
Real-World Outcomes of Ivacaftor Treatment in People with Cystic Fibrosis: A Systematic Review

Abstract
1. Introduction
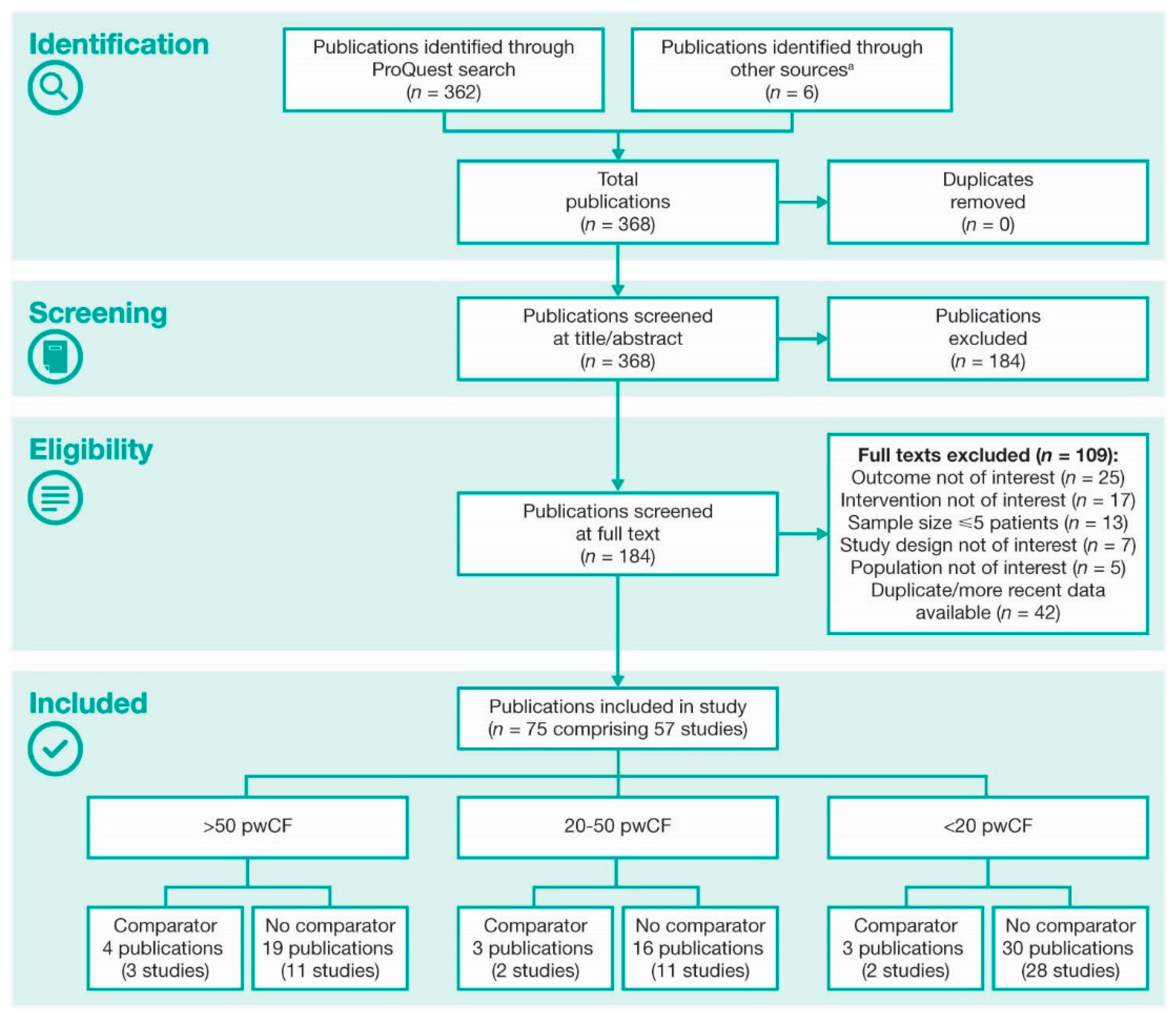
2. Methods
3. Results
3.1. Demographics, Patient Populations, and Study Designs
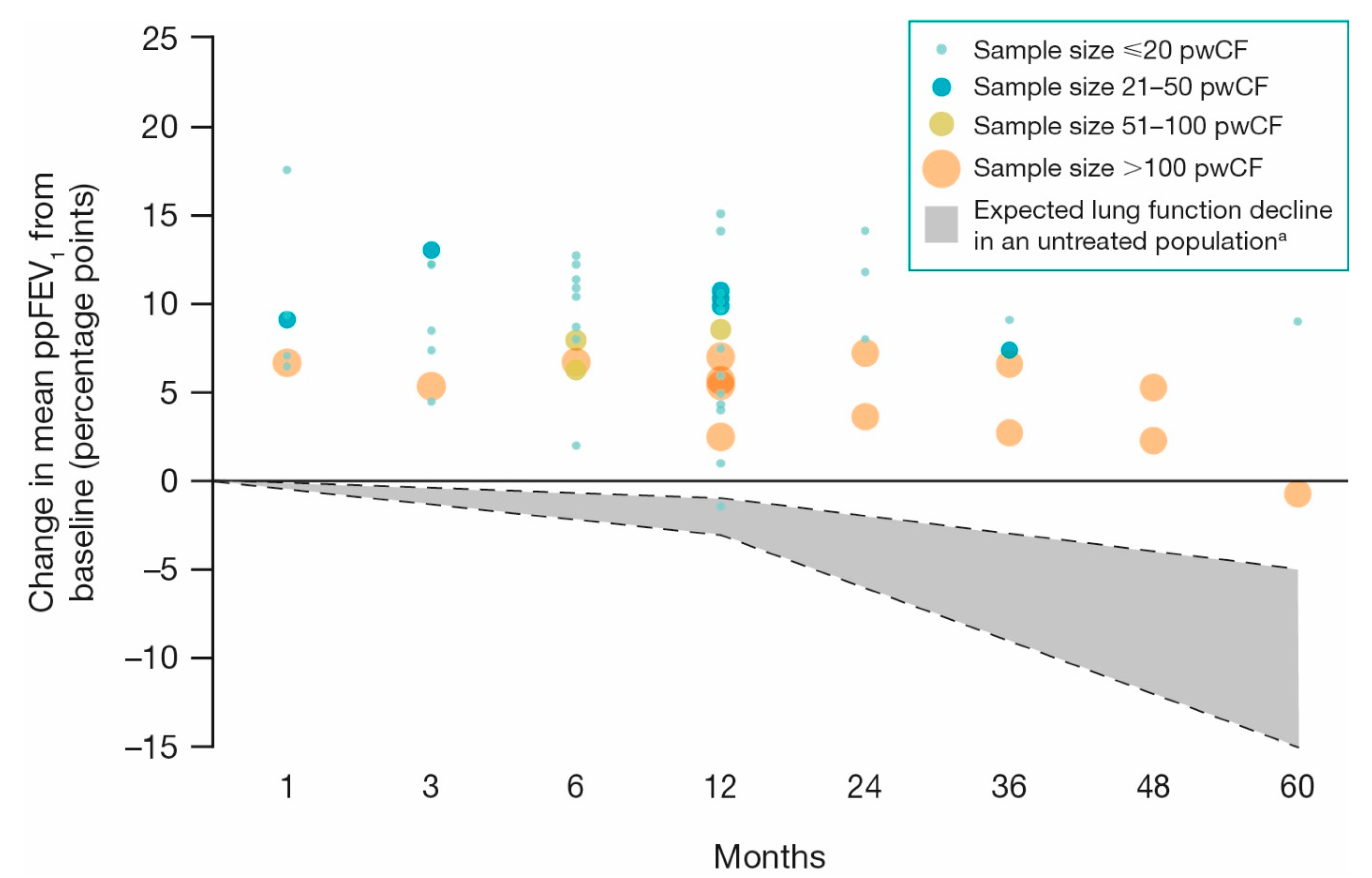
3.2. Lung Function and Pulmonary Exacerbations
3.2.1. ppFEV1
3.2.2. PEx
3.3. Nutritional Parameters
3.4. CFRD and Exocrine Pancreas
3.4.1. Exocrine Pancreas
3.4.2. CFRD
3.5. Healthcare Utilization
3.5.1. All-Cause Hospitalizations
3.5.2. PEx-Related Hospitalizations
3.5.3. Antibiotic Utilization and Duration
3.6. Lung Microbiome
3.6.1. Pseudomonas aeruginosa
3.6.2. Staphylococcus aureus
3.6.3. Other
3.7. PROs
3.7.1. CFQ and CFQ-R Respiratory Symptom Scores
3.7.2. Other PROs
3.8. Adverse Events
3.9. Other CF-Related Complications
3.10. Organ Transplants
3.11. Mortality
4. Discussion
4.1. Clinical Implications
4.2. Limitations
4.3. Future Research Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelly, J. Environmental scan of cystic fibrosis research worldwide. J. Cyst. Fibros. 2017, 16, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; White, R.; Tobin, P. Keep them breathing: Cystic fibrosis pathophysiology, diagnosis, and treatment. JAAPA 2017, 30, 23–27. [Google Scholar] [CrossRef]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Liou, T.G.; Elkin, E.P.; Pasta, D.J.; Jacobs, J.R.; Konstan, M.W.; Morgan, W.J.; Wagener, J.S. Year-to-year changes in lung function in individuals with cystic fibrosis. J. Cyst. Fibros. 2010, 9, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Bonafede, M.M.; Limone, B.L.; Hodgkins, P.; Sawicki, G.S. The burden of cystic fibrosis in the Medicaid population. Clinicoecon. Outcomes Res. 2018, 10, 423–431. [Google Scholar] [CrossRef]
- Sanders, D.B.; Bittner, R.C.L.; Rosenfeld, M.; Hoffman, L.R.; Redding, G.J.; Goss, C.H. