
Developing New Agents for Treatment of Childhood Cancer: Challenges and Opportunities for Preclinical Testing


Abstract
1. Introduction
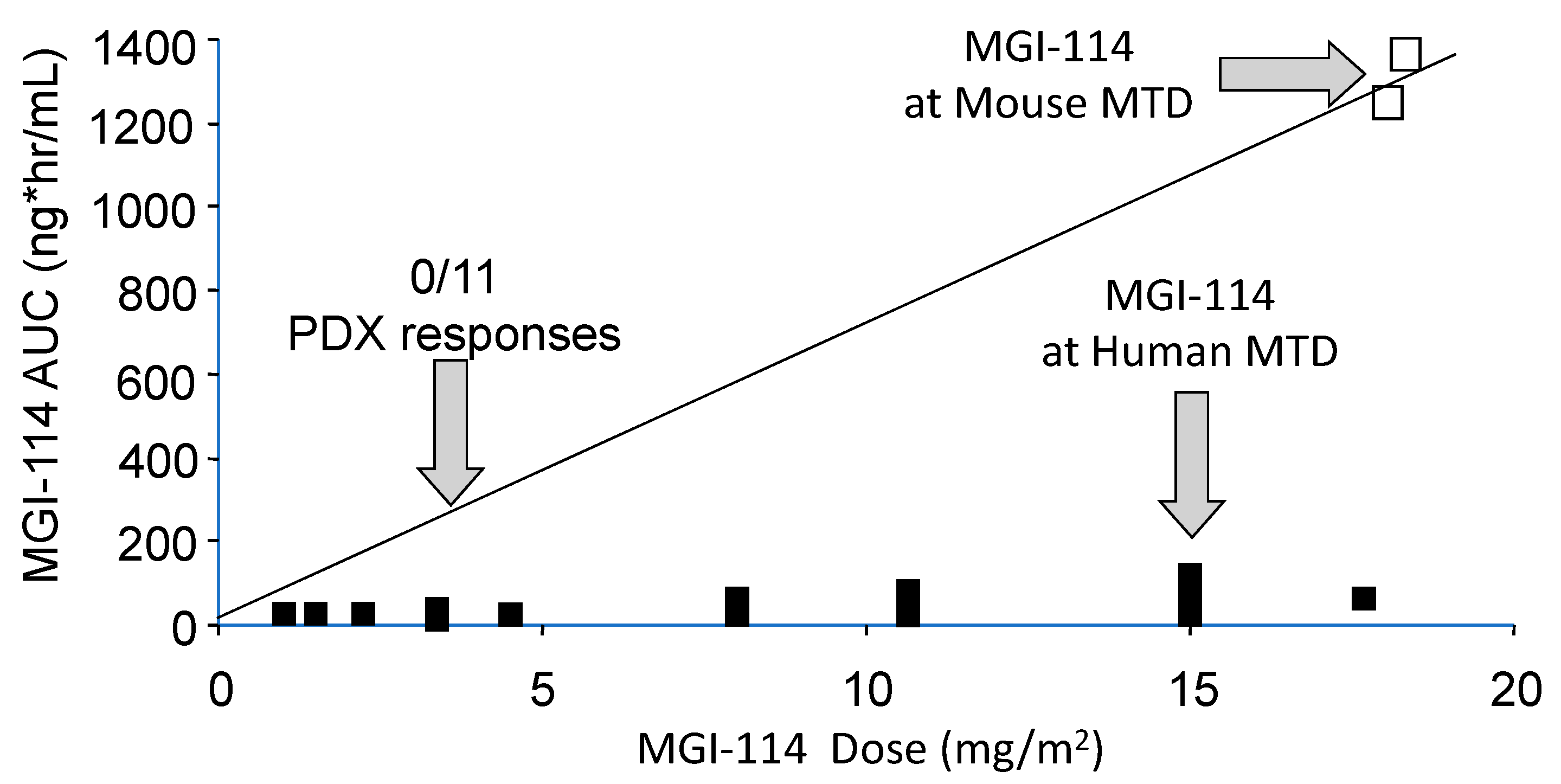
1.1. Differences in Species Tolerance
1.2. Preclinical Models Do Not Accurately Recapitulate Clinical Disease
1.3. Encompassing Genetic Heterogeneity of Clinical Cancer
1.4. Criteria for Defining ‘Antitumor Activity’ Is Less Stringent in Preclinical Studies Than in Clinical Trials
1.5. Unpredicted Toxicities in Patients
2. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Weigel, B.J.; Lyden, E.; Anderson, J.R.; Meyer, W.H.; Parham, D.M.; Rodeberg, D.A.; Michalski, J.M.; Hawkins, D.S.; Arndt, C.A. Intensive Multiagent Therapy, Including Dose-Compressed Cycles of Ifosfamide/Etoposide and Vincristine/Doxorubicin/Cyclophosphamide, Irinotecan, and Radiation, in Patients With High-Risk Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2016, 34, 117–122. [Google Scholar] [CrossRef]
- Balamuth, N.J.; Womer, R.B. Ewing’s sarcoma. Lancet Oncol. 2010, 11, 184–192. [Google Scholar] [CrossRef]
- Aljubran, A.H.; Griffin, A.; Pintilie, M.; Blackstein, M. Osteosarcoma in adolescents and adults: Survival analysis with and without lung metastases. Ann. Oncol. 2009, 20, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Stewart, M.; Blackman, S.; Rousseau, R.; Donoghue, M.; Cohen, K.; Seibel, N.; Fleury, M.; Benettaib, B.; Malik, R.; et al. Accelerating Pediatric Cancer Drug Development: Challenges and Opportunities for Pediatric Master Protocols. Ther. Innov. Regul. Sci. 2018. [Google Scholar] [CrossRef]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primers 2019, 5, 1. [Google Scholar] [CrossRef]
- Cooney, T.; Lane, A.; Bartels, U.; Bouffet, E.; Goldman, S.; Leary, S.E.S.; Foreman, N.K.; Packer, R.J.; Broniscer, A.; Minturn, J.E.; et al. Contemporary survival endpoints: An International Diffuse Intrinsic Pontine Glioma Registry study. Neuro-Oncology 2017, 19, 1279–1280. [Google Scholar] [CrossRef]
- Perkins, S.M.; Rubin, J.B.; Leonard, J.R.; Smyth, M.D.; El Naqa, I.; Michalski, J.M.; Simpson, J.R.; Limbrick, D.L.; Park, T.S.; Mansur, D.B. Glioblastoma in children: A single-institution experience. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 1117–1121. [Google Scholar] [CrossRef]
- Chow, E.J.; Chen, Y.; Hudson, M.M.; Feijen, E.A.M.; Kremer, L.C.; Border, W.L.; Green, D.M.; Meacham, L.R.; Mulrooney, D.A.; Ness, K.K.; et al. Prediction of Ischemic Heart Disease and Stroke in Survivors of Childhood Cancer. J. Clin. Oncol. 2018, 36, 44–52. [Google Scholar] [CrossRef]
- Eissa, H.M.; Lu, L.; Baassiri, M.; Bhakta, N.; Ehrhardt, M.J.; Triplett, B.M.; Green, D.M.; Mulrooney, D.A.; Robison, L.L.; Hudson, M.M.; et al. Chronic disease burden and frailty in survivors of childhood HSCT: A report from the St. Jude Lifetime Cohort Study. Blood Adv. 2017, 1, 2243–2246. [Google Scholar] [CrossRef]
