasis Rev. 2019, 38, 625–642. [Google Scholar] [CrossRef]
- Friedrich, P.; Ortiz, R.; Fuentes, S.; Gamboa, Y.; Ah Chu-Sanchez, M.S.; Arambu, I.C.; Montero, M.; Baez, F.; Rodriguez-Galindo, C.; Antillon-Klussmann, F.; et al. Barriers to effective treatment of pediatric solid tumors in middle-income countries: Can we make sense of the spectrum of nonbiologic factors that influence outcomes? Cancer 2014, 120, 112–125. [Google Scholar] [CrossRef]
- Totadri, S.; Bansal, D.; Rao, K.L.N.; Jain, R.; Saxena, A.K.; Kapoor, R.; Samujh, R.; Trehan, A. Challenges in the management of localized Ewing sarcoma in a developing country. Pediatr. Hematol. Oncol. 2020, 37, 610–619. [Google Scholar] [CrossRef]
- Howard, S.C.; Davidson, A.; Luna-Fineman, S.; Israels, T.; Chantada, G.; Lam, C.G.; Hunger, S.P.; Bailey, S.; Ribeiro, R.C.; Arora, R.S.; et al. A framework to develop adapted treatment regimens to manage pediatric cancer in low- and middle-income countries: The Pediatric Oncology in Developing Countries (PODC) Committee of the International Pediatric Oncology Society (SIOP). Pediatr. Blood Cancer 2017, 64 (Suppl. 5). [Google Scholar] [CrossRef]
- El Kababri, M.; Benmiloud, S.; Cherkaoui, S.; El Houdzi, J.; Maani, K.; Ansari, N.; Khoubila, N.; Kili, A.; El Khorassani, M.; Madani, A.; et al. Metro-SMHOP 01: Metronomics combination with cyclophosphamide-etoposide and valproic acid for refractory and relapsing pediatric malignancies. Pediatr. Blood Cancer 2020, 67, e28508. [Google Scholar] [CrossRef]
- Jain, S.; Kapoor, G. Chemotherapy in Ewing’s sarcoma. Indian J. Orthop. 2010, 44, 369–377. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Anderson, N.D.; de Borja, R.; Young, M.D.; Fuligni, F.; Rosic, A.; Roberts, N.D.; Hajjar, S.; Layeghifard, M.; Novokmet, A.; Kowalski, P.E.; et al. Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors. Science 2018, 361. [Google Scholar] [CrossRef]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. EBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef]
- Singh, V.K.; Rastogi, A.; Hu, X.; Wang, Y.; De, S. Mutational signature SBS8 predominantly arises due to late replication errors in cancer. Commun. Biol. 2020, 3, 421. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schonegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, J.; Shi, Q.; Song, X.; Shen, L.; Zhou, J.; Shao, J. The prognostic signature of the somatic mutations in Ewing sarcoma: From a network view. Jpn. J. Clin. Oncol. 2019, 49, 604–613. [Google Scholar] [CrossRef]
- Ohali, A.; Avigad, S.; Zaizov, R.; Ophir, R.; Horn-Saban, S.; Cohen, I.J.; Meller, I.; Kollender, Y.; Issakov, J.; Yaniv, I. Prediction of high risk Ewing’s sarcoma by gene expression profiling. Oncogene 2004, 23, 8997–9006. [Google Scholar] [CrossRef]
- Scotlandi, K.; Remondini, D.; Castellani, G.; Manara, M.C.; Nardi, F.; Cantiani, L.; Francesconi, M.; Mercuri, M.; Caccuri, A.M.; Serra, M.; et al. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J. Clin. Oncol. 2009, 27, 2209–2216. [Google Scholar] [CrossRef]
- Schaefer, K.L.; Eisenacher, M.; Braun, Y.; Brachwitz, K.; Wai, D.H.; Dirksen, U.; Lanvers-Kaminsky, C.; Juergens, H.; Herrero, D.; Stegmaier, S.; et al. Microarray analysis of Ewing’s sarcoma family of tumours reveals characteristic gene expression signatures associated with metastasis and resistance to chemotherapy. Eur. J. Cancer 2008, 44, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Bu, X.; Cao, M.; Zhang, X.; Huo, T.; Li, Z.; Gao, X.; Jing, L.; Luo, X.; Feng, H.; et al. Inactivation of ICAM1 inhibits metastasis and improves the prognosis of Ewing’s sarcoma. J. Cancer Res. Clin. Oncol. 2021, 147, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Volchenboum, S.L.; Andrade, J.; Huang, L.; Barkauskas, D.A.; Krailo, M.; Womer, R.B.; Ranft, A.; Potratz, J.; Dirksen, U.; Triche, T.J.; et al. Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children’s Oncology Group. J. Pathol. Clin. Res. 2015, 1, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Ren, E.H.; Deng, Y.J.; Yuan, W.H.; Wu, Z.L.; Zhang, G.Z.; Xie, Q.Q. An immune-related gene signature for determining Ewing sarcoma prognosis based on machine learning. J. Cancer Res. Clin. Oncol. 2021, 147, 153–165. [Google Scholar] [CrossRef]
- Stahl, D.; Gentles, A.J.; Thiele, R.; Gutgemann, I. Prognostic profiling of the immune cell microenvironment in Ewing s Sarcoma Family of Tumors. Oncoimmunology 2019, 8, e1674113. [Google Scholar] [CrossRef]
- Berghuis, D.; Santos, S.J.; Baelde, H.J.; Taminiau, A.H.; Egeler, R.M.; Schilham, M.W.; Hogendoorn, P.C.; Lankester, A.C. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T-lymphocyte infiltration and affect tumour progression. J. Pathol. 2011, 223, 347–357. [Google Scholar] [CrossRef]
- Bennani-Baiti, I.M.; Cooper, A.; Lawlor, E.R.; Kauer, M.; Ban, J.; Aryee, D.N.; Kovar, H. Intercohort gene expression co-analysis reveals chemokine receptors as prognostic indicators in Ewing’s sarcoma. Clin. Cancer Res. 2010, 16, 3769–3778. [Google Scholar] [CrossRef]
- Krook, M.A.; Nicholls, L.A.; Scannell, C.A.; Chugh, R.; Thomas, D.G.; Lawlor, E.R. Stress-induced CXCR4 promotes migration and invasion of ewing sarcoma. Mol. Cancer Res. 2014, 12, 953–964. [Google Scholar] [CrossRef]
- Liao, Y.X.; Zhou, C.H.; Zeng, H.; Zuo, D.Q.; Wang, Z.Y.; Yin, F.; Hua, Y.Q.; Cai, Z.D. The role of the CXCL12-CXCR4/CXCR7 axis in the progression and metastasis of bone sarcomas (Review). Int J. Mol. Med. 2013, 32, 1239–1246. [Google Scholar] [CrossRef]