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am. J. Respir. Crit. Care Med. 2010, 182, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.L.; Jacobs, D.R., Jr.; Milla, C.E. Longitudinal changes in growth parameters are correlated with changes in pulmonary function in children with cystic fibrosis. Pediatrics 2003, 112, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Zemel, B.S.; Jawad, A.F.; FitzSimmons, S.; Stallings, V.A. Longitudinal relationship among growth, nutritional status, and pulmonary function in children with cystic fibrosis: Analysis of the Cystic Fibrosis Foundation National CF Patient Registry. J. Pediatr. 2000, 137, 374–380. [Google Scholar] [CrossRef]
- Brennan, A.L.; Beynon, J. Clinical updates in cystic fibrosis-related diabetes. Semin. Respir. Crit. Care Med. 2015, 36, 236–250. [Google Scholar] [CrossRef]
- Hassan, M.; Bonafede, M.; Limone, B.; Hodgkins, P.; Sawicki, G.S. The burden of cystic fibrosis: Pulmonary exacerbations and healthcare resource utilization in a commercially insured US population. Poster presented at ISPOR 21st Annual International Meeting, Washington, DC, USA, 21–25 May 2016. [Google Scholar]
- Liou, T.G.; Adler, F.R.; FitzSimmons, S.C.; Cahill, B.C.; Hibbs, J.R.; Marshall, B.C. Predictive 5-year survivorship model of cystic fibrosis. Am. J. Epidemiol. 2001, 153, 345–352. [Google Scholar] [CrossRef]
- Liou, T.G.; Kartsonaki, C.; Keogh, R.H.; Adler, F.R. Evaluation of a five-year predicted survival model for cystic fibrosis in later time periods. Sci. Rep. 2020, 10, 6602. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation. 2018 Patient Registry: Annual Data Report. Available online: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2018-Patient-Registry-Annual-Data-Report.pdf (accessed on 17 December 2020).
- European Cystic Fibrosis Society. ECFS Patient Registry: Annual Data Report. Available online: https://www.ecfs.eu/sites/default/files/general-content-images/working-groups/ecfs-patient-registry/ECFSPR_Report2017_v1.3.pdf (accessed on 9 July 2020).
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Vertex Pharmaceuticals (Europe) Limited. Kalydeco 150 mg film-coated tablets SmPC. Available online: https://www.medicines.org.uk/emc/product/3040/smpc (accessed on 17 December 2020).
- Vertex Pharmaceuticals Incorporated. Kalydeco PI. Available online: https://www.drugs.com/pro/kalydeco.html. (accessed on 17 December 2020).