- Henderson, T.O.; Oeffinger, K.C. Paediatrics: Addressing the health burden of childhood cancer survivors—Improvements are needed. Nat. Rev. Clin. Oncol. 2018, 15, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, L.M.; Neglia, J.P.; Reulen, R.C.; Ronckers, C.M.; van Leeuwen, F.E.; Morton, L.M.; Hodgson, D.C.; Yasui, Y.; Oeffinger, K.C.; Henderson, T.O. Risk, Risk Factors, and Surveillance of Subsequent Malignant Neoplasms in Survivors of Childhood Cancer: A Review. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Houghton, P.J.; Kurmasheva, R.T. Challenges and Opportunities for Childhood Cancer Drug Development. Pharmacol. Rev. 2019, 71, 671–697. [Google Scholar] [CrossRef]
- Morton, C.L.; Houghton, P.J. Establishment of human tumor xenografts in immunodeficient mice. Nat. Protoc. 2007, 2, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.K.; Houghton, P.J. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur. J. Cancer 2004, 40, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.E.; Etcubanas, E.; Christensen, M.L.; Houghton, J.A.; George, S.L.; Green, A.A.; Houghton, P.J. Phase II testing of melphalan in children with newly diagnosed rhabdomyosarcoma: A model for anticancer drug development. J. Clin. Oncol. 1988, 6, 308–314. [Google Scholar] [CrossRef]
- Kurmasheva, R.T.; Bandyopadhyay, A.; Favours, E.; Del Pozo, V.; Ghilu, S.; Phelps, D.A.; Erickson, S.W.; Peer, C.J.; Figg, W.D.; Smith, M.A.; et al. Evaluation of entinostat alone and in combination with standard-of-care cytotoxic agents against rhabdomyosarcoma xenograft models. Pediatr. Blood Cancer 2019, 66, e27820. [Google Scholar] [CrossRef]
- Eckhardt, S.G.; Baker, S.D.; Britten, C.D.; Hidalgo, M.; Siu, L.; Hammond, L.A.; Villalona-Calero, M.A.; Felton, S.; Drengler, R.; Kuhn, J.G.; et al. Phase I and pharmacokinetic study of irofulven, a novel mushroom-derived cytotoxin, administered for five consecutive days every four weeks in patients with advanced solid malignancies. J. Clin. Oncol. 2000, 18, 4086–4097. [Google Scholar] [CrossRef]
- Leggas, M.; Stewart, C.F.; Woo, M.H.; Fouladi, M.; Cheshire, P.J.; Peterson, J.K.; Friedman, H.S.; Billups, C.; Houghton, P.J. Relation between Irofulven (MGI-114) systemic exposure and tumor response in human solid tumor xenografts. Clin. Cancer Res. 2002, 8, 3000–3007. [Google Scholar]
- Santana, V.M.; Furman, W.L.; Billups, C.A.; Hoffer, F.; Davidoff, A.M.; Houghton, P.J.; Stewart, C.F. Improved response in high-risk neuroblastoma with protracted topotecan administration using a pharmacokinetically guided dosing approach. J. Clin. Oncol. 2005, 23, 4039–4047. [Google Scholar] [CrossRef]
- Maris, J.M.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Wu, J.; et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr. Blood Cancer 2010, 55, 26–34. [Google Scholar] [CrossRef]