- Ban, J.; Bennani-Baiti, I.M.; Kauer, M.; Schaefer, K.L.; Poremba, C.; Jug, G.; Schwentner, R.; Smrzka, O.; Muehlbacher, K.; Aryee, D.N.; et al. EWS-FLI1 suppresses NOTCH-activated p53 in Ewing’s sarcoma. Cancer Res. 2008, 68, 7100–7109. [Google Scholar] [CrossRef]
- Przybyl, J.; Kozak, K.; Kosela, H.; Falkowski, S.; Switaj, T.; Lugowska, I.; Szumera-Cieckiewicz, A.; Ptaszynski, K.; Grygalewicz, B.; Chechlinska, M.; et al. Gene expression profiling of peripheral blood cells: New insights into Ewing sarcoma biology and clinical applications. Med. Oncol. 2014, 31, 109. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Invest. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Advances in immune checkpoint inhibitors for bone sarcoma therapy. J. Bone Oncol. 2019, 15, 100221. [Google Scholar] [CrossRef]
- Morales, E.; Olson, M.; Iglesias, F.; Dahiya, S.; Luetkens, T.; Atanackovic, D. Role of immunotherapy in Ewing sarcoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef]
- Charan, M.; Dravid, P.; Cam, M.; Audino, A.; Gross, A.C.; Arnold, M.A.; Roberts, R.D.; Cripe, T.P.; Pertsemlidis, A.; Houghton, P.J.; et al. GD2-directed CAR-T cells in combination with HGF-targeted neutralizing antibody (AMG102) prevent primary tumor growth and metastasis in Ewing sarcoma. Int. J. Cancer 2020, 146, 3184–3195. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Lesch, S.; Balbach, S.; Gottlich, C.; Kuhnemundt, J.; Mikesch, J.H.; Schelhaas, S.; Jamitzky, S.; Meltzer, J.; et al. EZH2 Inhibition in Ewing Sarcoma Upregulates GD2 Expression for Targeting with Gene-Modified T Cells. Mol. Ther. 2019, 27, 933–946. [Google Scholar] [CrossRef]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Hoffman, L.M.; Jensen, C.C.; Lin, Y.C.; Grossmann, A.H.; Randall, R.L.; Lessnick, S.L.; Welm, A.L.; Beckerle, M.C. Molecular dissection of the mechanism by which EWS/FLI expression compromises actin cytoskeletal integrity and cell adhesion in Ewing sarcoma. Mol. Biol. Cell 2014, 25, 2695–2709. [Google Scholar] [CrossRef]
- Katschnig, A.M.; Kauer, M.O.; Schwentner, R.; Tomazou, E.M.; Mutz, C.N.; Linder, M.; Sibilia, M.; Alonso, J.; Aryee, D.N.T.; Kovar, H. EWS-FLI1 perturbs MRTFB/YAP-1/TEAD target gene regulation inhibiting cytoskeletal autoregulatory feedback in Ewing sarcoma. Oncogene 2017. [Google Scholar] [CrossRef]
- Bailey, K.M.; Julian, C.M.; Klinghoffer, A.N.; Bernard, H.; Lucas, P.C.; McAllister-Lucas, L.M. EWS-FLI1 low Ewing sarcoma cells demonstrate decreased susceptibility to T-cell-mediated tumor cell apoptosis. Oncotarget 2019, 10, 3385–3399. [Google Scholar] [CrossRef]
- Hawkins, A.G.; Pedersen, E.A.; Treichel, S.; Temprine, K.; Sperring, C.; Read, J.A.; Magnuson, B.; Chugh, R.; Lawlor, E.R. Wnt/beta-catenin activated Ewing sarcoma cells promote the angiogenic switch. JCI Insight 2020. [Google Scholar] [CrossRef]
- Bierbaumer, L.; Katschnig, A.M.; Radic Sarikas, B.; Kauer, M.; Petro, J.R.; Högler, S.; Gurnhofer, E.; Pedot, G.; Schäfer, B.W.; Schwentner, R.; et al. YAP/TAZ inhibition reduces metastatic potential of Ewing sarcoma cells. Oncogenesis 2021, in press. [Google Scholar] [CrossRef]
- Rodriguez-Nunez, P.; Romero-Perez, L.; Amaral, A.T.; Puerto-Camacho, P.; Jordan, C.; Marcilla, D.; Grunewald, T.G.; Alonso, J.; de Alava, E.; Diaz-Martin, J. Hippo pathway effectors YAP1/TAZ induce an EWS-FLI1-opposing gene signature and associate with disease progression in Ewing sarcoma. J. Pathol. 2020, 250, 374–386. [Google Scholar] [CrossRef]
- Esiashvili, N.; Goodman, M.; Marcus, R.B., Jr. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J. Pediatr. Hematol. Oncol. 2008, 30, 425–430. [Google Scholar] [CrossRef]
- Miser, J.S.; Goldsby, R.E.; Chen, Z.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; et al. Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: Evaluation of increasing the dose intensity of chemotherapy--a report from the Children’s Oncology Group. Pediatr. Blood Cancer 2007, 49, 894–900. [Google Scholar] [CrossRef]
- Cotterill, S.J.; Ahrens, S.; Paulussen, M.; Jurgens, H.F.; Voute, P.A.; Gadner, H.; Craft, A.W. Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J. Clin. Oncol. 2000, 18, 3108–3114. [Google Scholar] [CrossRef]
- Rodriguez-Galindo, C.; Liu, T.; Krasin, M.J.; Wu, J.; Billups, C.A.; Daw, N.C.; Spunt, S.L.; Rao, B.N.; Santana, V.M.; Navid, F. Analysis of prognostic factors in ewing sarcoma family of tumors: Review of St. Jude Children’s Research Hospital studies. Cancer 2007, 110, 375–384. [Google Scholar] [CrossRef]
- Karski, E.E.; McIlvaine, E.; Segal, M.R.; Krailo, M.; Grier, H.E.; Granowetter, L.; Womer, R.B.; Meyers, P.A.; Felgenhauer, J.; Marina, N.; et al. Identification of Discrete Prognostic Groups in Ewing Sarcoma. Pediatr. Blood Cancer 2016, 63, 47–53. [Google Scholar] [CrossRef]
- Miser, J.S.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; Perlman, E.J.; Meyers, P.A.; et al. Treatment of metastatic Ewing’s sarcoma or primitive neuroectodermal tumor of bone: Evaluation of combination ifosfamide and etoposide--a Children’s Cancer Group and Pediatric Oncology Group study. J. Clin. Oncol. 2004, 22, 2873–2876. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Dai, M.; Zhang, B. Risk Factors for Metastasis at Initial Diagnosis With Ewing Sarcoma. Front. Oncol. 2019, 9, 1043. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, M.A.; Goldsby, R.; Neuhaus, J.; DuBois, S.G. Clinical features and outcomes in patients with Ewing sarcoma and regional lymph node involvement. Pediatr. Blood Cancer 2012, 59, 617–620. [Google Scholar] [CrossRef]
