
Bone Marrow Mastocytosis: A Diagnostic Challenge

 , , ,
, , ,
Abstract
1. Introduction
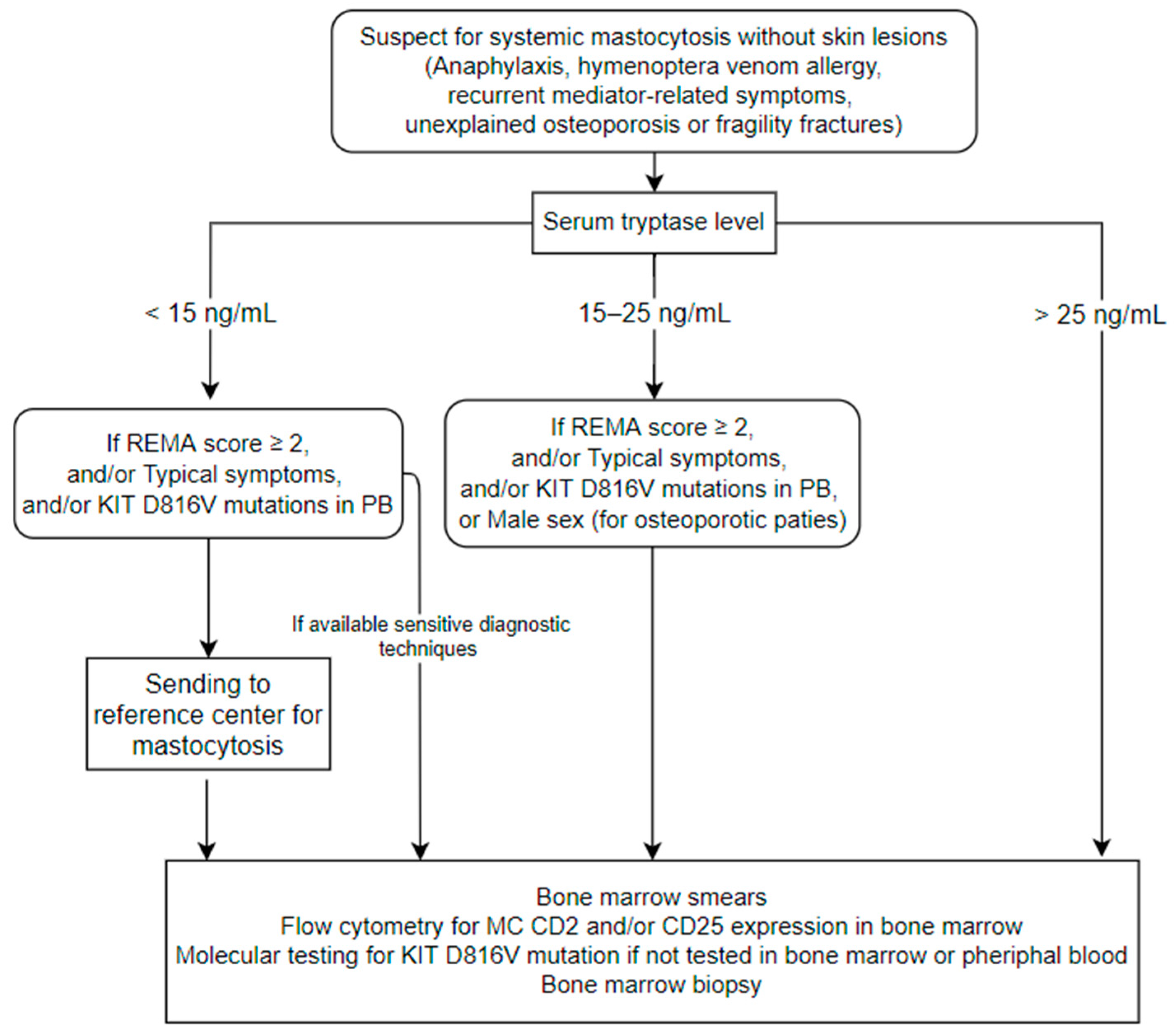
2. Bone Marrow Mastocytosis
3. Anaphylaxis and BMM
4. Osteoporosis and BMM
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valent, P.; Akin, C.; Escribano, L.; Födinger, M.; Hartmann, K.; Brockow, K.; Castells, M.; Sperr, W.R.; Kluin-Nelemans, H.C.; Hamdy, N.A.T.; et al. Standards and standardization in mastocytosis: Consensus statements on diagnostics, treatment recommendations and response criteria. Eur. J. Clin. Investig. 2007, 37, 435–453. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Montero, A.C.; Jara-Acevedo, M.; Teodosio, C.; Sanchez, M.L.; Nunez, R.; Prados, A.; Aldanondo, I.; Sanchez, L.; Dominguez, M.; Botana, L.M.; et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: A prospective study of the Spanish network on mastocytosis (REMA) in a series of 113 patients. Blood 2006, 108, 2366–2372. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, T.; Vestergaard, H.; Møller, M.B. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time QPCR assay. J. Mol. Diagn. 2011, 13, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Langenfeld, F.; Jeanningros, S.; Brenet, F.; Soucie, E.; Hermine, O.; Damaj, G.; Dubreuil, P.; Arock, M. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol. Allergy Clin. N. Am. 2014, 34, 239–262. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggendorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Kluger, S.; Naumann, N.; et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 Identify a high-risk group of patients with KIT D816V + advanced systemic mastocytosis. Leukemia 2016, 30, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.; Elala, Y.; Wassie, E.; Finke, C.; Reichard, K.K.; Chen, D.; Hanson, C.A.; Ketterling, R.P.; Tefferi, A. Next-generation sequencing in systemic mastocytosis: Derivation of a mutation-augmented clinical prognostic model for survival. Am. J. Hematol. 2016, 91, 888–893. [Google Scholar] [CrossRef]
- Muñoz-González, J.I.; Jara-Acevedo, M.; Alvarez-Twose, I.; Merker, J.D.; Teodosio, C.; Hou, Y.; Henriques, A.; Roskin, K.M.; Sanchez-Muñoz, L.; Tsai, A.G.; et al. Impact of somatic and germline mutations on the outcome of systemic mastocytosis. Blood Adv. 2018, 2, 2814–2828. [Google Scholar] [CrossRef]
- Cohen, S.S.; Skovbo, S.; Vestergaard, H.; Kristensen, T.; Møller, M.; Bindslev-Jensen, C.; Fryzek, J.P.; Broesby-Olsen, S. Epidemiology of systemic mastocytosis in Denmark. Br. J. Haematol. 2014, 166, 521–528. [Google Scholar] [CrossRef]
- Van Doormaal, J.J.; Arends, S.; Brunekreeft, K.L.; van der Wal, V.B.; Sietsma, J.; van Voorst Vader, P.C.; Oude Elberink, J.N.G.; Kluin-Nelemans, J.C.; van der Veer, E.; de Monchy, J.G.R. Prevalence of indolent systemic mastocytosis in a Dutch region. J. Allergy Clin. Immunol. 2013, 131, 1429–1431. [Google Scholar] [CrossRef]