- Carol, H.; Boehm, I.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Keir, S.T.; Wu, J.; et al. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother. Pharmacol. 2011, 68, 1291–1304. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Fox, E.; Teachey, D.T.; Reid, J.M.; Safgren, S.L.; Carol, H.; Lock, R.B.; Houghton, P.J.; Smith, M.A.; Hall, D.; et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children’s Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin. Cancer Res. 2019, 25, 3229–3238. [Google Scholar] [CrossRef]
- Carol, H.; Lock, R.; Houghton, P.J.; Morton, C.L.; Kolb, E.A.; Gorlick, R.; Reynolds, C.P.; Maris, J.M.; Keir, S.T.; Billups, C.A.; et al. Initial testing (stage 1) of the kinesin spindle protein inhibitor ispinesib by the pediatric preclinical testing program. Pediatr. Blood Cancer 2009, 53, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.B.; Carol, H.; Morton, C.L.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Wozniak, A.W.; Gorlick, R.; Kolb, E.A.; et al. Initial testing of the CENP-E inhibitor GSK923295A by the pediatric preclinical testing program. Pediatr. Blood Cancer 2012, 58, 916–923. [Google Scholar] [CrossRef]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Primers 2019, 5, 11. [Google Scholar] [CrossRef]
- Johnson, A.; Severson, E.; Gay, L.; Vergilio, J.A.; Elvin, J.; Suh, J.; Daniel, S.; Covert, M.; Frampton, G.M.; Hsu, S.; et al. Comprehensive Genomic Profiling of 282 Pediatric Low- and High-Grade Gliomas Reveals Genomic Drivers, Tumor Mutational Burden, and Hypermutation Signatures. Oncologist 2017, 22, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.; Federico, S.M.; Chen, X.; Shelat, A.A.; Bradley, C.; Gordon, B.; Karlstrom, A.; Twarog, N.R.; Clay, M.R.; Bahrami, A.; et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 2017, 549, 96–100. [Google Scholar] [CrossRef]
- Houghton, J.A.; Cook, R.L.; Lutz, P.J.; Houghton, P.J. Melphalan: A potential new agent in the treatment of childhood rhabdomyosarcoma. Cancer Treat. Rep. 1985, 69, 91–96. [Google Scholar] [PubMed]
- Houghton, J.A.; Houghton, P.J.; Brodeur, G.M.; Green, A.A. Development of resistance to vincristine in a childhood rhabdomyosarcoma growing in immune-deprived mice. Int. J. Cancer 1981, 28, 409–415. [Google Scholar] [CrossRef]
- Houghton, J.A.; Houghton, P.J.; Green, A.A. Chemotherapy of childhood rhabdomyosarcomas growing as xenografts in immune-deprived mice. Cancer Res. 1982, 42, 535–539. [Google Scholar]
- Houghton, J.A.; Houghton, P.J.; Webber, B.L. Growth and characterization of childhood rhabdomyosarcomas as xenografts. J. Natl. Cancer Inst. 1982, 68, 437–443. [Google Scholar] [PubMed]
- Lock, R.B.; Liem, N.; Farnsworth, M.L.; Milross, C.G.; Xue, C.; Tajbakhsh, M.; Haber, M.; Norris, M.D.; Marshall, G.M.; Rice, A.M. The nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and relapse. Blood 2002, 99, 4100–4108. [Google Scholar] [CrossRef]