- McKone, E.F.; Borowitz, D.; Drevinek, P.; Griese, M.; Konstan, M.W.; Wainwright, C.; Ratjen, F.; Sermet-Gaudelus, I.; Plant, B.; Munck, A.; et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: A phase 3, open-label extension study (PERSIST). Lancet Respir. Med. 2014, 2, 902–910. [Google Scholar] [CrossRef]
- Davies, J.C.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Robertson, S.; Green, Y.; Cooke, J.; et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir. Med. 2016, 4, 107–115. [Google Scholar] [CrossRef]
- Rosenfeld, M.; Wainwright, C.E.; Higgins, M.; Wang, L.T.; McKee, C.; Campbell, D.; Tian, S.; Schneider, J.; Cunningham, S.; Davies, J.C.; et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): A phase 3 single-arm study. Lancet Respir. Med. 2018, 6, 545–553. [Google Scholar] [CrossRef]
- Davies, J.; Wang, L.T.; Panorchan, P.; Campbell, D.; Tian, S.; Higgins, M.; Egbuna, O.; McKee, C.; Rosenfeld, M.; on behalf of the ARRIVAL Study Group. WS06-4 Ivacaftor (IVA) treatment in patients 6 to <12 months old with cystic fibrosis with a CFTR gating mutation: Results of a 2-part, single-arm, phase 3 study. J. Cyst. Fibros. 2019, 18, S11. [Google Scholar] [CrossRef]
- Davies, J.C.; Wainwright, C.E.; Sawicki, G.S.; Higgins, M.N.; Campbell, D.; Harris, C.; Panorchan, P.; Haseltine, E.; Tian, S.; Rosenfeld, M.; on behalf of the ARRIVAL Study Group. Ivacaftor in infants aged 4 to <12 months with cystic fibrosis and a gating mutation. Results of a two-part phase 3 clinical trial. Am. J. Respir. Crit. Care Med. 2021, 203, 585–593. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef]
- Moss, R.B.; Flume, P.A.; Elborn, J.S.; Cooke, J.; Rowe, S.M.; McColley, S.A.; Rubenstein, R.C.; Higgins, M.; on behalf of the VX11-770-110 (KONDUCT) Study Group. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: A double-blind, randomised controlled trial. Lancet Respir. Med. 2015, 3, 524–533. [Google Scholar] [CrossRef]
- Nick, J.A.; St Clair, C.; Jones, M.C.; Lan, L.; Higgins, M.; VX12-770-113 Study Team. Ivacaftor in cystic fibrosis with residual function: Lung function results from an N-of-1 study. J. Cyst. Fibros. 2020, 19, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; Mainz, J.G.; MacGregor, G.; Madge, S.; Macey, J.; Fridman, M.; Suthoff, E.D.; Narayanan, S.; Kinnman, N. Patient-reported outcomes in patients with cystic fibrosis with a G551D mutation on ivacaftor treatment: Results from a cross-sectional study. BMC Pulm. Med. 2019, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Bessonova, L.; Volkova, N.; Higgins, M.; Bengtsson, L.; Tian, S.; Simard, C.; Konstan, M.W.; Sawicki, G.S.; Sewall, A.; Nyangoma, S.; et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax 2018, 73, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Volkova, N.; Moy, K.; Evans, J.; Campbell, D.; Tian, S.; Simard, C.; Higgins, M.; Konstan, M.W.; Sawicki, G.S.; Elbert, A.; et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J. Cyst. Fibros. 2020, 19, 68–79. [Google Scholar] [CrossRef]
- Frost, F.J.; Nazareth, D.S.; Charman, S.C.; Winstanley, C.; Walshaw, M.J. Ivacaftor is associated with reduced lung infection by key cystic fibrosis pathogens: A cohort study using national registry data. Ann. Am. Thorac. Soc. 2019, 16, 1375–1382. [Google Scholar] [CrossRef]
- Barry, P.J.; Plant, B.J.; Nair, A.; Bicknell, S.; Simmonds, N.J.; Bell, N.J.; Shafi, N.T.; Daniels, T.; Shelmerdine, S.; Felton, I.; et al. Effects of ivacaftor in patients with cystic fibrosis who carry the G551D mutation and have severe lung disease. Chest 2014, 146, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.J.; Plant, B.J.; Simmonds, N.J.; Bicknell, S.; Bell, N.J.; Shafi, N.T.; Daniels, T.; Gunaratnam, C.; Horsley, A.; Jones, A.M. Ivacaftor decreases mortality in G551D patients with severe lung disease. Pediatr. Pulmonol. 2015, 50, 275–276. [Google Scholar] [CrossRef][Green Version]
- Emery, J.; Mullane, D.; Chroinin, M.N. The effects of ivacaftor on pancreatic function in paediatric patients with cystic fibrosis gating mutations. Arch. Dis. Child. 2019, 104, A149–A150. [Google Scholar] [CrossRef]
- McLearn-Montz, A.J.; Singh, S.B.; Larson Ode, K.; Fischer, A.J. Linear growth in children receiving ivacaftor or ivacaftor-lumacaftor for cystic fibrosis. Pediatr. Pulmonol. 2018, 53, 386. [Google Scholar] [CrossRef]
- Wainwright, C.; Bell, S.; Morton, J.; Ryan, G.; Serisier, D.; Bye, P.; Mulrennan, S.; Daley, C.; Greville, H. The effect of ivacaftor in individuals with cystic fibrosis and severe lung disease: Analysis of data from the Australian named patient programme. Respirology 2014, 19, 112. [Google Scholar] [CrossRef]
- Wainwright, C.; Bell, S.; Morton, J.; Ryan, G.; Serisier, D.; Greville, H.; Bye, P.; Mulrennan, S.; Daley, C. The effect of ivacaftor in individuals with CF and severe lung disease. Pediatr. Pulmonol. 2014, 49, 376–377. [Google Scholar] [CrossRef]
- Bonafede, M.M.; Limone, B.L.; Suthoff, E.D.; Cahill, J.R. Health resource utilization among patients with cystic fibrosis who initiate ivacaftor treatment. Pediatr. Pulmonol. 2014, 49, 287. [Google Scholar] [CrossRef]
- Castellani, C.; Colombo, C.; van der Ent, C.K.; Simmonds, N.J.; Kinnman, N.; Hassan, M.; DeSouza, C.; Kaviya, A. An observational study of ivacaftor in patients with cystic fibrosis and selected non-G551D gating mutations in UK, Italy and Netherlands: Healthcare resource utilization from the first interim analysis of the vocal study. Value Health 2018, 21, S255. [Google Scholar] [CrossRef]
- Castellani, C.; Colombo, C.; van der Ent, C.K.; Simmonds, N.J.; Kinnman, N.; Hassan, M.; DeSouza, C.; Kaviya, A. Clinical effectiveness results from the first interim analysis of the VOCAL study; an observational study of ivacaftor in patients with cystic fibrosis and selected non-G551D gating mutations. J. Cyst. Fibros. 2018, 17, S10. [Google Scholar] [CrossRef]
- Guimbellot, J.S.; Baines, A.; Khan, U.; Heltshe, S.; VanDalfsen, J.; Jain, M.; Rowe, S.M.; Sagel, S. Long term effects of ivacaftor in G551D patients: Five year follow-up data in GOAL-e2. Pediatr. Pulmonol. 2018, 53, 232. [Google Scholar] [CrossRef]
- Guimbellot, J.; Solomon, G.M.; Baines, A.; Heltshe, S.L.; VanDalfsen, J.; Joseloff, E.; Sagel, S.D.; Rowe, S.M. Effectiveness of ivacaftor in cystic fibrosis patients with non-G551D gating mutations. J. Cyst. Fibros. 2019, 18, 102–109. [Google Scholar] [CrossRef]
