
Inhibitors of Protein Convertase Subtilisin/Kexin 9 (PCSK9) and Acute Coronary Syndrome (ACS): The State-of-the-Art

 ,
,  , , ,
, , ,  ,
,  ,
,
Abstract
1. Introduction
2. PCSK9: Direct and Indirect Role on Coronary Plaques
3. Inhibitor of PCSK9
4. LDL Management in Patients after ACS
5. Pitfalls and Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; GBD-NHLBI-JACC Global Burden of Cardiovascular Diseases Writing Group; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Ralapanawa, U.; Sivakanesan, R. Epidemiology and the Magnitude of Coronary Artery Disease and Acute Coronary Syndrome: A Narrative Review. J. Epidemiol. Glob. Health 2021. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, T.; Hasvold, P.; Henriksson, M.; Hjelm, H.; Thuresson, M.; Janzon, M. Cardiovascular risk in post-myocardial infarction patients: Nationwide real world data demonstrate the importance of a long-term perspective. Eur. Heart J. 2015, 36, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Chiang, L.W.; Grenier, J.M.; Ettwiller, L.; Jenkins, L.P.; Ficenec, D.; Martin, J.; Jin, F.; DiStefano, P.S.; Wood, A. An orchestrated gene expression component of neuronal programmed cell death revealed by cDNA array analysis. Proc. Natl. Acad. Sci. USA 2001, 98, 2814–2819. [Google Scholar] [CrossRef]
- Piper, D.E.; Jackson, S.; Liu, Q.; Romanow, W.G.; Shetterly, S.; Thibault, S.T.; Shan, B.; Walker, N.P. The crystal structure of PCSK9: A regulator of plasma LDL-cholesterol. Structure 2007, 15, 545–552. [Google Scholar] [CrossRef]
- Navarese, E.P.; Kolodziejczak, M.; Kereiakes, D.J.; Tantry, U.S.; O’Connor, C.; Gurbel, P.A. Proprotein Convertase Subtilisin/Kexin Type 9 Monoclonal Antibodies for Acute Coronary Syndrome: A Narrative Review. Ann. Intern. Med. 2016, 164, 600–607. [Google Scholar] [CrossRef]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef]
- Kim, Y.U.; Kee, P.; Danila, D.; Teng, B.B. A Critical Role of PCSK9 in Mediating IL-17-Producing T Cell Responses in Hyperlipidemia. Immune. Netw. 2019, 19, e41. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Sun, C.; Wang, Y.; Mehta, J.L. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid. Redox Signal. 2015, 22, 760–771. [Google Scholar] [CrossRef]
- Ding, Z.; Pothineni, N.V.K.; Goel, A.; Lüscher, T.F.; Mehta, J.L. PCSK9 and inflammation: Role of shear stress, pro-inflammatory cytokines, and LOX-1. Cardiovasc. Res. 2019, 116, 908–915. [Google Scholar] [CrossRef]
- Dubuc, G.; Chamberland, A.; Wassef, H.; Davignon, J.; Seidah, N.G.; Bernier, L.; Prat, A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arter. Thromb. Vasc. Biol. 2004, 24, 1454–1459. [Google Scholar] [CrossRef]
- Roubtsova, A.; Munkonda, M.N.; Awan, Z.; Marcinkiewicz, J.; Chamberland, A.; Lazure, C.; Cianflone, K.; Seidah, N.G.; Prat, A. Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arter. Thromb. Vasc. Biol. 2011, 31, 785–791. [Google Scholar] [CrossRef]