- Escribano, L.; Diaz-Agustin, B.; López, A.; López, R.N.; García-Montero, A.; Almeida, J.; Prados, A.; Angulo, M.; Herrero, S.; Orfao, A. Immunophenotypic analysis of mast cells in mastocytosis: When and how to do it. Proposals of the Spanish network on mastocytosis (REMA). Cytom. B Clin. Cytom. 2004, 58, 1–8. [Google Scholar] [CrossRef]
- Perbellini, O.; Zamò, A.; Colarossi, S.; Zampieri, F.; Zoppi, F.; Bonadonna, P.; Schena, D.; Artuso, A.; Martinelli, G.; Chilosi, M.; et al. Primary role of multiparametric flow cytometry in the diagnostic work-up of indolent clonal mast cell disorders. Cytom. B Clin. Cytom. 2011, 80, 362–368. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, G.; Zanotti, R.; Colarossi, S.; de Benedittis, C.; Garcia-Montero, A.; Bonifacio, M.; Sartori, M.; Aprili, F.; Caruso, B.; Paviati, E.; et al. The impact of sensitive KIT D816V detection on recognition of indolent systemic mastocytosis. Leuk. Res. 2015, 39, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, P.; Perbellini, O.; Passalacqua, G.; Caruso, B.; Colarossi, S.; dal Fior, D.; Castellani, L.; Bonetto, C.; Frattini, F.; Dama, A.; et al. Clonal mast cell disorders in patients with systemic reactions to hymenoptera stings and increased serum tryptase levels. J. Allergy Clin. Immunol. 2009, 123, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, R.; Bonadonna, P.; Bonifacio, M.; Artuso, A.; Schena, D.; Rossini, M.; Perbellini, O.; Colarossi, S.; Chilosi, M.; Pizzolo, G. Isolated bone marrow mastocytosis: An underestimated subvariant of indolent systemic mastocytosis. Haematologica 2011, 96, 482–484. [Google Scholar] [CrossRef]
- Zanotti, R.; Lombardo, C.; Passalacqua, G.; Caimmi, C.; Bonifacio, M.; de Matteis, G.; Perbellini, O.; Rossini, M.; Schena, D.; Busa, M.; et al. Clonal mast cell disorders in patients with severe hymenoptera venom allergy and normal serum tryptase levels. J. Allergy Clin. Immunol. 2015, 136, 135–139. [Google Scholar] [CrossRef]
- Valent, P.; Escribano, L.; Broesby-Olsen, S.; Hartmann, K.; Grattan, C.; Brockow, K.; Niedoszytko, M.; Nedoszytko, B.; Oude Elberink, J.N.G.; Kristensen, T.; et al. Proposed diagnostic algorithm for patients with suspected mastocytosis: A proposal of the European competence network on mastocytosis. Allergy 2014, 69, 1267–1274. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Beau, M.M.L.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Valent, P.; Horny, H.P.; Escribano, L.; Longley, B.J.; Li, C.Y.; Schwartz, L.B.; Marone, G.; Nuñez, R.; Akin, C.; Sotlar, K.; et al. Diagnostic criteria and classification of mastocytosis: A consensus proposal. Leuk. Res. 2001, 25, 603–625. [Google Scholar] [CrossRef]
- Lim, K.H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.Y.; Pardanani, A. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef]