- Hathorne, H.; Brand, K.M.; Britton, L.J.; Jackson, T.; Heltshe, S.; Kohler, C.; Cheong, J.; Harrington, K.; Sorscher, E.J.; Rowe, S.M. The investigation of quality of life and adherence in patients with the G551D mutation receiving ivacaftor therapy. Pediatr. Pulmonol. 2015, 50, 427. [Google Scholar] [CrossRef][Green Version]
- Heltshe, S.L.; Mayer-Hamblett, N.; Burns, J.L.; Khan, U.; Baines, A.; Ramsey, B.W.; Rowe, M.; on behalf of the GOAL (the G551D Observation-AL). Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin. Infect. Dis. 2015, 60, 703–712. [Google Scholar] [CrossRef]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef]
- Sagel, S.D.; Heltshe, S.L.; Khan, U.; VanDalfsen, J.; Joseloff, E.; Rowe, S.M. Effect of ivacaftor in R117H patients following FDA approval: Early results of the G551D Observational-expanded and Extended (GOAL-E2) study. Pediatr. Pulmonol. 2015, 50, 261. [Google Scholar] [CrossRef][Green Version]
- van de Peppel, I.P.; Doktorova, M.; Berkers, G.; de Jonge, H.R.; Houwen, R.H.J.; Verkade, H.J.; Jonker, J.W.; Bodewes, F.A.J.A. IVACAFTOR restores FGF19 regulated bile acid homeostasis in cystic fibrosis patients with an S1251N or a G551D gating mutation. J. Cyst. Fibros. 2019, 18, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.B.; Grosse, S.D.; Green, R.F.; Fink, A.K.; Sawicki, G.S. Precision medicine in action: The impact of ivacaftor on cystic fibrosis-related hospitalizations. Health Aff. (Millwood) 2018, 37, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.; Sawicki, G.S.; Morgan, W.J.; Schechter, M.S.; Rosenfeld, M.; Marshall, B.C. Treatment response to ivacaftor in clinical practice: Analysis of the US CF foundation patient registry. Pediatr. Pulmonol. 2015, 50, 361. [Google Scholar] [CrossRef][Green Version]
- Hassan, M.; Bonafede, M.M.; Limone, B.L.; Hodgkins, P.; Suthoff, E.D.; Sawicki, G. Reduction in pulmonary exacerbations (PEx) after initiation of ivacaftor: A retrospective cohort study among patients with cystic fibrosis (CF) treated in real-world settings. J. Cyst. Fibros. 2016, 15, S58. [Google Scholar] [CrossRef]
- Hubert, D.; Dehillotte, C.; Munck, A.; David, V.; Baek, J.; Mely, L.; Dominique, S.; Ramel, S.; Danner Boucher, I.; Lefeuvre, S.; et al. Retrospective observational study of French patients with cystic fibrosis and a Gly551Asp-CFTR mutation after 1 and 2 years of treatment with ivacaftor in a real-world setting. J. Cyst. Fibros. 2018, 17, 89–95. [Google Scholar] [CrossRef]
- Hubert, D.; Fajac, I.; Munck, A.; Marguet, C.; Benichou, J.; Payen-Champenois, C.; Jha, L.; Hassan, M.; DeSouza, C.; Kinnman, N. An observational study of ivacaftor in patients with cystic fibrosis in France: First interim analysis of health care resource utilization from the BRIO study. Value Health 2018, 21, S342. [Google Scholar] [CrossRef]
- Hubert, D.; Fajac, I.; Munck, A.; Marguet, C.; Benichou, J.; Payen-Champenois, C.; Kaviya, A.; Hassan, M.; DeSouza, C.; Kinnman, N.; et al. Clinical effectiveness from the first interim analysis of the BRIO study: An observational study of cystic fibrosis patients treated with ivacaftor in France. Pediatr. Pulmonol. 2018, 53, 160. [Google Scholar] [CrossRef]
- Kirwan, L.; Fletcher, G.; Harrington, M.; Jeleniewska, P.; Zhou, S.; Casserly, B.; Gallagher, C.G.; Greally, P.; Gunaratnam, C.; Herzig, M.; et al. Longitudinal trends in real-world outcomes after initiation of ivacaftor: A cohort study from the Cystic Fibrosis Registry of Ireland. Ann. Am. Thorac. Soc. 2019, 16, 209–216. [Google Scholar] [CrossRef]
- Newsome, S.J.; Keogh, R.H.; Daniel, R.M.; CF-EpiNet. The effects of 3-year ivacaftor use on lung function and intravenous days seen in UK CF Registry Data. J. Cyst. Fibros. 2018, 17, S54. [Google Scholar] [CrossRef]
- Suthoff, E.D.; Bonafede, M.; Limone, B.; O’Callaghan, L.; Sawicki, G.S.; Wagener, J.S. Healthcare resource utilization associated with ivacaftor use in patients with cystic fibrosis. J. Med. Econ. 2016, 19, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Al Redha, K.Y.; Shebani, E.; Al Nuaimi, A.; Panickar, J. Comparison of FEV1 and BMI of the 4 common CF mutations in UAE. J. Cyst. Fibros. 2016, 15, S118. [Google Scholar] [CrossRef]
- Barry, P.J.; Jones, A.M.; Webb, A.K.; Horsley, A.R. Sweat chloride is not a useful marker of clinical response to Ivacaftor. Thorax 2014, 69, 586–587. [Google Scholar] [CrossRef][Green Version]
- Barry, P.J.; Banerjee, A.; Horsley, A.; Brennan, A.L. Impact of ivacaftor on glycaemic health in patients carrying the G551D mutation. J. Cyst. Fibros. 2015, 14, S104. [Google Scholar] [CrossRef]
- Banerjee, A.; Brennan, A.L.; Horsley, A.R.; Barry, P.J. Prospective examination of the effects of ivacaftor on glycaemic health. Thorax 2014, 69, A162. [Google Scholar] [CrossRef]
- Chassagnon, G.; Hubert, D.; Fajac, I.; Burgel, P.R.; Revel, M.P.; on behalf of the investigators. Long-term computed tomographic changes in cystic fibrosis patients treated with ivacaftor. Eur. Respir. J. 2016, 48, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Corvol, H.; Mésinèle, J.; Douksieh, I.H.; Strug, L.J.; Boëlle, P.Y.; Guillot, L. SLC26A9 gene is associated with lung function response to ivacaftor in patients with cystic fibrosis. Front. Pharmacol. 2018, 9, 828. [Google Scholar] [CrossRef]
- Deane, J.; Ronan, N.J.; O’Callaghan, G.P.; Fouhy, F.; Rea, M.C.; O’Sullivan, O.; Hill, C.J.; Shanahan, F.; Ross, R.P.; McCarthy, M.; et al. Clinical outcomes of real-world Kalydeco (CORK) study–investigating the impact of CFTR potentiation on the intestinal microbiota, exocrine pancreatic function and intestinal inflammation prospectively over 12 months. J. Cyst. Fibros. 2015, 14, S29. [Google Scholar] [CrossRef]
- Hickey, C.; Shanahan, P.; Ronan, N.; Shortt, C.; McCarthy, M.; Fleming, C.; Howlett, C.; Cronin, K.; O’Donovan, D.; Jennings, R.; et al. A retrospective analysis of patient chest physiotherapy practices and adherence to inhaled therapies before and after CFTR modulation with ivacaftor. Pediatr. Pulmonol. 2015, 50, 366. [Google Scholar] [CrossRef][Green Version]
- Ronan, G.; Ronan, N.J.; Shortt, C.; Fleming, C.; Cronin, K.; McCarthy, M.; Hickey, C.; Murphy, D.M.; Eustace, J.A.; O’Halloran, D.J.; et al. The metabolic consequences of CFTR modulation with ivacaftor in a single adult cystic fibrosis centre cohort. J. Cyst. Fibros. 2015, 14, S90. [Google Scholar] [CrossRef]