- Burdach, S.; Thiel, U.; Schoniger, M.; Haase, R.; Wawer, A.; Nathrath, M.; Kabisch, H.; Urban, C.; Laws, H.J.; Dirksen, U.; et al. Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant. 2010, 45, 483–489. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Paulino, A.C.; Mai, W.Y.; Teh, B.S. Radiotherapy in metastatic ewing sarcoma. Am. J. Clin. Oncol. 2013, 36, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, M.; Ahrens, S.; Burdach, S.; Craft, A.; Dockhorn-Dworniczak, B.; Dunst, J.; Frohlich, B.; Winkelmann, W.; Zoubek, A.; Jurgens, H. Primary metastatic (stage IV) Ewing tumor: Survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann. Oncol. 1998, 9, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Ewing Sarcoma Treatment (PDQ(R)): Health Professional Version. Updated 08/11/2020; In PDQ Cancer Information Summaries; U.S. Department of Health and Human Services, National Cancer Institute at the National Institutes of Health: Bethesda, MD, USA, 2002.
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef]
- Villasante, A.; Marturano-Kruik, A.; Ambati, S.R.; Liu, Z.; Godier-Furnemont, A.; Parsa, H.; Lee, B.W.; Moore, M.A.; Vunjak-Novakovic, G. Recapitulating the Size and Cargo of Tumor Exosomes in a Tissue-Engineered Model. Theranostics 2016, 6, 1119–1130. [Google Scholar] [CrossRef]
- Chen, G.; Dong, C.; Yang, L.; Lv, Y. 3D Scaffolds with Different Stiffness but the Same Microstructure for Bone Tissue Engineering. ACS Appl Mater. Interfaces 2015, 7, 15790–15802. [Google Scholar] [CrossRef]
- Panciera, T.; Citron, A.; Di Biagio, D.; Battilana, G.; Gandin, A.; Giulitti, S.; Forcato, M.; Bicciato, S.; Panzetta, V.; Fusco, S.; et al. Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nat. Mater. 2020. [Google Scholar] [CrossRef]
- Rogers, L.K.; Cismowski, M.J. Oxidative Stress in the Lung-The Essential Paradox. Curr. Opin. Toxicol. 2018, 7, 37–43. [Google Scholar] [CrossRef]
- Johnson, R.W.; Sowder, M.E.; Giaccia, A.J. Hypoxia and Bone Metastatic Disease. Curr. Osteoporos Rep. 2017, 15, 231–238. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef]
- Woelfle, U.; Cloos, J.; Sauter, G.; Riethdorf, L.; Janicke, F.; van Diest, P.; Brakenhoff, R.; Pantel, K. Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res. 2003, 63, 5679–5684. [Google Scholar] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Obenauf, A.C.; Massague, J. Surviving at a Distance: Organ-Specific Metastasis. Trends Cancer 2015, 1, 76–91. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- De Feo, A.; Sciandra, M.; Ferracin, M.; Felicetti, F.; Astolfi, A.; Pignochino, Y.; Picci, P.; Care, A.; Scotlandi, K. Exosomes from CD99-deprived Ewing sarcoma cells reverse tumor malignancy by inhibiting cell migration and promoting neural differentiation. Cell Death Dis. 2019, 10, 471. [Google Scholar] [CrossRef]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef]
- D’Oronzo, S.; Coleman, R.; Brown, J.; Silvestris, F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J. Bone Oncol. 2019, 15, 004. [Google Scholar] [CrossRef]
- Odri, G.A.; Dumoucel, S.; Picarda, G.; Battaglia, S.; Lamoureux, F.; Corradini, N.; Rousseau, J.; Tirode, F.; Laud, K.; Delattre, O.; et al. Zoledronic acid as a new adjuvant therapeutic strategy for Ewing’s sarcoma patients. Cancer Res. 2010, 70, 7610–7619. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Guan, H.; Duan, X.; Kleinerman, E.S. Zoledronic acid inhibits primary bone tumor growth in Ewing sarcoma. Cancer 2005, 104, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Chao, T.; Ruiqi, C.; Juan, S.; Zhihong, L. Patient-derived xenograft models in musculoskeletal malignancies. J. Transl. Med. 2018, 16, 107. [Google Scholar] [CrossRef] [PubMed]
- Ulz, P.; Thallinger, G.G.; Auer, M.; Graf, R.; Kashofer, K.; Jahn, S.W.; Abete, L.; Pristauz, G.; Petru, E.; Geigl, J.B.; et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 2016, 48, 1273–1278. [Google Scholar] [CrossRef]
- May, W.A.; Grigoryan, R.S.; Keshelava, N.; Cabral, D.J.; Christensen, L.L.; Jenabi, J.; Ji, L.; Triche, T.J.; Lawlor, E.R.; Reynolds, C.P. Characterization and drug resistance patterns of Ewing’s sarcoma family tumor cell lines. PLoS ONE 2013, 8, e80060. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Brodin, B.A.; Wennerberg, K.; Lidbrink, E.; Brosjo, O.; Potdar, S.; Wilson, J.N.; Ma, L.; Moens, L.N.; Hesla, A.; Porovic, E.; et al. Drug sensitivity testing on patient-derived sarcoma cells predicts patient response to treatment and identifies c-Sarc inhibitors as active drugs for translocation sarcomas. Br. J. Cancer 2019, 120, 435–443. [Google Scholar] [CrossRef]
- Fong, E.L.; Lamhamedi-Cherradi, S.E.; Burdett, E.; Ramamoorthy, V.; Lazar, A.J.; Kasper, F.K.; Farach-Carson, M.C.; Vishwamitra, D.; Demicco, E.G.; Menegaz, B.A.; et al. Modeling Ewing sarcoma tumors in vitro with 3D scaffolds. Proc. Natl. Acad. Sci. USA 2013, 110, 6500–6505. [Google Scholar] [CrossRef]
- Lamhamedi-Cherradi, S.E.; Santoro, M.; Ramammoorthy, V.; Menegaz, B.A.; Bartholomeusz, G.; Iles, L.R.; Amin, H.M.; Livingston, J.A.; Mikos, A.G.; Ludwig, J.A. 3D tissue-engineered model of Ewing’s sarcoma. Adv. Drug Deliv. Rev. 2014, 79–80, 155–171. [Google Scholar] [CrossRef]