- Georgin-Lavialle, S.; Lhermitte, L.; Dubreuil, P.; Chandesris, M.O.; Hermine, O.; Damaj, G. Mast cell leukemia. Blood 2013, 121, 1285–1295. [Google Scholar] [CrossRef]
- Metcalfe, D.D. Classification and diagnosis of mastocytosis: Current status. J. Investig. Dermatol. 1991, 96, S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Beau, M.M.L.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lim, K.H.; Lasho, T.L.; Finke, C.M.; McClure, R.F.; Li, C.Y.; Tefferi, A. WHO subvariants of indolent mastocytosis: Clinical details and prognostic evaluation in 159 consecutive adults. Blood 2010, 115, 150–151. [Google Scholar] [CrossRef]
- Florian, S.; Krauth, M.T.; Simonitsch-Klupp, I.; Sperr, W.R.; Fritsche-Polanz, R.; Sonneck, K.; Födinger, M.; Agis, H.; Böhm, A.; Wimazal, F.; et al. Indolent systemic mastocytosis with elevated serum tryptase, absence of skin lesions, and recurrent severe anaphylactoid episodes. Int. Arch. Allergy Immunol. 2005, 136, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Twose, I.; Zanotti, R.; González-de-Olano, D.; Bonadonna, P.; Vega, A.; Matito, A.; Sánchez-Muñoz, L.; Morgado, J.M.; Perbellini, O.; García-Montero, A.; et al. Nonaggressive systemic mastocytosis (SM) without skin lesions associated with insect-induced anaphylaxis shows unique features versus other indolent SM. J. Allergy Clin. Immunol. 2014, 133, 520–528. [Google Scholar] [CrossRef]
- Gülen, T.; Hägglund, H.; Sander, B.; Dahlén, B.; Nilsson, G. The presence of mast cell clonality in patients with unexplained anaphylaxis. Clin. Exp. Allergy 2014, 44, 1179–1187. [Google Scholar] [CrossRef]
- Akin, C.; Scott, L.M.; Kocabas, C.N.; Kushnir-Sukhov, N.; Brittain, E.; Noel, P.; Metcalfe, D.D. Demonstration of an aberrant mast-cell population with clonal markers in a subset of patients with “idiopathic” anaphylaxis. Blood 2007, 110, 2331–2333. [Google Scholar] [CrossRef] [PubMed]
- Pieri, L.; Bonadonna, P.; Elena, C.; Papayannidis, C.; Grifoni, F.I.; Rondoni, M.; Girlanda, S.; Mauro, M.; Magliacane, D.; Elli, E.M.; et al. Clinical presentation and management practice of systemic mastocytosis. A survey on 460 Italian patients. Am. J. Hematol. 2016, 91, 692–699. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, L.; Alvarez-Twose, I.; García-Montero, A.C.; Teodosio, C.; Jara-Acevedo, M.; Pedreira, C.E.; Matito, A.; Morgado, J.M.T.; Sánchez, M.L.; Mollejo, M.; et al. Evaluation of the WHO criteria for the classification of patients with mastocytosis. Mod. Pathol. 2011, 24, 1157–1168. [Google Scholar] [CrossRef]