- Ronan, N.J.; Einarsson, G.G.; Twomey, M.; Mooney, D.; Mullane, D.; NiChroinin, M.; O’Callaghan, G.; Shanahan, F.; Murphy, D.M.; O’Connor, O.J.; et al. CORK Study in cystic fibrosis: Sustained improvements in ultra-low-dose chest CT scores after CFTR modulation with ivacaftor. Chest 2018, 153, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Greenawald, L.; Shenoy, A.; Elidemir, O.; Livingston, F.; Schaeffer, D.; Chidekel, A. Real world effectiveness of ivacaftor in pediatric cystic fibrosis patients. Pediatr. Pulmonol. 2018, 53, 157. [Google Scholar] [CrossRef]
- Hassan, M.; Bonafede, M.M.; Limone, B.L.; Hodgkins, P. One-year evaluation of pulmonary exacerbation outcomes among patients with cystic fibrosis initiated on ivacaftor in a multistate Medicaid population. Pediatr. Pulmonol. 2016, 51, 355. [Google Scholar] [CrossRef]
- Looi, E.; Jones, A.; Barry, P.J. Ivacaftor therapy increases BMI but does not affect serum cholesterol in patients with gating mutations. Pediatr. Pulmonol. 2016, 51, 431. [Google Scholar] [CrossRef]
- McCullagh, M.; Wright, L.; Frost, F.; Greenwood, J.; Nazareth, D.; Walshaw, M. Long term microbiological outcomes of ivacaftor use. A single-centre retrospective study. Pediatr. Pulmonol. 2017, 52, 346. [Google Scholar] [CrossRef]
- Salvatore, D.; Braggion, C.; Calderazzo, M.A.; Cresta, F.; Majo, F.; Messore, B.; Pisi, G.; Pizzamiglio, G.; Biglia, C.; Botti, M.; et al. Ivacaftor treatment in patients with severe lung disease carrying CFTR mutations with residual function. J. Cyst. Fibros. 2019, 18, S128–S129. [Google Scholar] [CrossRef]
- Stallings, V.A.; Sainath, N.; Oberle, M.; Bertolaso, C.; Schall, J.I. Energy balance and mechanisms of weight gain with ivacaftor treatment of cystic fibrosis gating mutations. J. Pediatr. 2018, 201, 229–237.e4. [Google Scholar] [CrossRef]
- Al-Rashdi, Z.; Al-Busaidi, N. The effect of ivacaftor on adult cystic fibrosis patients at the Royal Hospital in Oman. J. Cyst. Fibros. 2019, 18, S129. [Google Scholar] [CrossRef]
- Aziz, A.; Borowicz-Klementowicz, J.; Barker, H.; Johnson, C.; Shafi, N.; Haworth, C.; Floto, A.; Hill, U. Ivacaftor-as effective in clinical practice? Eur. Respir. J. 2016, 48. [Google Scholar] [CrossRef]
- Carrion, A.; Borowitz, D.S.; Freedman, S.D.; Siracusa, C.M.; Goralski, J.L.; Hadjiliadis, D.; Srinivasan, S.; Stokes, D.C. Reduction of recurrence risk of pancreatitis in cystic fibrosis with ivacaftor: Case series. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 451–454. [Google Scholar] [CrossRef]
- Dagan, A.; Cohen-Cymberknoh, M.; Shteinberg, M.; Levine, H.; Vilozni, D.; Bezalel, Y.; Bar Aluma, B.E.; Sarouk, I.; Ashkenazi, M.; Lavie, M.; et al. Ivacaftor for the p.Ser549Arg (S549R) gating mutation—The Israeli experience. Respir. Med. 2017, 131, 225–228. [Google Scholar] [CrossRef]
- Ellemunter, H.; Hindinger, C.; Steinkamp, G. Long-term effects of ivacaftor in patients with G551D mutation and mild lung disease. J. Cyst. Fibros. 2018, 17, S54. [Google Scholar] [CrossRef]
- Ewence, A.E.; Eruchie, C.N.; Higton, A.M.; Ho, T.B.L. Does Ivacaftor improve objective measurements of health in patients with the G551D cystic fibrosis transmembrane conductance regulator (CFTR) protein mutation? The experience of a UK cystic fibrosis centre. Thorax 2013, 68, A120. [Google Scholar] [CrossRef][Green Version]
- Graeber, S.Y.; Hug, M.J.; Sommerburg, O.; Hirtz, S.; Hentschel, J.; Heinzmann, A.; Dopfer, C.; Schulz, A.; Mainz, J.G.; Tümmler, B.; et al. Intestinal current measurements detect activation of mutant CFTR in patients with cystic fibrosis with the G551D mutation treated with ivacaftor. Am. J. Respir. Crit. Care Med. 2015, 192, 1252–1255. [Google Scholar] [CrossRef]
- Grasemann, H.; Gonska, T.; Avolio, J.; Klingel, M.; Tullis, E.; Ratjen, F. Effect of ivacaftor therapy on exhaled nitric oxide in patients with cystic fibrosis. J. Cyst. Fibros. 2015, 14, 727–732. [Google Scholar] [CrossRef]
- Grasemann, H.; Klingel, M.; Avolio, J.; Gonska, T.; Tullis, E.; Ratjen, F. Effect of ivacaftor therapy on nitric oxide/l-arginine metabolism in airways of patients with cystic fibrosis. Pediatr. Pulmonol. 2018, 53, 233–234. [Google Scholar] [CrossRef]
- Green, H.D.; Barry, P.J.; Paisey, C.; Smith, A.; Flight, W.G.; Marchesi, J.; Jones, A.M.; Horsley, A.; Mahenthiralingam, E. The effect of ivacaftor therapy on the microbial diversity of cystic fibrosis lung infection. Thorax 2014, 69, A162. [Google Scholar] [CrossRef]
- Guhaniyogi, L.; Speight, L.; Lea-Davies, M.; Prosser, A.; Lau, D.; Ketchell, R.I.; Duckers, J. Transformational care at the All Wales Adult CF Centre (AWACFC—The impact of ivacaftor (Kalydeco®) one year on. J. Cyst. Fibros. 2015, 14, S92. [Google Scholar] [CrossRef]
- Hebestreit, H.; Sauer-Heilborn, A.; Fischer, R.; Käding, M.; Mainz, J.G. Effects of ivacaftor on severely ill patients with cystic fibrosis carrying a G551D mutation. J. Cyst. Fibros. 2013, 12, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Iacotucci, P.; Salvatore, D.; Carnovale, V.; d’Ippolito, M.; Buonaurio, S.; Celardo, A.; Colangelo, C.; Smaldore, G.; Ferrara, N. Effects of ivacaftor in cystic fibrosis patients carrying a non-G551D gating mutation. J. Cyst. Fibros. 2016, 15, S59. [Google Scholar] [CrossRef]
- Jenkins, L.E.; Reid, A.; Downey, D.G.; Elborn, J.S.; Rendall, J.C. The use of LCI as an effective tool for monitoring clinical response to ivacaftor therapy in CF patients with at least one G551D-allele. J. Cyst. Fibros. 2014, 13, S40. [Google Scholar] [CrossRef]
- Kane, M.; Gonska, T.; Jensen, R.; Avolio, J.; Klingel, M.; Ratjen, F. Lung clearance index response in CF patients with class III CFTR mutations. Pediatr. Pulmonol. 2015, 50, 278. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.I.; de Winter-de Groot, K.M.; Berkers, G.; de Graaf, E.; Arets, H.G.; Bogaert, D.; van der Ent, C.K. The effect of treatment with ivacaftor on the respiratory microbial composition in the upper and lower airways. Pediatr. Pulmonol. 2016, 51, 306. [Google Scholar] [CrossRef]