- Santoro, M.; Lamhamedi-Cherradi, S.E.; Menegaz, B.A.; Ludwig, J.A.; Mikos, A.G. Flow perfusion effects on three-dimensional culture and drug sensitivity of Ewing sarcoma. Proc. Natl. Acad. Sci. USA 2015, 112, 10304–10309. [Google Scholar] [CrossRef]
- Nanni, P.; Landuzzi, L.; Manara, M.C.; Righi, A.; Nicoletti, G.; Cristalli, C.; Pasello, M.; Parra, A.; Carrabotta, M.; Ferracin, M.; et al. Bone sarcoma patient-derived xenografts are faithful and stable preclinical models for molecular and therapeutic investigations. Sci. Rep. 2019, 9, 12174. [Google Scholar] [CrossRef]
- Stebbing, J.; Paz, K.; Schwartz, G.K.; Wexler, L.H.; Maki, R.; Pollock, R.E.; Morris, R.; Cohen, R.; Shankar, A.; Blackman, G.; et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer 2014, 120, 2006–2015. [Google Scholar] [CrossRef]
- Murakami, T.; Singh, A.S.; Kiyuna, T.; Dry, S.M.; Li, Y.; James, A.W.; Igarashi, K.; Kawaguchi, K.; DeLong, J.C.; Zhang, Y.; et al. Effective molecular targeting of CDK4/6 and IGF-1R in a rare FUS-ERG fusion CDKN2A-deletion doxorubicin-resistant Ewing’s sarcoma patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget 2016, 7, 47556–47564. [Google Scholar] [CrossRef]
- Miyake, K.; Murakami, T.; Kiyuna, T.; Igarashi, K.; Kawaguchi, K.; Miyake, M.; Li, Y.; Nelson, S.D.; Dry, S.M.; Bouvet, M.; et al. The combination of temozolomide-irinotecan regresses a doxorubicin-resistant patient-derived orthotopic xenograft (PDOX) nude-mouse model of recurrent Ewing’s sarcoma with a FUS-ERG fusion and CDKN2A deletion: Direction for third-line patient therapy. Oncotarget 2017, 8, 103129–103136. [Google Scholar] [CrossRef]
- Murakami, T.; Li, S.; Han, Q.; Tan, Y.; Kiyuna, T.; Igarashi, K.; Kawaguchi, K.; Hwang, H.K.; Miyake, K.; Singh, A.S.; et al. Recombinant methioninase effectively targets a Ewing’s sarcoma in a patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget 2017, 8, 35630–35638. [Google Scholar] [CrossRef]
- Miyake, K.; Kiyuna, T.; Li, S.; Han, Q.; Tan, Y.; Zhao, M.; Oshiro, H.; Kawaguchi, K.; Higuchi, T.; Zhang, Z.; et al. Combining Tumor-Selective Bacterial Therapy with Salmonella typhimurium A1-R and Cancer Metabolism Targeting with Oral Recombinant Methioninase Regressed an Ewing’s Sarcoma in a Patient-Derived Orthotopic Xenograft Model. Chemotherapy 2018, 63, 278–283. [Google Scholar] [CrossRef]
- Miyake, K.; Kiyuna, T.; Kawaguchi, K.; Higuchi, T.; Oshiro, H.; Zhang, Z.; Wangsiricharoen, S.; Razmjooei, S.; Li, Y.; Nelson, S.D.; et al. Regorafenib regressed a doxorubicin-resistant Ewing’s sarcoma in a patient-derived orthotopic xenograft (PDOX) nude mouse model. Cancer Chemother. Pharmacol. 2019, 83, 809–815. [Google Scholar] [CrossRef]
- Hoffman, R.M. Patient-derived orthotopic xenografts: Better mimic of metastasis than subcutaneous xenografts. Nat. Rev. Cancer 2015, 15, 451–452. [Google Scholar] [CrossRef]
- Steinestel, K.; Trautmann, M.; Jansen, E.P.; Dirksen, U.; Rehkamper, J.; Mikesch, J.H.; Gerke, J.S.; Orth, M.F.; Sannino, G.; Arteaga, M.F.; et al. Focal adhesion kinase confers pro-migratory and antiapoptotic properties and is a potential therapeutic target in Ewing sarcoma. Mol. Oncol. 2020, 14, 248–260. [Google Scholar] [CrossRef]
- Ban, J.; Aryee, D.N.; Fourtouna, A.; van der Ent, W.; Kauer, M.; Niedan, S.; Machado, I.; Rodriguez-Galindo, C.; Tirado, O.M.; Schwentner, R.; et al. Suppression of deacetylase SIRT1 mediates tumor-suppressive NOTCH response and offers a novel treatment option in metastatic Ewing sarcoma. Cancer Res. 2014, 74, 6578–6588. [Google Scholar] [CrossRef]
- van der Ent, W.; Jochemsen, A.G.; Teunisse, A.F.; Krens, S.F.; Szuhai, K.; Spaink, H.P.; Hogendoorn, P.C.; Snaar-Jagalska, B.E. Ewing sarcoma inhibition by disruption of EWSR1-FLI1 transcriptional activity and reactivation of p53. J. Pathol. 2014, 233, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Berning, P.; Hennemann, C.; Tulotta, C.; Schaefer, C.; Lechtape, B.; Hotfilder, M.; El Gourari, Y.; Jurgens, H.; Snaar-Jagalska, E.; Hempel, G.; et al. The Receptor Tyrosine Kinase RON and Its Isoforms as Therapeutic Targets in Ewing Sarcoma. Cancers 2020, 12, 904. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hwang, E.E.; Guha, R.; O’Neill, A.F.; Melong, N.; Veinotte, C.J.; Conway Saur, A.; Wuerthele, K.; Shen, M.; McKnight, C.; et al. High-throughput Chemical Screening Identifies Focal Adhesion Kinase and Aurora Kinase B Inhibition as a Synergistic Treatment Combination in Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 4552–4566. [Google Scholar] [CrossRef] [PubMed]
- Sainz-Jaspeado, M.; Lagares-Tena, L.; Lasheras, J.; Navid, F.; Rodriguez-Galindo, C.; Mateo-Lozano, S.; Notario, V.; Sanjuan, X.; Garcia Del Muro, X.; Fabra, A.; et al. Caveolin-1 modulates the ability of Ewing’s sarcoma to metastasize. Mol. Cancer Res. 2010, 8, 1489–1500. [Google Scholar] [CrossRef]
- von Heyking, K.; Calzada-Wack, J.; Gollner, S.; Neff, F.; Schmidt, O.; Hensel, T.; Schirmer, D.; Fasan, A.; Esposito, I.; Muller-Tidow, C.; et al. The endochondral bone protein CHM1 sustains an undifferentiated, invasive phenotype, promoting lung metastasis in Ewing sarcoma. Mol. Oncol. 2017, 11, 1288–1301. [Google Scholar] [CrossRef]
- Garcia-Monclus, S.; Lopez-Alemany, R.; Almacellas-Rabaiget, O.; Herrero-Martin, D.; Huertas-Martinez, J.; Lagares-Tena, L.; Alba-Pavon, P.; Hontecillas-Prieto, L.; Mora, J.; de Alava, E.; et al. EphA2 receptor is a key player in the metastatic onset of Ewing sarcoma. Int. J. Cancer 2018, 143, 1188–1201. [Google Scholar] [CrossRef]