- Escribano, L.; Alvarez-Twose, I.; Sánchez-Muñoz, L.; Garcia-Montero, A.; Núñez, R.; Almeida, J.; Jara-Acevedo, M.; Teodósio, C.; García-Cosío, M.; Bellas, C.; et al. Prognosis in adult indolent systemic mastocytosis: A long-term study of the Spanish network on mastocytosis in a Series of 145 Patients. J. Allergy Clin. Immunol. 2009, 124, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Twose, I.; González de Olano, D.; Sánchez-Muñoz, L.; Matito, A.; Esteban-López, M.I.; Vega, A.; Mateo, M.B.; Alonso Díaz de Durana, M.D.; de la Hoz, B.; del Pozo Gil, M.D.; et al. Clinical, biological, and molecular characteristics of clonal mast cell disorders presenting with systemic mast cell activation symptoms. J. Allergy Clin. Immunol. 2010, 125, 1269–1278. [Google Scholar] [CrossRef]
- Rossini, M.; Zanotti, R.; Bonadonna, P.; Artuso, A.; Caruso, B.; Schena, D.; Vecchiato, D.; Bonifacio, M.; Viapiana, O.; Gatti, D.; et al. Bone mineral density, bone turnover markers and fractures in patients with indolent systemic mastocytosis. Bone 2011, 49, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Sonneck, K.; Florian, S.; Müllauer, L.; Wimazal, F.; Födinger, M.; Sperr, W.R.; Valent, P. Diagnostic and subdiagnostic accumulation of mast cells in the bone marrow of patients with anaphylaxis: Monoclonal mast cell activation syndrome. Int. Arch. Allergy Immunol. 2007, 142, 158–164. [Google Scholar] [CrossRef]
- Pardanani, A.; Chen, D.; Abdelrahman, R.A.; Reichard, K.K.; Zblewski, D.; Wood, A.J.; McClure, R.F.; Butterfield, J.H.; Hanson, C.A.; Tefferi, A. Clonal mast cell disease not meeting WHO criteria for diagnosis of mastocytosis: Clinicopathologic features and comparison with indolent mastocytosis. Leukemia 2013, 27, 2091–2094. [Google Scholar] [CrossRef] [PubMed]
- Sperr, W.R.; Jordan, J.H.; Fiegl, M.; Escribano, L.; Bellas, C.; Dirnhofer, S.; Semper, H.; Simonitsch-Klupp, I.; Horny, H.P.; Valent, P. Serum tryptase levels in patients with mastocytosis: Correlation with mast cell burden and implication for defining the category of disease. Int. Arch. Allergy Immunol. 2002, 128, 136–141. [Google Scholar] [CrossRef]
- Lyons, J.J.; Chovanec, J.; O’Connell, M.P.; Liu, Y.; Šelb, J.; Zanotti, R.; Bai, Y.; Kim, J.; Le, Q.T.; DiMaggio, T.; et al. Heritable risk for severe anaphylaxis associated with increased α-tryptase-encoding germline copy number at TPSAB1. J. Allergy Clin. Immunol. 2021, 147, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Greiner, G.; Sprinzl, B.; Górska, A.; Ratzinger, F.; Gurbisz, M.; Witzeneder, N.; Schmetterer, K.G.; Gisslinger, B.; Uyanik, G.; Hadzijusufovic, E.; et al. Hereditary α tryptasemia is a valid genetic biomarker for severe mediator-related symptoms in mastocytosis. Blood 2021, 137, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Gülen, T.; Hägglund, H.; Dahlén, B.; Nilsson, G. High prevalence of anaphylaxis in patients with systemic mastocytosis—A single-centre experience. Clin. Exp. Allergy 2014, 44, 121–129. [Google Scholar] [CrossRef] [PubMed]
- González de Olano, D.; de la Hoz Caballer, B.; Núñez López, R.; Sánchez Muñoz, L.; Cuevas Agustín, M.; Diéguez, M.C.; Alvarez Twose, I.; Castells, M.C.; Escribano Mora, L. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: A study of the Spanish network on mastocytosis (REMA). Clin. Exp. Allergy 2007, 37, 1547–1555. [Google Scholar] [CrossRef]