- Norata, G.D.; Tavori, H.; Pirillo, A.; Fazio, S.; Catapano, A.L. Biology of proprotein convertase subtilisin kexin 9: Beyond low-density lipoprotein cholesterol lowering. Cardiovasc. Res. 2016, 112, 429–442. [Google Scholar] [CrossRef]
- Qi, Z.; Hu, L.; Zhang, J.; Yang, W.; Liu, X.; Jia, D.; Yao, Z.; Chang, L.; Pan, G.; Zhong, H.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) Enhances Platelet Activation, Thrombosis, and Myocardial Infarct Expansion by Binding to Platelet CD36. Circulation 2021, 143, 45–61. [Google Scholar] [CrossRef]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef]
- Yang, M.; Silverstein, R.L. CD36 signaling in vascular redox stress. Free. Radic. Biol. Med. 2019, 136, 159–171. [Google Scholar] [CrossRef]
- Yang, M.; Kholmukhamedov, A.; Schulte, M.L.; Cooley, B.C.; Scoggins, N.O.; Wood, J.P.; Cameron, S.J.; Morrell, C.N.; Jobe, S.M.; Silverstein, R.L. Platelet CD36 signaling through ERK5 promotes caspase-dependent procoagulant activity and fibrin deposition in vivo. Blood Adv. 2018, 2, 2848–2861. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Navarese, E.P.; Tantry, U.S. Exploration of PCSK9 as a Cardiovascular Risk Factor: Is There a Link to the Platelet? J. Am. Coll. Cardiol. 2017, 70, 1463–1466. [Google Scholar] [CrossRef]
- Navarese, E.P.; Kolodziejczak, M.; Winter, M.P.; Alimohammadi, A.; Lang, I.M.; Buffon, A.; Lip, G.Y.; Siller-Matula, J.M. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: The PCSK9-REACT study. Int. J. Cardiol. 2017, 227, 644–649. [Google Scholar] [CrossRef]
- Pastori, D.; Nocella, C.; Farcomeni, A.; Bartimoccia, S.; Santulli, M.; Vasaturo, F.; Carnevale, R.; Menichelli, D.; Violi, F.; ATHERO-AF Study Group; et al. Relationship of PCSK9 and Urinary Thromboxane Excretion to Cardiovascular Events in Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1455–1462. [Google Scholar] [CrossRef]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgözoğlu, L.; Nordestgaard, B.G.; et al. European Atherosclerosis Society Consensus Panel. Statin-associated muscle symptoms: Impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Hear. J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef]
- Stein, E.A.; Mellis, S.; Yancopoulos, G.D.; Stahl, N.; Logan, D.; Smith, W.B.; Lisbon, E.; Gutierrez, M.; Webb, C.; Wu, R.; et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 2012, 366, 1108–1118. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef]
- The Emerging Risk Factors Collaboration; Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009, 302, 412–423. [Google Scholar] [CrossRef]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.L.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. ORION-10 and ORION-11 Investigators. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; ESC Scientific Document Group; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Grundy, S.M.; Arai, H.; Barter, P.; Bersot, T.P.; Betteridge, D.J.; Carmena, R.; Cuevas, A.; Davidson, M.H.; Genest, J.; Kesäniemi, Y.A.; et al. An International Atherosclerosis Society Position Paper: Global recommendations for the management of dyslipidemia--full report. J. Clin. Lipidol. 2014, 8, 29–60. [Google Scholar] [CrossRef]