- Lock, R.B.; Liem, N.L.; Papa, R.A. Preclinical testing of antileukemic drugs using an in vivo model of systemic disease. Methods Mol. Med. 2005, 111, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Liem, N.L.; Papa, R.A.; Milross, C.G.; Schmid, M.A.; Tajbakhsh, M.; Choi, S.; Ramirez, C.D.; Rice, A.M.; Haber, M.; Norris, M.D.; et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood 2004, 103, 3905–3914. [Google Scholar] [CrossRef]
- Jones, L.; Carol, H.; Evans, K.; Richmond, J.; Houghton, P.J.; Smith, M.A.; Lock, R.B. A review of new agents evaluated against pediatric acute lymphoblastic leukemia by the Pediatric Preclinical Testing Program. Leukemia 2016, 30, 2133–2141. [Google Scholar] [CrossRef]
- Samuels, A.L.; Beesley, A.H.; Yadav, B.D.; Papa, R.A.; Sutton, R.; Anderson, D.; Marshall, G.M.; Cole, C.H.; Kees, U.R.; Lock, R.B. A pre-clinical model of resistance to induction therapy in pediatric acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e232. [Google Scholar] [CrossRef]
- Yadav, B.D.; Samuels, A.L.; Wells, J.E.; Sutton, R.; Venn, N.C.; Bendak, K.; Anderson, D.; Marshall, G.M.; Cole, C.H.; Beesley, A.H.; et al. Heterogeneity in mechanisms of emergent resistance in pediatric T-cell acute lymphoblastic leukemia. Oncotarget 2016, 7, 58728–58742. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Morton, C.L.; Tucker, C.; Payne, D.; Favours, E.; Cole, C.; Gorlick, R.; Kolb, E.A.; Zhang, W.; Lock, R.; et al. The pediatric preclinical testing program: Description of models and early testing results. Pediatr. Blood Cancer 2007, 49, 928–940. [Google Scholar] [CrossRef]
- Geier, B.; Kurmashev, D.; Kurmasheva, R.T.; Houghton, P.J. Preclinical Childhood Sarcoma Models: Drug Efficacy Biomarker Identification and Validation. Front. Oncol. 2015, 5, 193. [Google Scholar] [CrossRef] [PubMed]
- Kurmasheva, R.T.; Houghton, P.J. Identifying novel therapeutic agents using xenograft models of pediatric cancer. Cancer Chemother. Pharmacol. 2016, 78, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Long, Y.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef]
- Ducreux, M.; Chamseddine, A.; Laurent-Puig, P.; Smolenschi, C.; Hollebecque, A.; Dartigues, P.; Samallin, E.; Boige, V.; Malka, D.; Gelli, M. Molecular targeted therapy of BRAF-mutant colorectal cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919856494. [Google Scholar] [CrossRef]
- Fangusaro, J.; Onar-Thomas, A.; Young Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Goldman, S.; et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262. [Google Scholar] [CrossRef]
- Murphy, B.; Yin, H.; Maris, J.M.; Kolb, E.A.; Gorlick, R.; Reynolds, C.P.; Kang, M.H.; Keir, S.T.; Kurmasheva, R.T.; Dvorchik, I.; et al. Evaluation of Alternative In Vivo Drug Screening Methodology: A Single Mouse Analysis. Cancer Res. 2016, 76, 5798–5809. [Google Scholar] [CrossRef]