- Brockow, K.; Jofer, C.; Behrendt, H.; Ring, J. Anaphylaxis in patients with mastocytosis: A study on history, clinical features and risk factors in 120 patients. Allergy 2008, 63, 226–232. [Google Scholar] [CrossRef]
- Muraro, A.; Roberts, G.; Worm, M.; Bilò, M.B.; Brockow, K.; Fernández Rivas, M.; Santos, A.F.; Zolkipli, Z.Q.; Bellou, A.; Beyer, K.; et al. Anaphylaxis: Guidelines from the European academy of allergy and clinical immunology. Allergy 2014, 69, 1026–1045. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.; Camargo, C.A.; Bohlke, K.; Jick, H.; Miller, R.L.; Sheikh, A.; Simons, F.E.R. Epidemiology of anaphylaxis: Findings of the American college of allergy, asthma and immunology epidemiology of anaphylaxis working group. Ann. Allergy Asthma Immunol. 2006, 97, 596–602. [Google Scholar] [CrossRef]
- Jara-Acevedo, M.; Teodosio, C.; Sanchez-Muñoz, L.; Álvarez-Twose, I.; Mayado, A.; Caldas, C.; Matito, A.; Morgado, J.M.; Muñoz-González, J.I.; Escribano, L.; et al. Detection of the KIT D816V mutation in peripheral blood of systemic mastocytosis: Diagnostic implications. Mod. Pathol. 2015, 28, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Biló, B.M.; Rueff, F.; Mosbech, H.; Bonifazi, F.; Oude-Elberink, J.N.G.; EAACI Interest Group on Insect Venom Hypersensitivity. Diagnosis of hymenoptera venom allergy. Allergy 2005, 60, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Niedoszytko, M.; de Monchy, J.; van Doormaal, J.J.; Jassem, E.; Oude Elberink, J.N.G. Mastocytosis and insect venom allergy: Diagnosis, safety and efficacy of venom immunotherapy. Allergy 2009, 64, 1237–1245. [Google Scholar] [CrossRef]
- Bonadonna, P.; Lombardo, C.; Zanotti, R. Mastocytosis and allergic diseases. J. Investig. Allergol. Clin. Immunol. 2014, 24, 288–297. [Google Scholar]
- Bonadonna, P.; Scaffidi, L. Hymenoptera anaphylaxis as a clonal mast cell disorder. Immunol. Allergy Clin. North Am. 2018, 38, 455–468. [Google Scholar] [CrossRef]
- Dubois, A.E.J. Mastocytosis and hymenoptera allergy. Curr. Opin. Allergy Clin. Immunol. 2004, 4, 291–295. [Google Scholar] [CrossRef]
- Haeberli, G.; Brönnimann, M.; Hunziker, T.; Müller, U. Elevated basal serum tryptase and hymenoptera venom allergy: Relation to severity of sting reactions and to safety and efficacy of venom immunotherapy. Clin. Exp. Allergy 2003, 33, 1216–1220. [Google Scholar] [CrossRef]
- Ruëff, F.; Placzek, M.; Przybilla, B. Mastocytosis and hymenoptera venom allergy. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 284–288. [Google Scholar] [CrossRef]