- Tanaka, K.; Kawano, M.; Itonaga, I.; Iwasaki, T.; Miyazaki, M.; Ikeda, S.; Tsumura, H. Tumor suppressive microRNA-138 inhibits metastatic potential via the targeting of focal adhesion kinase in Ewing’s sarcoma cells. Int. J. Oncol. 2016, 48, 1135–1144. [Google Scholar] [CrossRef]
- Vormoor, B.; Knizia, H.K.; Batey, M.A.; Almeida, G.S.; Wilson, I.; Dildey, P.; Sharma, A.; Blair, H.; Hide, I.G.; Heidenreich, O.; et al. Development of a preclinical orthotopic xenograft model of ewing sarcoma and other human malignant bone disease using advanced in vivo imaging. PLoS ONE 2014, 9, e85128. [Google Scholar] [CrossRef]
- Odri, G.; Kim, P.P.; Lamoureux, F.; Charrier, C.; Battaglia, S.; Amiaud, J.; Heymann, D.; Gouin, F.; Redini, F. Zoledronic acid inhibits pulmonary metastasis dissemination in a preclinical model of Ewing’s sarcoma via inhibition of cell migration. BMC Cancer 2014, 14, 169. [Google Scholar] [CrossRef]
- Bierbaumer, L.; Katschnig, A.M.; Radic-Sarikas, B.; Kauer, M.O.; Petro, J.A.; Hogler, S.; Gurnhofer, E.; Pedot, G.; Schafer, B.W.; Schwentner, R.; et al. YAP/TAZ inhibition reduces metastatic potential of Ewing sarcoma cells. Oncogenesis 2021, 10, 2. [Google Scholar] [CrossRef]
- Hayashi, M.; Chu, D.; Meyer, C.F.; Llosa, N.J.; McCarty, G.; Morris, C.D.; Levin, A.S.; Wolinsky, J.P.; Albert, C.M.; Steppan, D.A.; et al. Highly personalized detection of minimal Ewing sarcoma disease burden from plasma tumor DNA. Cancer 2016, 122, 3015–3023. [Google Scholar] [CrossRef]
- Hesketh, A.J.; Maloney, C.; Behr, C.A.; Edelman, M.C.; Glick, R.D.; Al-Abed, Y.; Symons, M.; Soffer, S.Z.; Steinberg, B.M. The Macrophage Inhibitor CNI-1493 Blocks Metastasis in a Mouse Model of Ewing Sarcoma through Inhibition of Extravasation. PLoS ONE 2015, 10, e0145197. [Google Scholar] [CrossRef]
- Tong, A.A.; Hashem, H.; Eid, S.; Allen, F.; Kingsley, D.; Huang, A.Y. Adoptive natural killer cell therapy is effective in reducing pulmonary metastasis of Ewing sarcoma. Oncoimmunology 2017, 6, e1303586. [Google Scholar] [CrossRef]
- Minas, T.Z.; Surdez, D.; Javaheri, T.; Tanaka, M.; Howarth, M.; Kang, H.J.; Han, J.; Han, Z.Y.; Sax, B.; Kream, B.E.; et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget 2017, 8, 34141–34163. [Google Scholar] [CrossRef]
- Leacock, S.W.; Basse, A.N.; Chandler, G.L.; Kirk, A.M.; Rakheja, D.; Amatruda, J.F. A zebrafish transgenic model of Ewing’s sarcoma reveals conserved mediators of EWS-FLI1 tumorigenesis. Dis. Model. Mech 2012, 5, 95–106. [Google Scholar] [CrossRef]
- Shukla, N.; Schiffman, J.; Reed, D.; Davis, I.J.; Womer, R.B.; Lessnick, S.L.; Lawlor, E.R. Biomarkers in Ewing Sarcoma: The Promise and Challenge of Personalized Medicine. A Report from the Children’s Oncology Group. Front. Oncol. 2013, 3, 141. [Google Scholar] [CrossRef]
- Grunewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef]
- Franzius, C.; Sciuk, J.; Brinkschmidt, C.; Jurgens, H.; Schober, O. Evaluation of chemotherapy response in primary bone tumors with F-18 FDG positron emission tomography compared with histologically assessed tumor necrosis. Clin. Nucl. Med. 2000, 25, 874–881. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Schuetze, S.M.; Butrynski, J.E.; Rajendran, J.G.; Vernon, C.B.; Conrad, E.U., 3rd; Eary, J.F. [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. J. Clin. Oncol. 2005, 23, 8828–8834. [Google Scholar] [CrossRef]
- Pfleiderer, C.; Zoubek, A.; Gruber, B.; Kronberger, M.; Ambros, P.F.; Lion, T.; Fink, F.M.; Gadner, H.; Kovar, H. Detection of tumour cells in peripheral blood and bone marrow from Ewing tumour patients by RT-PCR. Int. J. Cancer 1995, 64, 135–139. [Google Scholar] [CrossRef]
- West, D.C.; Grier, H.E.; Swallow, M.M.; Demetri, G.D.; Granowetter, L.; Sklar, J. Detection of circulating tumor cells in patients with Ewing’s sarcoma and peripheral primitive neuroectodermal tumor. J. Clin. Oncol. 1997, 15, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, I.; Cohen, I.J.; Stein, J.; Zilberstein, J.; Liberzon, E.; Atlas, O.; Grunshpan, A.; Sverdlov, Y.; Ash, S.; Zaizov, R.; et al. Tumor cells are present in stem cell harvests of Ewings sarcoma patients and their persistence following transplantation is associated with relapse. Pediatr. Blood Cancer 2004, 42, 404–409. [Google Scholar] [CrossRef] [PubMed]
- de Alava, E.; Lozano, M.D.; Patino, A.; Sierrasesumaga, L.; Pardo-Mindan, F.J. Ewing family tumors: Potential prognostic value of reverse-transcriptase polymerase chain reaction detection of minimal residual disease in peripheral blood samples. Diagn. Mol. Pathol. 1998, 7, 152–157. [Google Scholar] [CrossRef]
- Avigad, S.; Cohen, I.J.; Zilberstein, J.; Liberzon, E.; Goshen, Y.; Ash, S.; Meller, I.; Kollender, Y.; Issakov, J.; Zaizov, R.; et al. The predictive potential of molecular detection in the nonmetastatic Ewing family of tumors. Cancer 2004, 100, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, M.; Casini, B.; Mandoj, C.; Benini, S.; Alberti, L.; Novello, M.; Melucci, E.; Conti, L.; Covello, R.; Pescarmona, E.; et al. Precision diagnostics of Ewing’s sarcoma by liquid biopsy: Circulating EWS-FLI1 fusion transcripts. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef]
- Thomson, B.; Hawkins, D.; Felgenhauer, J.; Radich, J. RT-PCR evaluation of peripheral blood, bone marrow and peripheral blood stem cells in children and adolescents undergoing VACIME chemotherapy for Ewing’s sarcoma and alveolar rhabdomyosarcoma. Bone Marrow Transplant. 1999, 24, 527–533. [Google Scholar] [CrossRef][Green Version]