- Millar, B.C.; McCaughan, J.; Rendall, J.C.; Downey, D.G.; Moore, J.E. Pseudomonas aeruginosa in cystic fibrosis patients with c.1652G›A (G551D)-CFTR treated with ivacaftor—Changes in microbiological parameters. J. Clin. Pharm. Ther. 2018, 43, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.M.; Horsley, A.R.; Jones, A.M.; Barry, P.J. Ivacaftor therapy in patients with severe baseline lung disease carrying a residual function mutation. J. Cyst. Fibros. 2018, 17, S55. [Google Scholar] [CrossRef]
- Mouzaki, M.; Avolio, J.; Griffin, K.; Tullis, D.E.; Ratjen, F.; Gonska, T. Weight increase in CF patients on Kalydeco is due to decrease in resting energy expenditure and associated with increase in adipose tissue. Pediatr. Pulmonol. 2017, 52, 448. [Google Scholar] [CrossRef]
- Robson, E.A.; Feltbower, R.; Lee, T. Real world ivacaftor efficacy in children: Five years on. J. Cyst. Fibros. 2019, 18, S128. [Google Scholar] [CrossRef]
- Salvatore, D.; Braggion, C.; Messore, B.; Pisi, G.; Tuccio, G.; Bena, C.; Colangelo, C. Effects of ivacaftor in patients with cystic fibrosis and severe lung disease carrying CFTR mutations with residual function. J. Cyst. Fibros. 2018, 17, S54–S55. [Google Scholar] [CrossRef]
- Salvatore, D.; Braggion, C.; Messore, B.; Pisi, G.; Calderazzo, M.A.; Francalanci, M.; Colangelo, C.; De Gregorio, F.; Clivati, E.; Biglia, C.; et al. Long-term effectiveness of ivacaftor in patients with Cystic Fibrosis carrying CFTR mutations with residual function and severe lung disease. Ital. J. Pediatr. 2019, 45, 6–7. [Google Scholar] [CrossRef]
- Salvatore, D.; Carnovale, V.; Iacotucci, P.; Braggion, C.; Castellani, C.; Cimino, G.; Colangelo, C.; Francalanci, M.; Leonetti, G.; Lucidi, V.; et al. Effectivenesss of ivacaftor in severe cystic fibrosis patients and non-G551D gating mutations. Pediatr. Pulmonol. 2019, 54, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Sermet-Gaudelus, I.; Delion, M.; Durieu, I.; Jacquot, J.; Hubert, D. Bone demineralization is improved by ivacaftor in patients with cystic fibrosis carrying the p.Gly551Asp mutation. J. Cyst. Fibros. 2016, 15, e67–e69. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.I.; Long, F.R.; McCoy, K.S.; Johnson, T.; Ryan-Wenger, N.A.; Hayes, D., Jr. Ivacaftor improves appearance of sinus disease on computerised tomography in cystic fibrosis patients with G551D mutation. Clin. Otolaryngol. 2015, 40, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.I.; Long, F.R.; McCoy, K.S.; Johnson, T.; Ryan-Wenger, N.A.; Hayes, D., Jr. Computed tomography correlates with improvement with ivacaftor in cystic fibrosis patients with G551D mutation. J. Cyst. Fibros. 2015, 14, 84–89. [Google Scholar] [CrossRef]
- Spoletini, G.; Shaw, N.; Wood, A.; Gillgrass, L.; Etherington, C.; Whitaker, P.; Clifton, I.; Peckham, D. Use of ivacaftor (IVA) in patients heterozygous for R117H mutation: Real-life experience in a large UK adult CF centre. J. Cyst. Fibros. 2019, 18, S128. [Google Scholar] [CrossRef]
- Tierney, A.; King, S.J.; Edgeworth, D.; Williams, E.; Finlayson, F.; Keating, D.; Clark, D.; Button, B.M.; Kotsimbos, T.; Wilson, J.W. An increase in weight and fat mass observed following five months of ivacaftor treatment plateaus at 24 months in adults with G551D-related cystic fibrosis. J. Cyst. Fibros. 2018, 17, S117. [Google Scholar] [CrossRef]
- Trinh, I.; Bermudez, E.; Abbas, N.; Viguier, F.; Hubert, D.; Guérin, C.; Chast, F. Ivacaftor in adults with cystic fibrosis: One-year experience in the real world setting. Int. J. Clin. Pharm. 2013, 35, 1331. [Google Scholar] [CrossRef]
- McKone, E.F.; Emerson, S.S.; Edwards, K.L.; Aitken, M.L. Effect of genotype on phenotype and mortality in cystic fibrosis: A retrospective cohort study. Lancet 2003, 361, 1671–1676. [Google Scholar] [CrossRef]
- Sawicki, G.S.; McKone, E.F.; Millar, S.J.; Pasta, D.J.; Konstan, M.W.; Lubarsky, B.; Wagener, J.S. Patients with cystic fibrosis and a G551D or homozygous F508del mutation: Similar lung function decline. Am. J. Respir. Crit. Care Med. 2017, 195, 1673–1676. [Google Scholar] [CrossRef]
- Quittner, A.L.; Sawicki, G.S.; McMullen, A.; Rasouliyan, L.; Pasta, D.J.; Yegin, A.; Konstan, M.W. Psychometric evaluation of the Cystic Fibrosis Questionnaire-Revised in a national sample. Qual. Life Res. 2012, 21, 1267–1278. [Google Scholar] [CrossRef]
- Piccirillo, J.F.; Merritt, M.G., Jr.; Richards, M.L. Psychometric and clinimetric validity of the 20-Item Sino-Nasal Outcome Test (SNOT-20). Otolaryngol. Head Neck Surg. 2002, 126, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Whiting, P.; Al, M.; Burgers, L.; Westwood, M.; Ryder, S.; Hoogendoorn, M.; Armstrong, N.; Allen, A.; Severens, H.; Kleijnen, J. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: A systematic review and cost-effectiveness analysis. Health Technol. Assess. 2014, 18, 1–106. [Google Scholar] [CrossRef]
- Gramegna, A.; Contarini, M.; Aliberti, S.; Casciaro, R.; Blasi, F.; Castellani, C. From ivacaftor to triple combination: A systematic review of efficacy and safety of CFTR modulators in people with cystic fibrosis. Int. J. Mol. Sci. 2020, 21, 5882. [Google Scholar] [CrossRef]
- Shteinberg, M.; Taylor-Cousar, J.L. Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur. Respir. Rev. 2020, 29, 190112. [Google Scholar] [CrossRef]
- Dagenais, R.V.E.; Su, V.C.; Quon, B.S. Real-world safety of CFTR modulators in the treatment of cystic fibrosis: A systematic review. J. Clin. Med. 2021, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Canada. The Canadian Cystic Fibrosis Registry. 2017 Annual Data Report. Available online: https://www.cysticfibrosis.ca/uploads/Registry%20Report%202017/2017%20Registry%20Annual%20Data%20Report.pdf (accessed on 17 December 2020).
- French Cystic Fibrosis Registry. Annual Data Report 2016. Available online: https://www.vaincrelamuco.org/sites/default/files/french_cf_patient_registry_-_report_on_2016_data.pdf (accessed on 17 December 2020).
- UK Cystic Fibrosis Registry. Annual Data Report 2017. Available online: https://www.cysticfibrosis.org.uk/~/media/documents/the-work-we-do/uk-cf-registry/2017-registry-annual-data-report-updated-291018.ashx?la=en (accessed on 17 December 2020).
- Ruseckaite, R.; Ahern, S.; Ranger, T.; Dean, J.; Gardam, M.; Bell, S.; Burke, N.; on behalf of the Australian Cystic Fibrosis Data Registry. The Australian Cystic Fibrosis Data Registry Annual Report. 2017. Available online: https://www.cysticfibrosis.org.au/getmedia/24e94d66-29fa-4e3f-8e65-21ee24ed2e5a/ACFDR-2017-Annual-Report_highres_singles.pdf.aspx (accessed on 17 December 2020).