- Potier, A.; Lavigne, C.; Chappard, D.; Verret, J.L.; Chevailler, A.; Nicolie, B.; Drouet, M. Cutaneous manifestations in hymenoptera and diptera anaphylaxis: Relationship with basal serum tryptase. Clin. Exp. Allergy 2009, 39, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Guenova, E.; Volz, T.; Eichner, M.; Hoetzenecker, W.; Caroli, U.; Griesinger, G.; Burow, G.; Mitev, V.; Biedermann, T. Basal serum tryptase as risk assessment for severe hymenoptera sting reactions in elderly. Allergy 2010, 65, 919–923. [Google Scholar] [CrossRef]
- Schwartz, L.B. Diagnostic value of tryptase in anaphylaxis and mastocytosis. Immunol. Allergy Clin. North Am. 2006, 26, 451–463. [Google Scholar] [CrossRef]
- Bonadonna, P.; Gonzalez-de-Olano, D.; Zanotti, R.; Riccio, A.; de Ferrari, L.; Lombardo, C.; Rogkakou, A.; Escribano, L.; Alvarez-Twose, I.; Matito, A.; et al. Venom immunotherapy in patients with clonal mast cell disorders: Efficacy, safety, and practical considerations. J. Allergy Clin. Immunol. Pract. 2013, 1, 474–478. [Google Scholar] [CrossRef]
- Sturm, G.J.; Varga, E.M.; Roberts, G.; Mosbech, H.; Bilò, M.B.; Akdis, C.A.; Antolín-Amérigo, D.; Cichocka-Jarosz, E.; Gawlik, R.; Jakob, T.; et al. EAACI guidelines on allergen immunotherapy: Hymenoptera venom allergy. Allergy 2018, 73, 744–764. [Google Scholar] [CrossRef] [PubMed]
- Incorvaia, C.; Mauro, M.; Gritti, B.L.; Makri, E.; Ridolo, E. Venom immunotherapy in patients with allergic reactions to insect stings. Expert Rev. Clin. Immunol. 2018, 14, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Bilò, M.B.; Cichocka-Jarosz, E.; Pumphrey, R.; Oude-Elberink, J.N.; Lange, J.; Jakob, T.; Bonadonna, P.; Fernandez, J.; Kosnik, M.; Helbling, A.; et al. Self-medication of anaphylactic reactions due to hymenoptera stings-an EAACI task force consensus statement. Allergy 2016, 71, 931–943. [Google Scholar] [CrossRef]
- Hermans, M.A.W.; Rietveld, M.J.A.; van Laar, J.A.M.; Dalm, V.A.S.H.; Verburg, M.; Pasmans, S.G.M.A.; Gerth van Wijk, R.; van Hagen, P.M.; van Daele, P.L.A. Systemic mastocytosis: A cohort study on clinical characteristics of 136 patients in a large tertiary centre. Eur. J. Intern. Med. 2016, 30, 25–30. [Google Scholar] [CrossRef]
- Barete, S.; Assous, N.; de Gennes, C.; Grandpeix, C.; Feger, F.; Palmerini, F.; Dubreuil, P.; Arock, M.; Roux, C.; Launay, J.M.; et al. Systemic mastocytosis and bone involvement in a cohort of 75 patients. Ann. Rheum. Dis. 2010, 69, 1838–1841. [Google Scholar] [CrossRef]
- Van der Veer, E.; Arends, S.; van der Hoek, S.; Versluijs, J.B.; de Monchy, J.G.R.; Oude Elberink, J.N.G.; van Doormaal, J.J. Predictors of new fragility fractures after diagnosis of indolent systemic mastocytosis. J. Allergy Clin. Immunol. 2014, 134, 1413–1421. [Google Scholar] [CrossRef]