- Lee, S.Y.; Lim, S.; Cho, D.H. Personalized genomic analysis based on circulating tumor cells of extra-skeletal Ewing sarcoma of the uterus: A case report of a 16-year-old Korean female. Exp. Ther. Med. 2018, 16, 1343–1349. [Google Scholar] [CrossRef]
- Krumbholz, M.; Hellberg, J.; Steif, B.; Bauerle, T.; Gillmann, C.; Fritscher, T.; Agaimy, A.; Frey, B.; Juengert, J.; Wardelmann, E.; et al. Genomic EWSR1 Fusion Sequence as Highly Sensitive and Dynamic Plasma Tumor Marker in Ewing Sarcoma. Clin. Cancer Res. 2016, 22, 4356–4365. [Google Scholar] [CrossRef]
- Schmidkonz, C.; Krumbholz, M.; Atzinger, A.; Cordes, M.; Goetz, T.I.; Prante, O.; Ritt, P.; Schaefer, C.; Agaimy, A.; Hartmann, W.; et al. Assessment of treatment responses in children and adolescents with Ewing sarcoma with metabolic tumor parameters derived from (18)F-FDG-PET/CT and circulating tumor DNA. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1564–1575. [Google Scholar] [CrossRef]
- Vo, K.T.; Edwards, J.V.; Epling, C.L.; Sinclair, E.; Hawkins, D.S.; Grier, H.E.; Janeway, K.A.; Barnette, P.; McIlvaine, E.; Krailo, M.D.; et al. Impact of Two Measures of Micrometastatic Disease on Clinical Outcomes in Patients with Newly Diagnosed Ewing Sarcoma: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2016, 22, 3643–3650. [Google Scholar] [CrossRef]
- Shukla, N.N.; Patel, J.A.; Magnan, H.; Zehir, A.; You, D.; Tang, J.; Meng, F.; Samoila, A.; Slotkin, E.K.; Ambati, S.R.; et al. Plasma DNA-based molecular diagnosis, prognostication, and monitoring of patients with EWSR1 fusion-positive sarcomas. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- Klega, K.; Imamovic-Tuco, A.; Ha, G.; Clapp, A.N.; Meyer, S.; Ward, A.; Clinton, C.; Nag, A.; Van Allen, E.; Mullen, E.; et al. Detection of Somatic Structural Variants Enables Quantification and Characterization of Circulating Tumor DNA in Children With Solid Tumors. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef]
- Shulman, D.S.; Klega, K.; Imamovic-Tuco, A.; Clapp, A.; Nag, A.; Thorner, A.R.; Van Allen, E.; Ha, G.; Lessnick, S.L.; Gorlick, R.; et al. Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: A report from the Children’s Oncology Group. Br. J. Cancer 2018, 119, 615–621. [Google Scholar] [CrossRef]
- Mc Connell, L.; Gazdova, J.; Beck, K.; Srivastava, S.; Harewood, L.; Stewart, J.P.; Hubschmann, D.; Stenzinger, A.; Glimm, H.; Heilig, C.E.; et al. Detection of Structural Variants in Circulating Cell-Free DNA from Sarcoma Patients Using Next Generation Sequencing. Cancers 2020, 12, 3627. [Google Scholar] [CrossRef]
- Yu, M.; Wan, Y.F.; Zou, Q.H. Cell-free circulating mitochondrial DNA in the serum: A potential non-invasive biomarker for Ewing’s sarcoma. Arch. Med. Res. 2012, 43, 389–394. [Google Scholar] [CrossRef]
- Nie, C.L.; Ren, W.H.; Ma, Y.; Xi, J.S.; Han, B. Circulating miR-125b as a biomarker of Ewing’s sarcoma in Chinese children. Genet. Mol. Res. 2015, 14, 19049–19056. [Google Scholar] [CrossRef]
- Sciandra, M.; De Feo, A.; Parra, A.; Landuzzi, L.; Lollini, P.L.; Manara, M.C.; Mattia, G.; Pontecorvi, G.; Baricordi, C.; Guerzoni, C.; et al. Circulating miR34a levels as a potential biomarker in the follow-up of Ewing sarcoma. J. Cell Commun. Signal. 2020, 14, 335–347. [Google Scholar] [CrossRef]
- Miller, I.V.; Raposo, G.; Welsch, U.; Prazeres da Costa, O.; Thiel, U.; Lebar, M.; Maurer, M.; Bender, H.U.; von Luettichau, I.; Richter, G.H.; et al. First identification of Ewing’s sarcoma-derived extracellular vesicles and exploration of their biological and potential diagnostic implications. Biol. Cell 2013, 105, 289–303. [Google Scholar] [CrossRef]
- Samuel, G.; Crow, J.; Klein, J.B.; Merchant, M.L.; Nissen, E.; Koestler, D.C.; Laurence, K.; Liang, X.; Neville, K.; Staggs, V.; et al. Ewing sarcoma family of tumors-derived small extracellular vesicle proteomics identify potential clinical biomarkers. Oncotarget 2020, 11, 2995–3012. [Google Scholar] [CrossRef]
- de Groot, S.; Gelderblom, H.; Fiocco, M.; Bovee, J.V.; van der Hoeven, J.J.; Pijl, H.; Kroep, J.R. Serum levels of IGF-1 and IGF-BP3 are associated with event-free survival in adult Ewing sarcoma patients treated with chemotherapy. Onco. Targets Ther. 2017, 10, 2963–2970. [Google Scholar] [CrossRef][Green Version]
- Rutkowski, P.; Kaminska, J.; Kowalska, M.; Ruka, W.; Steffen, J. Cytokine and cytokine receptor serum levels in adult bone sarcoma patients: Correlations with local tumor extent and prognosis. J. Surg. Oncol. 2003, 84, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Lissat, A.; Joerschke, M.; Shinde, D.A.; Braunschweig, T.; Meier, A.; Makowska, A.; Bortnick, R.; Henneke, P.; Herget, G.; Gorr, T.A.; et al. IL6 secreted by Ewing sarcoma tumor microenvironment confers anti-apoptotic and cell-disseminating paracrine responses in Ewing sarcoma cells. BMC Cancer 2015, 15, 552. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Koefoed, P.; Hansen, T.; Stauffer, E.; Rauch, D.; Nielsen, F.C.; Rehfeld, J.F. Procholecystokinin as marker of human Ewing sarcomas. Clin. Cancer Res. 2004, 10, 5523–5530. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Katagiri, H.; Takahashi, M.; Ishida, Y.; Ono, A.; Takahashi, T.; Ohshima, K.; Mochizuki, T.; Urakami, K.; Muramatsu, K.; et al. ProGRP is a possible tumor marker for patients with Ewing sarcoma. Biomed. Res. 2015, 36, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Katagiri, H.; Takahashi, M.; Murata, H.; Wasa, J.; Hosaka, S.; Ishida, Y.; Ito, I.; Muramatsu, K.; Mochizuki, T.; et al. Pro-gastrin-releasing peptide as a marker for the Ewing sarcoma family of tumors. Int. J. Clin. Oncol. 2019, 24, 1468–1478. [Google Scholar] [CrossRef] [PubMed]