- Sawicki, G.S.; McKone, E.F.; Pasta, D.J.; Millar, S.J.; Wagener, J.S.; Johnson, C.A.; Konstan, M.W. Sustained benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am. J. Respir. Crit. Care Med. 2015, 192, 836–842. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference(s) (Study Name) | Country/ Geographic Region | Data Type | Source | Follow-Up, Months | pwCF Treated With Ivacaftor, n | CFTR Mutation/Class of Mutation | pwCF Aged ≥18 Years, % | Mean (SD) Baseline ppFEV1, % |
|---|---|---|---|---|---|---|---|---|
| Studies with non-ivacaftor-treated comparator | ||||||||
| >50pwCF treated with ivacaftor (3 studies) | ||||||||
| Bell 2019 [28] | Interna-tional | Multicenter | NR (international) | 22 (mean) | IVA: 72 | G551D | 58.3 | 79.8 (25.6) |
| COMP: 137 | F508del | 69.3 | 70.7 (28.8) | |||||
| Bessonova 2018 (LTSS) [29] | UK, US | Registry | US CF Foundation Patient Registry (CFFPR), UK CF Registry | UK, ≤24 a US, ≤36 a | IVA: 411 (UK), 1256 (US) | Class I-VI and VI b | 62.3 (UK), 58.2 (US) | UK, 70.6 US, 79.8 |
| COMP: 2069 (UK), 6200 (US) | 61.8 (UK), 58.2 (US) | UK, 71.4 US, 80.4 | ||||||
| Volkova 2020 (LTSS) [30] | UK, ≤48 US, ≤60 | IVA: 247 (UK), 635 (US) | Class I-III b | 69.2 (UK), 61.1 (US) | UK, 73.0 (23.6) US, 79.0 (25.3) | |||
| COMP: 1230 (UK), 1874 (US) | 66.6 (UK), 57.3 (US) | UK, 73.4 (22.4) US, 81.7 (23.7) | ||||||
| Frost 2019 [31] | UK | Registry | UK CF Registry | 36 | IVA: 276 | G551D | NR | 81.07 (22.5) |
| COMP: 5296 | Rest of CF population not treated with IVA | 72.91 (23.3) | ||||||
| 20–50pwCF treated with ivacaftor (2 studies) | ||||||||
| Barry 2014 [32], Barry 2015 c [33] | UK, Ireland | Multicenter | CF centers in UK and Ireland (Compassionate-use program) | 37 (median) | IVA:21 | G551D | 100 | 26.5 (7.2) [32] |
| COMP: 35 | Non-G551D gating | 30.3 (7.5) [32] | ||||||
| Emery 2019 c [34] | Ireland | Single center | Cork University Hospital | 12 | 28 d | NR | 0 | NR |
| <20pwCF treated with ivacaftor (2 studies) | ||||||||
| McLearn-Montz 2018 c [35] | US | Single center | University of Iowa Children’s Center | NR e | IVA: 8 | NR | 0 | NR |
| COMP: 16 | NR | |||||||
| Wainwright 2014 c [36], Wainwright 2014 c [37] | Australia | Multicenter (IVA), Registry (COMP) | NR (IVA), ACFDR (COMP) | 12 | IVA: 17 | G551D | 100 | 38.3 (12.4) [37] |
| COMP: 314 | NR | 45.4 (14.5) [37] | ||||||
| Studies without a non-ivacaftor-treated comparator | ||||||||
| >50 pwCF treated with ivacaftor (11 studies) | ||||||||
| Bonafede 2014 c [38] | US | Administra-tive claims data | Truven Health MarketScan Commercial Database | 6 | 102 | NR | 60 | NR |
| Castellani 2018 c [39], Castellani 2018 c [40] (VOCAL) | UK, Nether-lands, Italy | Multicenter | 15 sites in Italy, Netherlands, and UK (interim analysis) | 12 | 71 | Non-G551D gating | 68 | 64.7 (24.5) [39] |
| Guimbellot 2018 c [41] (GOAL/GOAL-e2) | US | Multicenter | 28 centers within the Cystic Fibrosis Therapeutics Development Network (GOAL study) | 66 | 96 | G551D | 46 | 82 (NR) |
| Guimbellot 2019 [42] | 6 | 21 f | Non-G551D gating | 48 | 68.0 (28.4) | |||
| Hathorne 2015 c [43], | 12 | 18 f | G551D | NR | NR | |||
| Heltshe 2015 [44] | 12 | 151 | G551D | 54 | 82.6 g (25.6) | |||
| Rowe 2014 [45] | 6 | 151 | G551D | 54 | 82.6 (25.6) | |||
| Sagel 2015 c [46] | 6 | 54 | R117H | 61 | 85 (25) | |||
| van de Peppel 2019 [47] | 6 | 99 | G551D | NR | 93.4 (median) | |||
| Feng 2018 [48] | US | Administra-tive claims data | Truven Health MarketScan Commercial Database | 12 | 143 | NR | 63 | NR |
| Fink 2015 c [49] | US | Registry | US CF Foundation Patient Registry (CFFPR) | 12 | 403 | G551D | NR | NR |
| Hassan 2016 c [50] | US | Administra-tive claims data | Truven MarketScan Medicaid Multi-State database | 12 | 84 | NR | 56 | NR |
| Hubert 2018 [51] | France | Registry | NR | 24 | 57 | Gating | 47 | 72.3 (26.4) |
| Hubert 2018 c [52], Hubert 2018 c [53] (BRIO) | France | Multicenter | 35 French CF centers (interim analysis) | 12 | 107 | Gating | 48 | 72.6 (24.4) [53] |
| Kirwan 2019 [54] | Ireland | Registry | CF Registry of Ireland | 36 | 80 | G551D | 44 | 71.53 (26.09) |
| Newsome 2018 c [55] | UK | Registry | UK CF Registry | 36 | 361 | NR | NR | NR |
| Suthoff 2016 [56] | US | Database | Truven Health MarketScan Commercial Database | 12 | 79 | NR | 48 | NR |
| 20–50 pwCF treated with ivacaftor (11 studies) | ||||||||
| Al Redha 2016 c [57] | UAE | Single center | UAE Paediatric CF Centre | 12 | 12 | S549R | 0 | S549R/S549R, 70 (NR) |
| Barry 2014 [58], Barry 2015 c [59], Banerjee 2014 c [60] | UK | Single center | Manchester Adult CF Centre | 12 | 24 | G551D | 100 | 64.3 (NR) [58] |
| Chassagnon 2016 [61] | France | Multicenter | 8 French CF centers | 37 (median) | 22 | Gating | 100 | 39.5 (NR) |
| Corvol 2018 [62] | France | Multicenter | French CF Modifier Gene Study | 12 | 30 | Gating | NR | NR |
| Deane 2015 c [63] (CORK) | Ireland | Single center | Cork CF Centre | 12 | 20 | G551D | 100 | NR |
| Hickey 2015 c [64] (CORK) | 20 (mean) | 36 | NR | NR | ||||
| Ronan 2015 c [65] (CORK) | NR | 24 | 100 | NR | ||||
| Ronan 2018 [66] (CORK) | 12 | 33 | 61.0 | 75.21 (20.7) | ||||
| Greenawald 2018 c [67] | US | Multicenter | Nemours CF centers | 12 | 26 | Class III-V | 0 | NR |