- Hingorani, P.; Zhang, W.; Kurmasheva, R.; Zhang, Z.; Wang, Y.; Xu, Z.; Roth, M.; Gill, J.; Harrison, D.; Erickson, S.; et al. Abstract LB-217: Preclinical evaluation of trastuzumab deruxtecan (T-DXd; DS-8201a), a HER2 antibody-drug conjugate, in pediatric solid tumors by the Pediatric Preclinical Testing Consortium (PPTC). Cancer Res. 2020, 80, LB-217. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, L.; Meyer, W.H.; Lyden, E.; Rodeberg, D.A.; Indelicato, D.J.; Linardic, C.M.; Anderson, J.R.; Hawkins, D.S.; Soft Tissue Sarcoma Committee; Children’s Oncology Group. Randomized phase 2 trial of bevacizumab and temsirolimus in combination with vinorelbine and cyclophosphamide for first relapse/disease progression in rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2014, 32, 10003. [Google Scholar] [CrossRef]
- Grabowski, B. “P < 0.05” Might Not Mean What You Think: American Statistical Association Clarifies P Values. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: Status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef]
- Stegmaier, K.; Wong, J.S.; Ross, K.N.; Chow, K.T.; Peck, D.; Wright, R.D.; Lessnick, S.L.; Kung, A.L.; Golub, T.R. Signature-based small molecule screening identifies cytosine arabinoside as an EWS/FLI modulator in Ewing sarcoma. PLoS Med. 2007, 4, e122. [Google Scholar] [CrossRef]
- DuBois, S.G.; Krailo, M.D.; Lessnick, S.L.; Smith, R.; Chen, Z.; Marina, N.; Grier, H.E.; Stegmaier, K.; Children’s Oncology, G. Phase II study of intermediate-dose cytarabine in patients with relapsed or refractory Ewing sarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2009, 52, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | AUC @ Mouse MTD AUC @ Human MTD | Effective Dose Range from MTD in Mouse |
|---|---|---|
| Clinically Not Active | ||
| DMP-840 | 15–20 | ~2–3 |
| Carzelesin | ~80 | <2 |
| Sulophenur | ~8 | ~3 |
| MGI-114 | >15 | ~3 |
| Alisertib (MLN8237) | ~8 | ~3 |
| Clinically Active | ||
| Melphalan | 1 | ~2–3 |
| Topotecan | ~3 | ~10 |
| Irinotecan | 16 | ~100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghilu, S.; Kurmasheva, R.T.; Houghton, P.J. Developing New Agents for Treatment of Childhood Cancer: Challenges and Opportunities for Preclinical Testing. J. Clin. Med. 2021, 10, 1504. https://doi.org/10.3390/jcm10071504
Ghilu S, Kurmasheva RT, Houghton PJ. Developing New Agents for Treatment of Childhood Cancer: Challenges and Opportunities for Preclinical Testing. Journal of Clinical Medicine. 2021; 10(7):1504. https://doi.org/10.3390/jcm10071504
Chicago/Turabian StyleGhilu, Samson, Raushan T. Kurmasheva, and Peter J. Houghton. 2021. "Developing New Agents for Treatment of Childhood Cancer: Challenges and Opportunities for Preclinical Testing" Journal of Clinical Medicine 10, no. 7: 1504. https://doi.org/10.3390/jcm10071504
APA StyleGhilu, S., Kurmasheva, R. T., & Houghton, P. J. (2021). Developing New Agents for Treatment of Childhood Cancer: Challenges and Opportunities for Preclinical Testing. Journal of Clinical Medicine, 10(7), 1504. https://doi.org/10.3390/jcm10071504