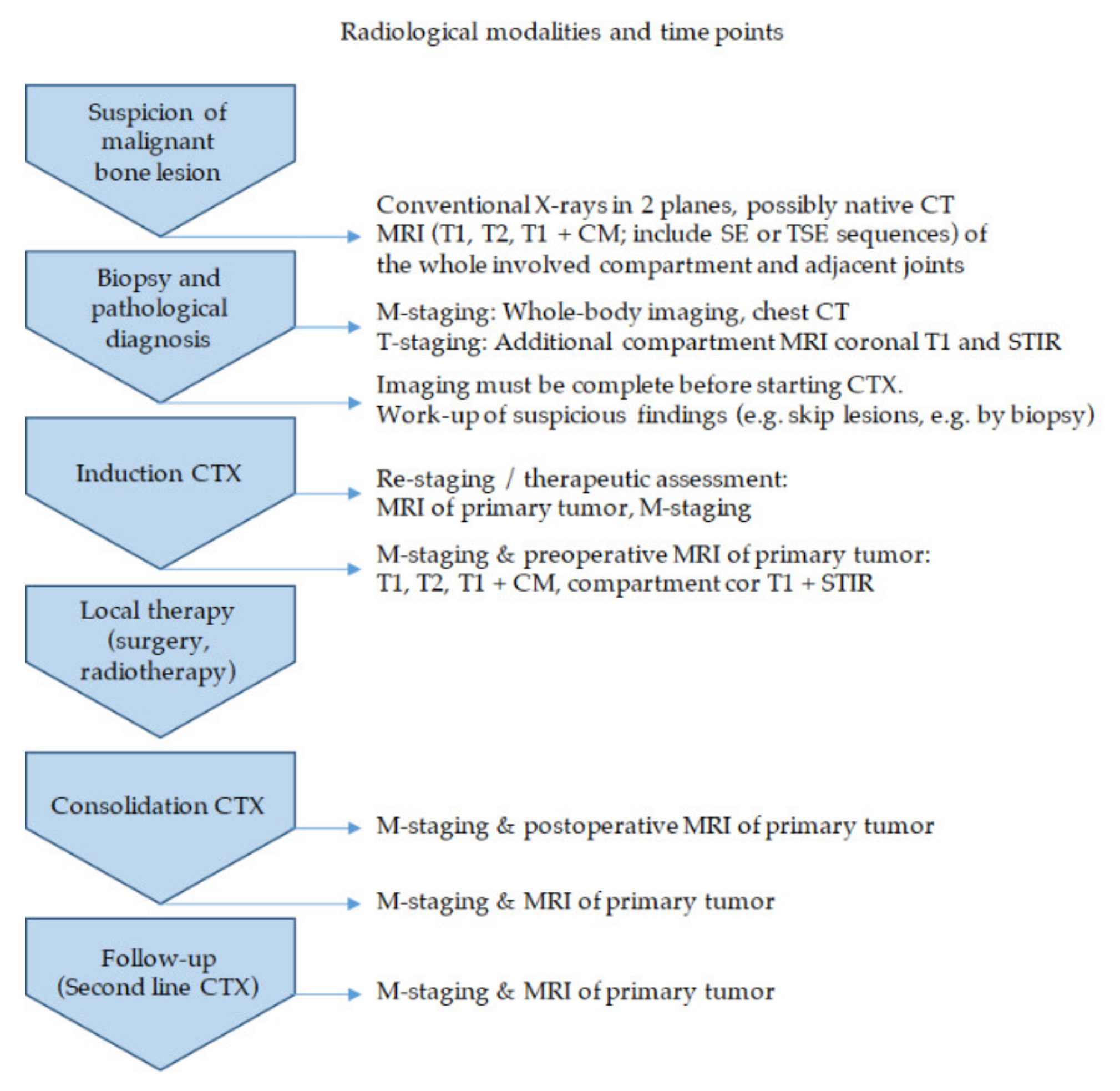{kind=link}
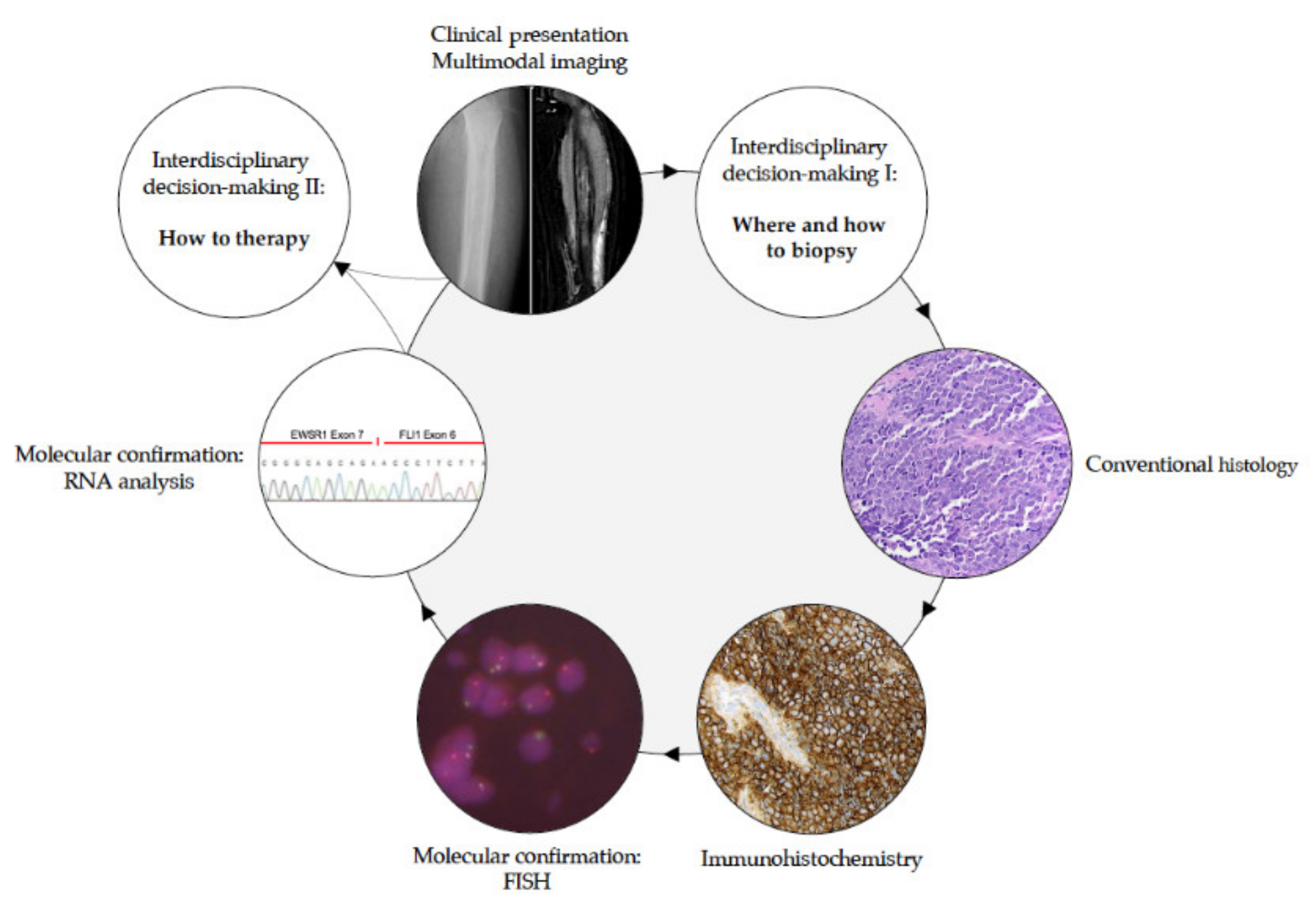{kind=link}
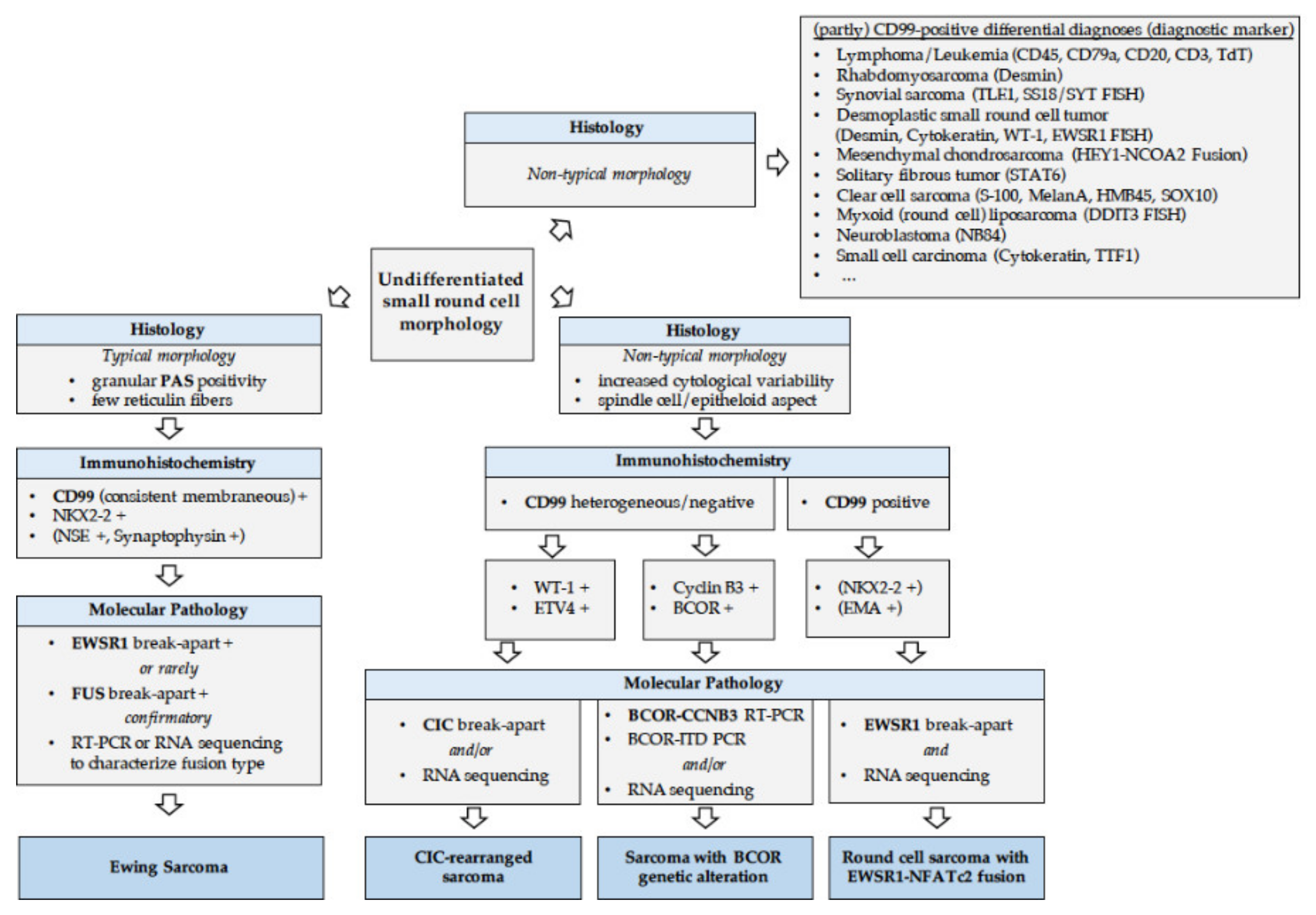{kind=link}
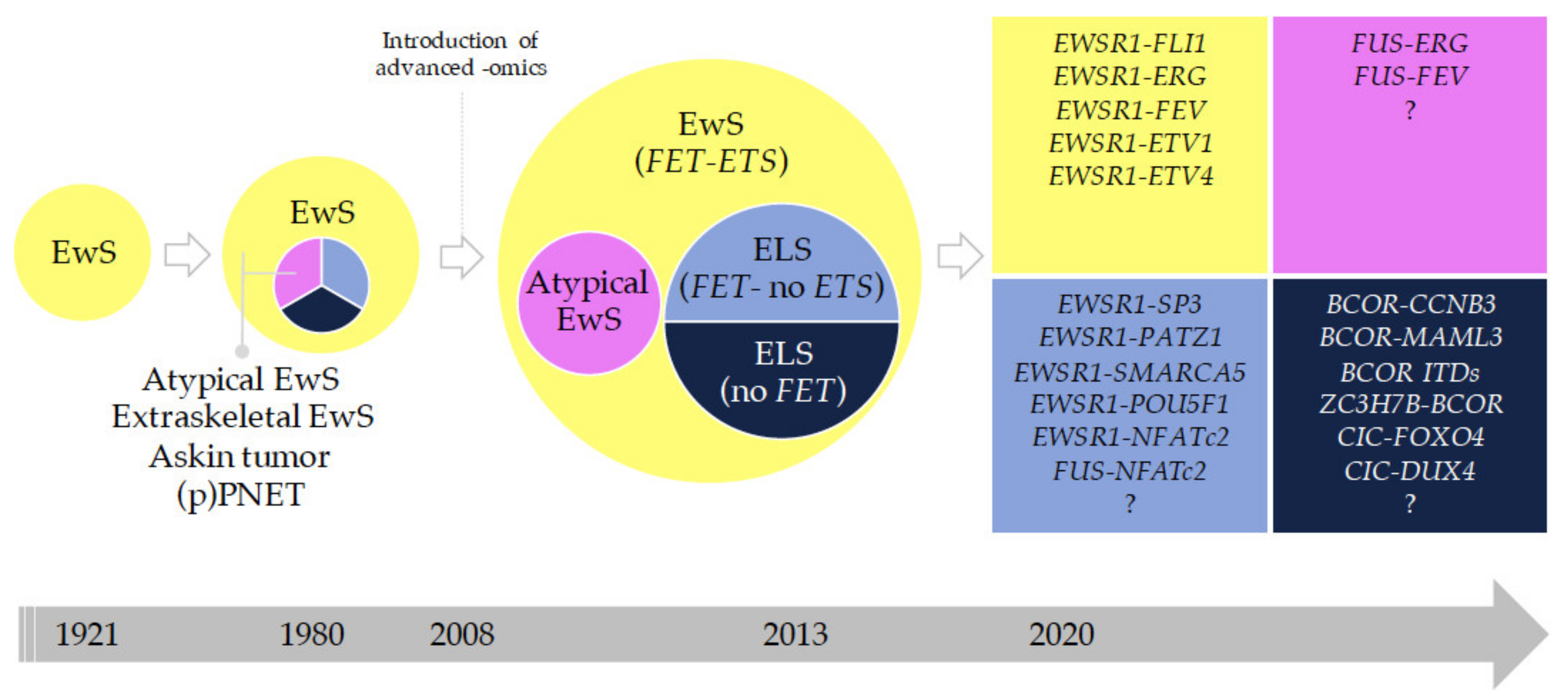{kind=link}
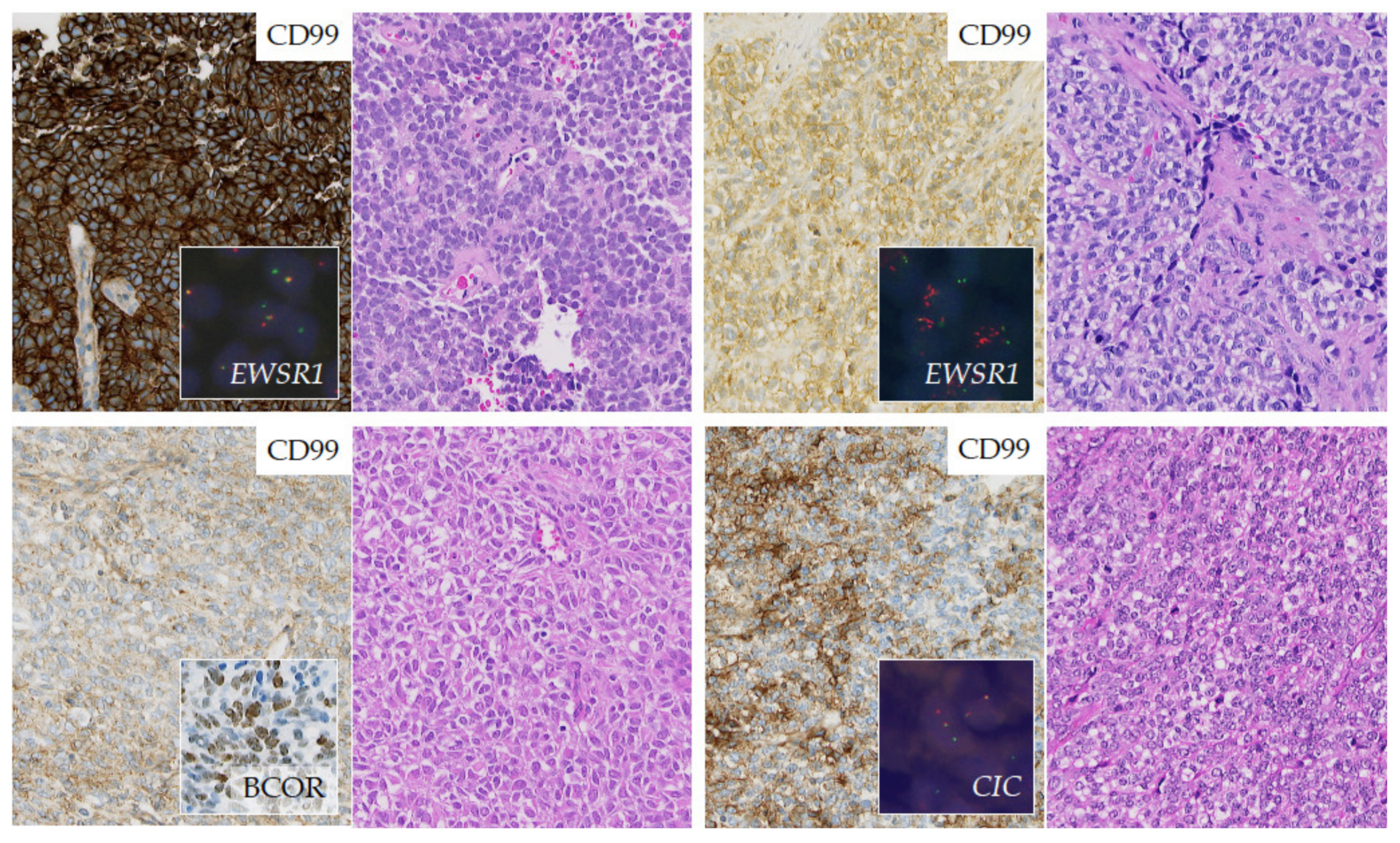{kind=link}
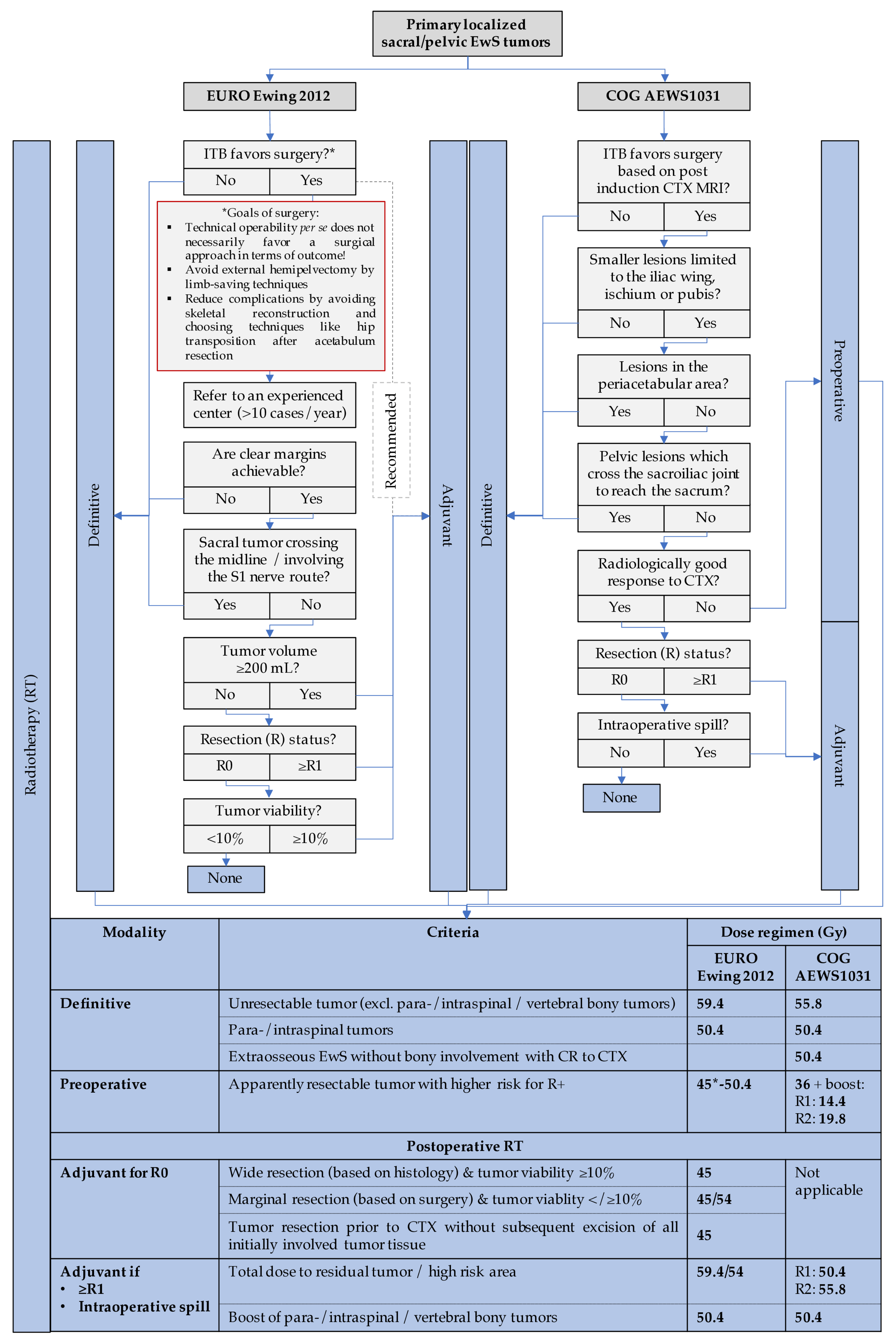{kind=link}
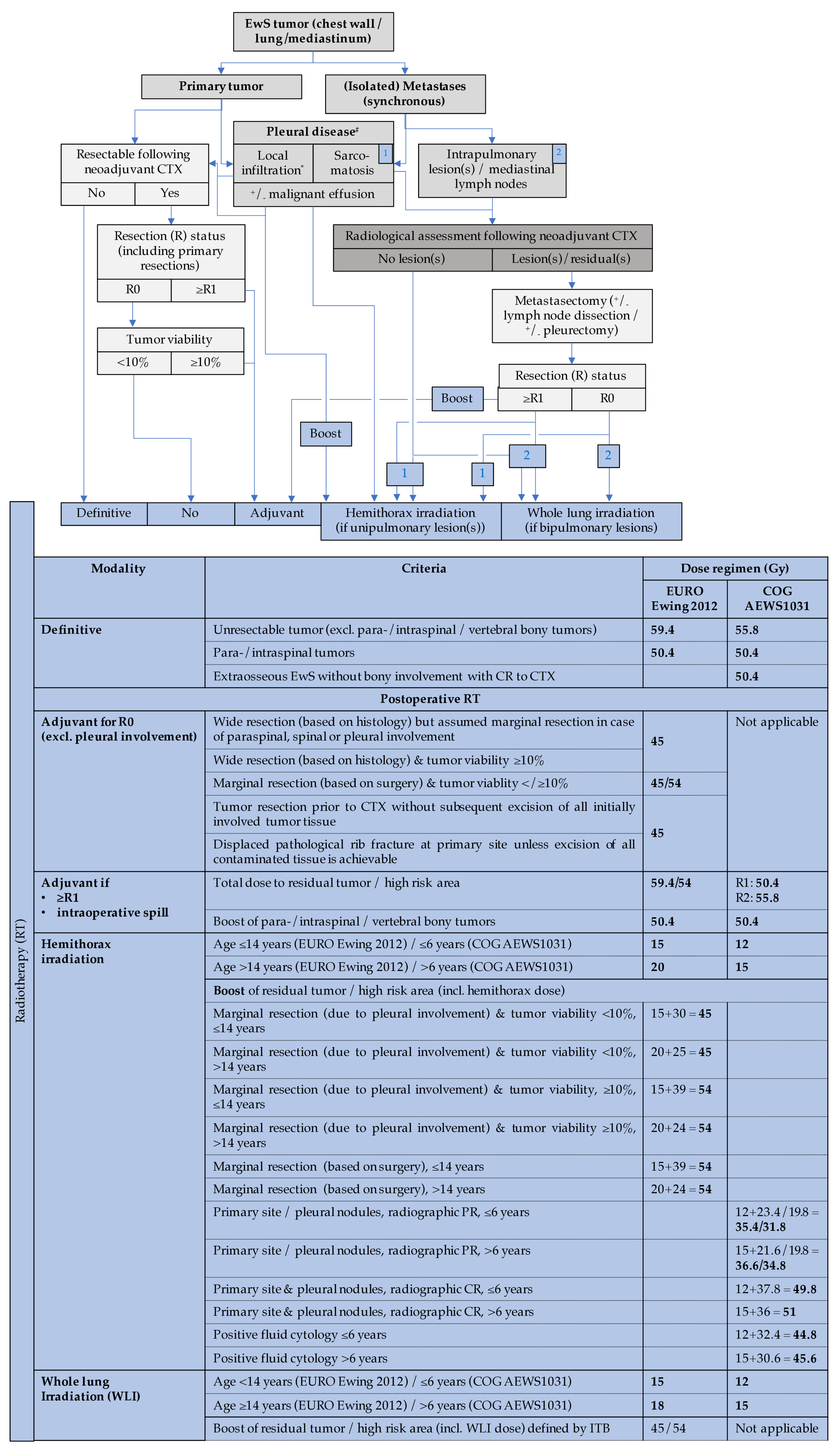{kind=link}
| Reference | Year of Publication | Chemotherapy Backbone Evaluated | Trial Name | Key Findings |
|---|---|---|---|---|
| [219] | 2003 | VDC vs. VDC/IE | INT-0091 | Five-year EFS improved from 54% to 69% with the addition of IE to VDC for patients with localized EwS |
| [222] | 2003 | P6 (VDC/IE with augmented Cy) | MSK | Four-year EFS 82% for patients with localized EwS |
| [105] | 2009 | VDC/IE with augmented alkylator dosing vs. standard VDC/IE | INT-0154 | No improvement in outcomes with alkylator dose intensification |
| [223] | 2012 | IC-VDC/IE | AEWS0031 | Six-year EFS improved to 73% from 65% with interval compressed chemotherapy for localized EwS |
| [7] | 2018 | VAI vs. VAI/HD-BuMel | Euro-E.W.I.N.G. 99 and Ewing 2008 | Eight-year EFS improved to 60.7% from 47.1% for localized high-risk EwS |
| [229] | 2019 | VDC/IE vs. VDC/IE/VTC | AEWS1031 | No benefit to the addition of VCT cycles for localized EwS |
| [232] | 2019 | VDC/IE vs. VDC/IE/ganitumab | AEWS1221 | No improvement in outcomes for metastatic EwS with the addition of ganitumab |
| [6] | 2019 | VIDE induction + VAI/VAC (or VIA/HD-BuMel) vs. VDC/IE induction + IE/VC (or VAI/HD-BuMel) | EURO Ewing 2012 | VDC/IE induction was found on preliminary analysis to have superior PFS and OS compared to VIDE induction |
| Reference | Year of Publication | Agents Evaluated | Key Findings |
|---|---|---|---|
| Trials of conventional chemotherapy for relapse | |||
| [236] | 2009 | High-dose ifosfamide | High-dose ifosfamide is active in relapsed EwS previously treated with standard-dose ifosfamide |
| [242] | 2006 | Topotecan/cyclophosphamide | 33% of patients with PR and 27% with SD |
| [243] | 2000 | Topotecan/high-dose cyclophosphamide | Responses seen in patients with EwS |
| [244] | 2004 | Irinotecan/temozolomide | Responses seen in patients with EwS |
| [245] | 2009 | Irinotecan/temozolomide | 63% ORR |
| [246] | 2012 | Gemcitabine/docetaxel | Limited activity seen in patients with EwS |
| [238] | 2020 | High-dose ifosfamide vs. topotecan/cyclophosphamide vs. irinotecan/temozolomide vs. gemcitabine/docetaxel | High-dose ifosfamide and topotecan/cyclophosphamide arms superior to irinotecan/temozolomide and gemcitabine/docetaxel. Enrollment is ongoing. |
| Trials of targeted therapies for relapse | |||
| [250] | 2017 | Regorafenib (REGO) | 10% objective response rate and a median PFS of 3.6 months |
| [251] | 2020 | Cabozantinib (CABONE) | 26% ORR |
| [252] | 2010 | Figitumumab | 2/16 patients with PR |
| [253] | 2011 | Figitumumab | 14% of patients with PR |
| [254] | 2011 | R1507 | 10% ORR |
| [255] | 2012 | Ganitumab | 6% ORR |
| [256] | 2014 | Olaparib | No patients with objective response |
| [257] | 2020 | Talazoparib/temozolomide | No patients with objective response, 15 with SD |
| [258] | 2020 | Talazoparib/irinotecan +/− temozolomide | 73% of patients had a clinical response (1 CR; 4 PR; 11 SD) |
| [259] | 2020 | TK216 | Two CR have been reported Enrollment is ongoing |
| [260] | 2020 | Seclidemstat | Enrollment is ongoing |
| Patient Cohort (Number of Patients) | Genes/Pathways/Cell Infiltration in Patient Tumors with Poor Prognosis | Reference | |
|---|---|---|---|
| Enriched | Downregulated | ||
| Localized tumors– non-progression (n = 7) vs. progression (n = 7) |
|
| [314] |
| Localized tumors – non-progression (n = 13) vs. progression (n = 17) additional 12 tumors for validation | ▪ MGST1 | [315] | |
| Metastatic and localized tumors (n = 27)- non-regression (n = 7) vs. regression (n = 20) by chemotherapy |
| [316] | |
| Primary tumors (n = 5) vs. unrelated metastasis samples (n = 6) | ▪ ICAM1 | [317] | |
| Primary tumors (n = 56; additional n = 39 as validation cohort)– non-survivors vs. survivors |
| [318] | |
| Primary tumors (n = 88; additional n = 57 as validation cohort)– relapse vs. relapse-free survival |
|
| [319] |
| Primary tumors (n = 197)– non-survivors vs. survivors |
|
| [320] |
| (Therapy naïve) Primary tumors (n = 27)– progression vs. non-progression |
| [321] | |
| Biomarker Characteristics | Patient Characteristics | Study Details | |||||
|---|---|---|---|---|---|---|---|
| Type | Category | Biomaterial | Number | Disease Status | Detection Rate (% Positive) | Conclusion | Reference |
| EwS-specific biomarkers | |||||||
| Circulating tumor cells | Diagnostic/ therapeutic | PB, BM | 16 | PB, BM at diagnosis (1 pts, 6 pts) BM during therapy (2 pts) | EwS cells in BM or PB can be identified by RT-PCR | [406] | |
| Diagnostic/ therapeutic | PB, BM | 28 | Primary and recurrent | PB, BM in non-metastatic pts (25) PB, BM in metastatic/ relapsed pts (50) | RT-PCR could serve as a EwS-specific marker of residual disease during CTX | [407] | |