- Guedeney, P.; Giustino, G.; Sorrentino, S.; Claessen, B.E.; Camaj, A.; Kalkman, D.N.; Vogel, B.; Sartori, S.; De Rosa, S.; Baber, U.; et al. Efficacy and safety of alirocumab and evolocumab: A systematic review and meta-analysis of randomized controlled trials. Eur. Heart J. 2019. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, e285–e350. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Ezekowitz, M.D.; Ganz, P.; Oliver, M.F.; Waters, D.; Zeiher, A.; Chaitman, B.R.; Leslie, S.; Stern, T. Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) Study Investigators. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: The MIRACL study: A randomized controlled trial. JAMA 2001, 285, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Blazing, M.A.; Braunwald, E. Ezetimibe plus a Statin after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 373, 1473–1477. [Google Scholar] [CrossRef]
- Gencer, B.; Mach, F. Lipid management in ACS: Should we go lower faster? Atherosclerosis 2018, 275, 368–375. [Google Scholar] [CrossRef]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. JAMA 2016, 316, 1289–1297. [Google Scholar] [CrossRef]
- Li, S.; Peng, Y.; Wang, X.; Qian, Y.; Xiang, P.; Wade, S.W.; Guo, H.; Lopez, J.A.G.; Herzog, C.A.; Handelsman, Y. Cardiovascular events and death after myocardial infarction or ischemic stroke in an older Medicare population. Clin. Cardiol. 2019, 42, 391–399. [Google Scholar] [CrossRef]
- Abtan, J.; Bhatt, D.L.; Elbez, Y.; Sorbets, E.; Eagle, K.; Ikeda, Y.; Wu, D.; Hanson, M.E.; Hannachi, H.; Singhal, P.K.; et al. REACH Registry Investigators. Residual Ischemic Risk and Its Determinants in Patients with Previous Myocardial Infarction and Without Prior Stroke or TIA: Insights From the REACH Registry. Clin. Cardiol. 2016, 39, 670–677. [Google Scholar] [CrossRef]
- Murphy, A.; Hamilton, G.; Andrianopoulos, N.; Yudi, M.B.; Farouque, O.; Duffy, S.J.; Lefkovits, J.; Brennan, A.; Reid, C.M.; Melbourne Interventional Group; et al. One-Year Outcomes of Patients with Established Coronary Artery Disease Presenting With Acute Coronary Syndromes. Am. J. Cardiol. 2019, 123, 1387–1392. [Google Scholar] [CrossRef]
- Leucker, T.M.; Blaha, M.J.; Jones, S.R.; Vavuranakis, M.A.; Williams, M.S.; Lai, H.; Schindler, T.H.; Latina, J.; Schulman, S.P.; Gerstenblith, G. Effect of Evolocumab on Atherogenic Lipoproteins During the Peri- and Early Postinfarction Period: A Placebo-Controlled, Randomized Trial. Circulation 2020, 142, 419–421. [Google Scholar] [CrossRef]