- Degboé, Y.; Eischen, M.; Nigon, D.; Apoil, P.A.; Mailhol, C.; Tournier, E.; Laurent, C.; Hanssens, K.; Hermine, O.; Paul, C.; et al. Prevalence and risk factors for fragility fracture in systemic mastocytosis. Bone 2017, 105, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Broesby-Olsen, S.; Farkas, D.K.; Vestergaard, H.; Hermann, A.P.; Møller, M.B.; Mortz, C.G.; Kristensen, T.K.; Bindslev-Jensen, C.; Sørensen, H.T.; Frederiksen, H. Risk of solid cancer, cardiovascular disease, anaphylaxis, osteoporosis and fractures in patients with systemic mastocytosis: A nationwide population-based study. Am. J. Hematol. 2016, 91, 1069–1075. [Google Scholar] [CrossRef]
- Orsolini, G.; Viapiana, O.; Rossini, M.; Bonifacio, M.; Zanotti, R. Bone disease in mastocytosis. Immunol. Allergy Clin. North Am. 2018, 38, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Carosi, G.; Guabello, G.; Longhi, M.; Grifoni, F.; Passeri, E.; Corbetta, S. Hypertryptasemia and mast cell-related disorders in severe osteoporotic patients. Mediat. Inflamm. 2020, 2020, 5785378. [Google Scholar] [CrossRef] [PubMed]
- Riffel, P.; Schwaab, J.; Lutz, C.; Naumann, N.; Metzgeroth, G.; Fabarius, A.; Schoenberg, S.O.; Hofmann, W.K.; Valent, P.; Reiter, A.; et al. An increased bone mineral density is an adverse prognostic factor in patients with systemic mastocytosis. J. Cancer Res. Clin. Oncol. 2020, 146, 945–951. [Google Scholar] [CrossRef]
- Leone, A.; Criscuolo, M.; Gullì, C.; Petrosino, A.; Carlo Bianco, N.; Colosimo, C. Systemic mastocytosis revisited with an emphasis on skeletal manifestations. Radiol. Med. 2020. [Google Scholar] [CrossRef]


{kind=link}
| DIAGNOSTIC CRITERIA | B FINDING III | C FINDING IV | |
|---|---|---|---|
| ONE MAJOR I CRITERIA AND ONE MINOR II OR THREE MINORS | |||
| BMM | YES | ˂2 | NO |
| ISM | YES | ˂2 | NO |
| SSM | YES | ≥2 | NO |
| ASM | YES | ≥1 | |
| SM—AHN | YES | ASSOCIATED HEMATOLOGIC NEOPLASM (non MC-lineage) | |
| MCL | ≥20% MAST CELL IN BONE MARROW SMEAR |
| VARIABLE | SCORE | |
|---|---|---|
| GENDER | Male | +1 |
| Female | −1 | |
| CLINICAL SYMPTOMS | Absence of hives, pruritus and angioedema | +1 |
| Hives, pruritus and angioedema | −2 | |
| Presyncope and/or syncope | +3 | |
| S-BASAL TRYPTASE | <15 ng/mL | −1 |
| >25 ng/mL | +1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanotti, R.; Tanasi, I.; Bernardelli, A.; Orsolini, G.; Bonadonna, P. Bone Marrow Mastocytosis: A Diagnostic Challenge. J. Clin. Med. 2021, 10, 1420. https://doi.org/10.3390/jcm10071420
Zanotti R, Tanasi I, Bernardelli A, Orsolini G, Bonadonna P. Bone Marrow Mastocytosis: A Diagnostic Challenge. Journal of Clinical Medicine. 2021; 10(7):1420. https://doi.org/10.3390/jcm10071420
Chicago/Turabian StyleZanotti, Roberta, Ilaria Tanasi, Andrea Bernardelli, Giovanni Orsolini, and Patrizia Bonadonna. 2021. "Bone Marrow Mastocytosis: A Diagnostic Challenge" Journal of Clinical Medicine 10, no. 7: 1420. https://doi.org/10.3390/jcm10071420
APA StyleZanotti, R., Tanasi, I., Bernardelli, A., Orsolini, G., & Bonadonna, P. (2021). Bone Marrow Mastocytosis: A Diagnostic Challenge. Journal of Clinical Medicine, 10(7), 1420. https://doi.org/10.3390/jcm10071420