| Diagnostic/ prognostic | Stem cell harvest; PB, BM | 11 | Stem cell harvest (100) | Number of cells may correlate with relapse after transplantation | [408] | ||
| Circulating tumor RNA (ctRNA) | Diagnostic/ prognostic | PB | 28 | 68 | Detection preceded progression; specific transcript types may affect progression | [409] | |
| Diagnostic/ prognostic | PB, BM | 26 | BM at diagnosis (43) PB, BM during follow up (58) | Occult tumor cells are strong predictors of recurrent disease in non-metastatic pts | [410] | ||
| Diagnostic/ therapeutic | Tissue, PB | 10 | Tissue (83), PB (100) | EWSR1-FLI1 molecular diagnosis possible in PB even in absence of tissue; ctRNA correlated with 18F-FDG-PET parameters | [411] | ||
| Circulating tumor cells + circulating tumor RNA | Diagnostic/ therapeutic | PB, BM, PBSC | 12 | Metastatic | PBSC (2.5) | RT-PCR signal declines in PB and BM during CTX | [412] |
| Diagnostic | PB | 1 | [413] | ||||
| Circulating tumor DNA (by EWSR1-FLI1 ctDNA PCR) | Diagnostic/ therapeutic | 1 | ddPCR to detect ctDNA could serve as a EwS-specific marker of recurrence | [397] | |||
| Diagnostic/ therapeutic | PB | 20 | Localized and metastatic | Kinetics of EWSR1-FLI1 ctDNA correlated with tumor volume | [414] | ||
| Diagnostic/ therapeutic | PB | 20 | 100 | Combination of 18F-FDG-PET/CT and ctDNA quantification could serve as a EwS-specific marker for CTX response and relapse | [415] | ||
| Circulating tumor cells + ctDNA PCR | Diagnostic/ prognostic | PB, BM | Flow cytometry (109) PCR (225) | Flow cytometry (CD99(+), CD45(-)) (12.8) PCR (19.6) | Detection of micrometastatic disease by flow cytometry or RT-PCR is not associated with outcome | [416] | |
| Circulating tumor DNA (by WGS) | Diagnostic | Tissue, PB | 11 | WGS (100) | ctDNA by both NGS and ddPCR could serve as a EwS-specific marker | [417] | |
| Diagnostic/ therapeutic/ prognostic | PB | 11 | ctDNA levels corresponded to CTX response | [418] | |||
| Diagnostic/ prognostic | PB | 94 | ctDNA in localized pts (53) | ctDNA detection associated with inferior outcomes | [419] | ||
| Diagnostic | Tissue, PB | 2 | [420] | ||||
| Circulating cell-free mitochondrial DNA (ccf mtDNA) | Diagnostic/ prognostic | PB | 25 | ccf mtDNA levels associated with metastatic disease | [421] | ||
| MicroRNA– miR-125b | Diagnostic/ therapeutic | PB | 63 | miR-125b elevated in pts compared to healthy controls; miR-125b downregulation correlated with poor response to CTX | [422] | ||
| MicroRNA– miR34a | Diagnostic/ therapeutic/ prognostic | PB | 31 | Localized and metastatic | High miR34a inversely correlated with tumor volume; miR34a elevated in localized compared to metastatic pts; miR34a increased in localized pts at diagnosis and after end of CTX | [423] | |
| Extracellular vesicles (EV)– EwS-specific transcripts | Diagnostic | Preclinical model | EVs could serve as a EwS-specific marker | [424] | |||
| Extracellular vesicles (EV)– EwS soluble EV proteome | Diagnostic/ prognostic | PB | 10 | Localized and metastatic | [425] | ||
| EwS-non-specific biomarkers (cytokines and other secreted peptides) | |||||||
| IGF-1 and IGF-BP3 | Prognostic | PB | 22 | Localized and metastatic | High baseline IGF-1 and IGF-BP3 associated with improved EFS; IGF-BP3 and IGF-2 increased during CTX | [426] | |
| IL-6 and IL-8 | Diagnostic/ therapeutic/ prognostic | PB | 13 | Localized | [427] | ||
| Diagnostic/ prognostic | Tissue, PB | 14 | IL-6 elevated in some pts with poor prognosis | [428] | |||
| (Pro)cholecystokinin ((pro)CCK) | Diagnostic/ therapeutic/ prognostic | Tissue, PB | 12 | Primary and recurrent | ProCCK elevated in pts at primary Dx/ recurrence compared to pts during CTX; ProCCK correlated with tumor size | [429] | |
| Pro-gastrin-releasing peptide (ProGRP) and neuron- specific enolase (NSE) | Diagnostic | Tissue, PB | 9 | ProGRP could serve as a EwS-specific marker | [430] | ||
| Diagnostic/ prognostic | PB | 16 | ProGRP elevated in half of the pts; ProGRP reflected therapeutic response | [431] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zöllner, S.K.; Amatruda, J.F.; Bauer, S.; Collaud, S.; de Álava, E.; DuBois, S.G.; Hardes, J.; Hartmann, W.; Kovar, H.; Metzler, M.; et al. Ewing Sarcoma—Diagnosis, Treatment, Clinical Challenges and Future Perspectives. J. Clin. Med. 2021, 10, 1685. https://doi.org/10.3390/jcm10081685
Zöllner SK, Amatruda JF, Bauer S, Collaud S, de Álava E, DuBois SG, Hardes J, Hartmann W, Kovar H, Metzler M, et al. Ewing Sarcoma—Diagnosis, Treatment, Clinical Challenges and Future Perspectives. Journal of Clinical Medicine. 2021; 10(8):1685. https://doi.org/10.3390/jcm10081685
Chicago/Turabian StyleZöllner, Stefan K., James F. Amatruda, Sebastian Bauer, Stéphane Collaud, Enrique de Álava, Steven G. DuBois, Jendrik Hardes, Wolfgang Hartmann, Heinrich Kovar, Markus Metzler, and et al. 2021. "Ewing Sarcoma—Diagnosis, Treatment, Clinical Challenges and Future Perspectives" Journal of Clinical Medicine 10, no. 8: 1685. https://doi.org/10.3390/jcm10081685
APA StyleZöllner, S. K., Amatruda, J. F., Bauer, S., Collaud, S., de Álava, E., DuBois, S. G., Hardes, J., Hartmann, W., Kovar, H., Metzler, M., Shulman, D. S., Streitbürger, A., Timmermann, B., Toretsky, J. A., Uhlenbruch, Y., Vieth, V., Grünewald, T. G. P., & Dirksen, U. (2021). Ewing Sarcoma—Diagnosis, Treatment, Clinical Challenges and Future Perspectives. Journal of Clinical Medicine, 10(8), 1685. https://doi.org/10.3390/jcm10081685