- Koskinas, K.C.; Windecker, S.; Pedrazzini, G.; Mueller, C.; Cook, S.; Matter, C.M.; Muller, O.; Häner, J.; Gencer, B.; Crljenica, C.; et al. Evolocumab for Early Reduction of LDL Cholesterol Levels in Patients With Acute Coronary Syndromes (EVOPACS). J. Am. Coll. Cardiol. 2019, 74, 2452–2462. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. ODYSSEY OUTCOMES Committees and Investigators. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Gentile, M.; Iannuzzo, G.; Simeon, V.; Mattiello, A.; Rubba, F.; Panico, C.; Panico, S.; Rubba, P. Association between atherogenic index of plasma and carotid intima-media thickness in a cohort of Mediterranean women. Acta. Cardiol. 2020, 11, 1–6. [Google Scholar] [CrossRef]
- Gentile, M.; Simeon, V.; Iannuzzo, G.; Mattiello, A.; Donata di Taranto, M.; Panico, S.; Rubba, P. Lipoprotein (a) is an independent predictor of cardiovascular events in Mediterranean women (Progetto Atena). Eur. J. Prev. Cardiol. 2020, 27, 2248–2250. [Google Scholar] [CrossRef]
- Qamar, A.; Giugliano, R.P.; Keech, A.C.; Kuder, J.F.; Murphy, S.A.; Kurtz, C.E.; Wasserman, S.M.; Sever, P.S.; Pedersen, T.R.; Sabatine, M.S. Interindividual Variation in Low-Density Lipoprotein Cholesterol Level Reduction With Evolocumab: An Analysis of FOURIER Trial Data. JAMA Cardiol. 2019, 4, 59–63. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Miles, J.; Tavori, H.; Fazio, S. Diagnosing Resistance to a Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitor. Ann. Intern. Med. 2017, 168, 376. [Google Scholar] [CrossRef]
- Bays, H.E.; Rosenson, R.S.; Baccara-Dinet, M.T.; Louie, M.J.; Thompson, D.; Hovingh, G.K. Assessment of the 1% of Patients with Consistent <15% Reduction in Low-Density Lipoprotein Cholesterol: Pooled Analysis of 10 Phase 3 ODYSSEY Alirocumab Trials. Cardiovasc. Drugs Ther. 2018, 32, 175–180. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Hegele, R.A.; Fazio, S.; Cannon, C.P. The Evolving Future of PCSK9 Inhibitors. J. Am. Coll. Cardiol. 2018, 72, 314–329. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ. Res. 2018, 122, 1420–1438. [Google Scholar] [CrossRef]
- Di Minno, A.; Gentile, M.; Iannuzzo, G.; Calcaterra, I.; Tripaldella, M.; Porro, B.; Cavalca, V.; Di Taranto, M.D.; Tremoli, E.; Fortunato, G.; et al. Endothelial function improvement in patients with familial hypercholesterolemia receiving PCSK-9 inhibitors on top of maximally tolerated lipid lowering therapy. Thromb. Res. 2020, 194, 229–236. [Google Scholar] [CrossRef]
- Di Minno, M.N.D.; Gentile, M.; Di Minno, A.; Iannuzzo, G.; Calcaterra, I.; Buonaiuto, A.; Di Taranto, M.D.; Giacobbe, C.; Fortunato, G.; Rubba, P.O.F. Changes in carotid stiffness in patients with familial hypercholesterolemia treated with Evolocumab®: A prospective cohort study. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 996–1004. [Google Scholar] [CrossRef]


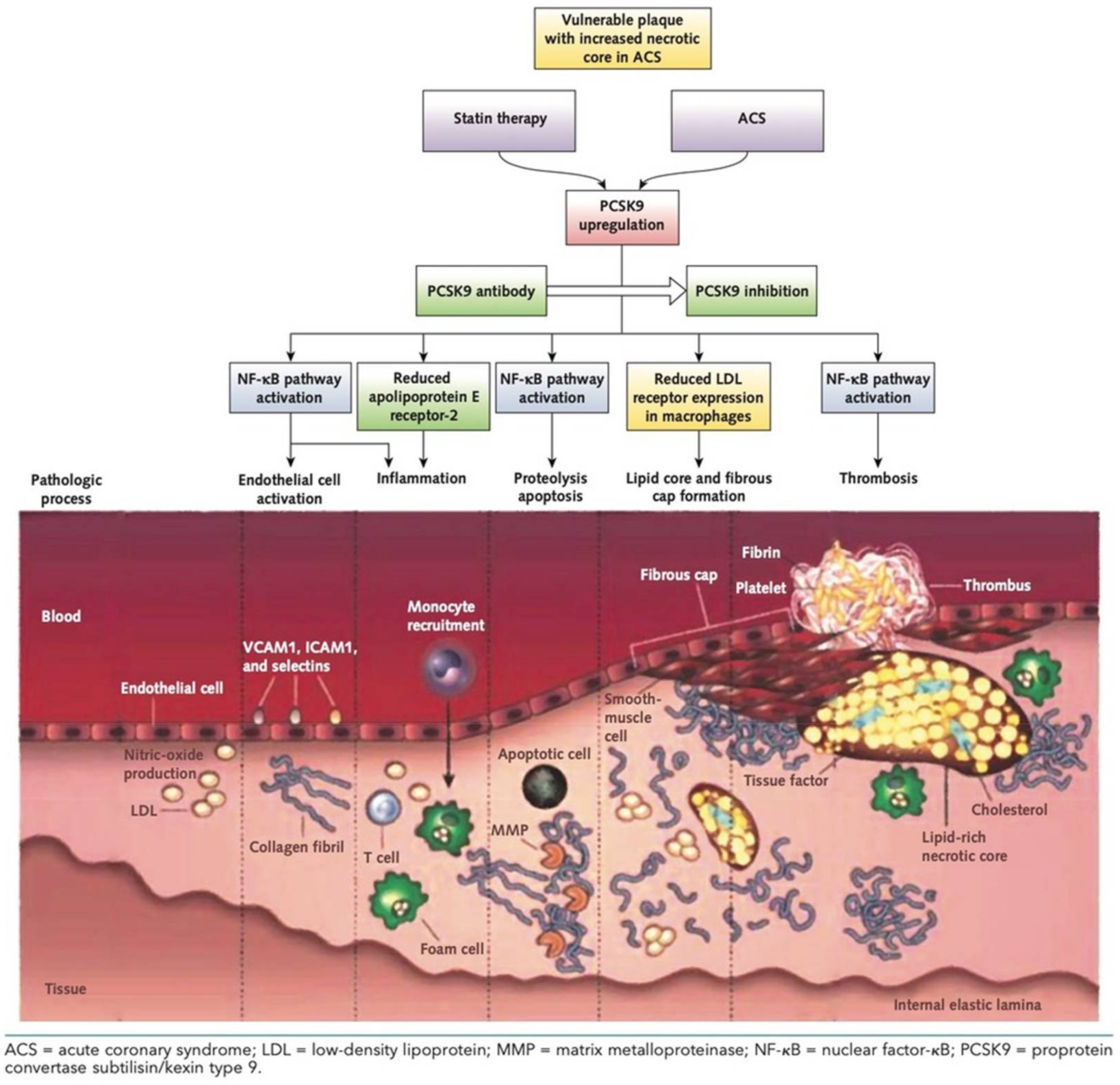{kind=link}
| Study ID | Study Population/Design | Changes in LDL (%) | Efficacy | Safety |
|---|---|---|---|---|