| Hassan 2016 c [68] | US | Administra-tive claims data | Truven MarketScan Medicaid Multi-State Database | 12 | 44 | NR | 14 | NR |
| Looi 2016 c [69] | UK | Single center | Manchester Adult CF Centre | 6 | 30 | Gating | NR | NR |
| McCullagh 2017 c [70] | UK | Single center | Liverpool Heart & Chest Hospital | 36 | 22 | G551D, S549N | 100 | 83.1 (NR) |
| Salvatore 2019 c [71] | Italy | Multicenter | NR | 12 | 25 | Class IV-V | 100 | 31.5 (14.5) |
| Stallings 2018 [72] | US | Single center | Children’s Hospital of Philadelphia | 3 | 23 | Gating | NR | 86 (21) |
| <20 pwCF treated with ivacaftor (28 studies) | ||||||||
| Al-Rashdi 2019 c [73] | Oman | Single center | The Royal Hospital | 12 | 15 | P.ser 549 Arg Del | NR | 54.27 (25.46) |
| Aziz 2016 c [74] | UK | Single center | Cambridge Centre for Lung Infection | 24 | 15 | G551D | 100 | 59.6 (NR) |
| Carrion 2018 [75] | US | Multicenter | 4 CF care centers | 12 | 6 | Class III-IV | 67 | 49.5 (median) |
| Dagan 2017 [76] | Israel | Multicenter | NR | 12 | 8 | S549R | 50 | 74 (23) |
| Ellemunter 2018 c [77] | Austria | Single center | CF Centre Innsbruck | 24 | 7 | G551D | NR | 93.2 (NR) |
| Ewence 2013 c [78] | UK | Single center | Frimley Park Hospital NHS Foundation Trust | 3 | 10 | G551D | NR | 58.2 (19.8) |
| Graeber 2015 [79] | Germany | Multicenter | NR | 3 | 12 | G551D | NR | 88.8 (21.7) |
| Grasemann 2015 [80] | Canada | Multicenter | Hospital for Sick Children, St. Michael’s Hospital | 1 | 15 | Gating | 53 | 69.7 (16.7) |
| Grasemann 2018 c [81] | 24 | 20 | 65 | Pediatric, 80 (NR) Adults, 65 (NR) | ||||
| Green 2014 c [82] | UK | Single center | Manchester Adult CF Centre | 1 | 13 | NR | 100 | 56.0 (NR) |
| Guhaniyogi 2015 c [83] | UK | Single center | All Wales Adult CF Centre | 12 | 11 | G551D | 100 | 63.5 (26.2) |
| Hebestreit 2013 [84] | Germany | Multicenter | German CF centers | 8 (mean) | 14 | G551D | 100 | 25.0 (7.5) |
| Hisert 2017 [85] | Interna-tional | Multicenter | NR | 24 | 12 | G551D | 100 | 64.2 (NR) |
| Iacotucci 2016 c [86] | Italy | Multicenter | Adult CF Center, University of Naples; CF Center, Hospital San Carlo | 6 | 18 | Non-G551D gating | NR | 55.8 (23.6) |
| Jenkins 2014 c [87] | UK | Multicenter | Royal Belfast Hospital for Sick Children, Belfast Adult CF Centre | 6 | 14 | G551D | 50 | 84.1 (NR) |
| Kane 2015 c [88] | Canada | Single center | Hospital for Sick Children | 1 (median) | 10 | G551D, G178R | NR | 70.0 (NR) |
| Kristensen 2016 c [89] | Multiple countries | Multicenter | HIT-CF Program | 2 | 15 | S1251N | NR | NR |
| Millar 2018 [90] | UK | Multicenter | Northern Ireland Regional Adult CF Center; Centre for Experimental Medicine, Queens University | 24 (mean) | 15 | G551D | NR | NR |
| Mitchell 2018 c [91] | UK | Single center | Manchester Adult CF Centre | 12 | 12 | Class IV–V | NR | 31.3 (5.4) |
| Mouzaki 2017 c [92] | Canada | Multicenter | Hospital for Sick Children, St. Michael’s Hospital | 24 | 18 | Gating | NR | 76 (20) |
| Robson 2019 c [93] | UK | Single center | Leeds Regional Paediatric CF Centre | 60 | 10 | Gating | 0 | 87.0 (median) |
| Salvatore 2018 c [94] | Italy | Multicenter | NR | 6 | 9 | Class IV-VI | 100 | 37.3 (9.4) |
| Salvatore 2019 c [95] | Italy | Multicenter | Compassionate-use program | 12 | 10 | Class IV-VI | 100 | NR |
| Salvatore 2019 [96] | Italy | Multicenter | Compassionate-use program | 12 | 13 | Non-G551D gating | 92 | 35.1 (14.3) |
| Sermet-Gaudelus 2016 [97] | France | Single center | NR | 20 (mean) | 7 | Gly551Asp | 100 | 48 (9) |
| Sheikh 2015 [98], Sheikh 2015 [99] | US | Single center | CF Center at Nationwide Children’s Hospital | 12 | 12 | G551D | 50 | 82.5 (26) [98] |
| Spoletini 2019 c [100] | UK | Single center | Adult CF Unit, Leeds | 24 | 9 | R117H | NR | NR |
| Tierney 2018 c [101] | Australia | Single center | Alfred Health CF Service | 24 | 11 | G551D | 100 | 61.8 (22.9) |
| Trinh 2013 c [102] | France | Single center | Pulmonary Department, Hopitaux Universitaires Paris Centre | 11 | 7 | G551D | 100 | 44.1 (NR) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duckers, J.; Lesher, B.; Thorat, T.; Lucas, E.; McGarry, L.J.; Chandarana, K.; De Iorio, F. Real-World Outcomes of Ivacaftor Treatment in People with Cystic Fibrosis: A Systematic Review. J. Clin. Med. 2021, 10, 1527. https://doi.org/10.3390/jcm10071527
Duckers J, Lesher B, Thorat T, Lucas E, McGarry LJ, Chandarana K, De Iorio F. Real-World Outcomes of Ivacaftor Treatment in People with Cystic Fibrosis: A Systematic Review. Journal of Clinical Medicine. 2021; 10(7):1527. https://doi.org/10.3390/jcm10071527
Chicago/Turabian StyleDuckers, Jamie, Beth Lesher, Teja Thorat, Eleanor Lucas, Lisa J. McGarry, Keval Chandarana, and Fosca De Iorio. 2021. "Real-World Outcomes of Ivacaftor Treatment in People with Cystic Fibrosis: A Systematic Review" Journal of Clinical Medicine 10, no. 7: 1527. https://doi.org/10.3390/jcm10071527
APA StyleDuckers, J., Lesher, B., Thorat, T., Lucas, E., McGarry, L. J., Chandarana, K., & De Iorio, F. (2021). Real-World Outcomes of Ivacaftor Treatment in People with Cystic Fibrosis: A Systematic Review. Journal of Clinical Medicine, 10(7), 1527. https://doi.org/10.3390/jcm10071527