| EVACS [39] | 56 patients: 29 evolocumab, 27 placebo; Mean age 55 ± 13; Male 59%; | −61% after 30 days. | LDL-C in patients treated with evolocumab was 28.6 mg/dL lower than in the placebo group after 30 days; >75% patients have LDL-C < 55 mg/dL after 30 days. | |
| EVOPACS [40] | 308 patients: 155 evolocumab, 153 placebo; Mean age 60.5 ± 12 (evolocumab), 61.0 ± 10.7 (placebo); Male 83% (evolocumab), 80% (placebo) | −77.1 ± 15.8% after 8 weeks | LDL mean difference reduction of −40.7% between evolocumab and placebo group (95% CI: −45.2 to −36.2%; p < 0.001). Lp(a) mean difference reduction of −10.4% (p = 0.47) | Similar between two groups. Local injection site reactions: 3.2% (evolocumab), 2% (placebo); All-cause death: 1.3% (evolocumab), 0% (placebo) (p = 0.5) |
| ODYSSEY [41] | 18924 patients: 9462 alirocumab, 9462 placebo; Mean age: 58.5 ± 9.3 (alirocumab), 58.6 ± 9.4 (placebo); Male: 74.7% (alirocumab), 74.9% (placebo); | −62.7% after 4 months,−54.7% at 48 months after randomization | Reduction of 15% of MACE (HR: 0.85, 95%CI: 0.78–0.93, p = 0.0003, absolute risk reduction: 1.7% CHD death was not significantly reduced (HR: 0.92, 95%CI: 0.76–1.11, p = 0.38) Non-fatal MI: HR: 0.86 (95%CI: 0.77–0.96, p = 0.006) Ischemic stroke: HR: 0.73 (95%CI: 0.57–0.93, p = 0.01) Unstable angina: HR: 0.61 (95%CI: 0.41–0.92, p = 0.02) | Similar between two groups. Local injection site reactions: 3.8% (alirocumab), 2.1% (placebo). ALT > 3 times U.L.N.: 2.3 (alirocumab), 2.4 (placebo); Adverse event that led to death: 1.9% (alirocumab), 2.4% (placebo). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannuzzo, G.; Gentile, M.; Bresciani, A.; Mallardo, V.; Di Lorenzo, A.; Merone, P.; Cuomo, G.; Pacileo, M.; Sarullo, F.M.; Venturini, E.; et al. Inhibitors of Protein Convertase Subtilisin/Kexin 9 (PCSK9) and Acute Coronary Syndrome (ACS): The State-of-the-Art. J. Clin. Med. 2021, 10, 1510. https://doi.org/10.3390/jcm10071510
Iannuzzo G, Gentile M, Bresciani A, Mallardo V, Di Lorenzo A, Merone P, Cuomo G, Pacileo M, Sarullo FM, Venturini E, et al. Inhibitors of Protein Convertase Subtilisin/Kexin 9 (PCSK9) and Acute Coronary Syndrome (ACS): The State-of-the-Art. Journal of Clinical Medicine. 2021; 10(7):1510. https://doi.org/10.3390/jcm10071510
Chicago/Turabian StyleIannuzzo, Gabriella, Marco Gentile, Alessandro Bresciani, Vania Mallardo, Anna Di Lorenzo, Pasquale Merone, Gianluigi Cuomo, Mario Pacileo, Filippo M. Sarullo, Elio Venturini, and et al. 2021. "Inhibitors of Protein Convertase Subtilisin/Kexin 9 (PCSK9) and Acute Coronary Syndrome (ACS): The State-of-the-Art" Journal of Clinical Medicine 10, no. 7: 1510. https://doi.org/10.3390/jcm10071510
APA StyleIannuzzo, G., Gentile, M., Bresciani, A., Mallardo, V., Di Lorenzo, A., Merone, P., Cuomo, G., Pacileo, M., Sarullo, F. M., Venturini, E., D’Andrea, A., Vigorito, C., & Giallauria, F. (2021). Inhibitors of Protein Convertase Subtilisin/Kexin 9 (PCSK9) and Acute Coronary Syndrome (ACS): The State-of-the-Art. Journal of Clinical Medicine, 10(7), 1510. https://doi.org/10.3390/jcm10071510