
Comparison of Clinical Characteristics and Outcomes between Idiopathic and Secondary Pleuroparenchymal Fibroelastosis

 ,
,
Abstract
1. Introduction
2. Materials and Methods
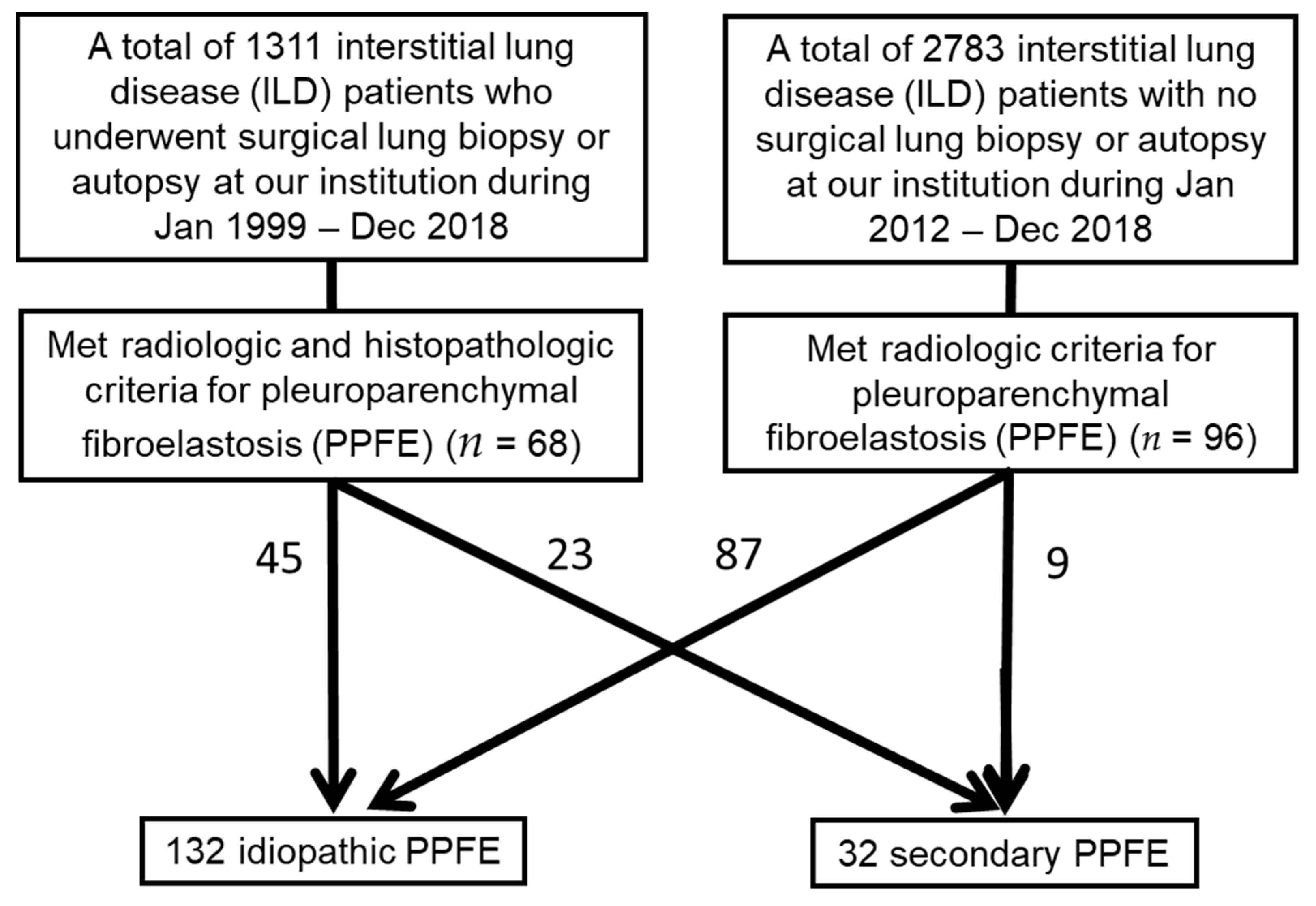
2.1. Patient Selection and Data Collection
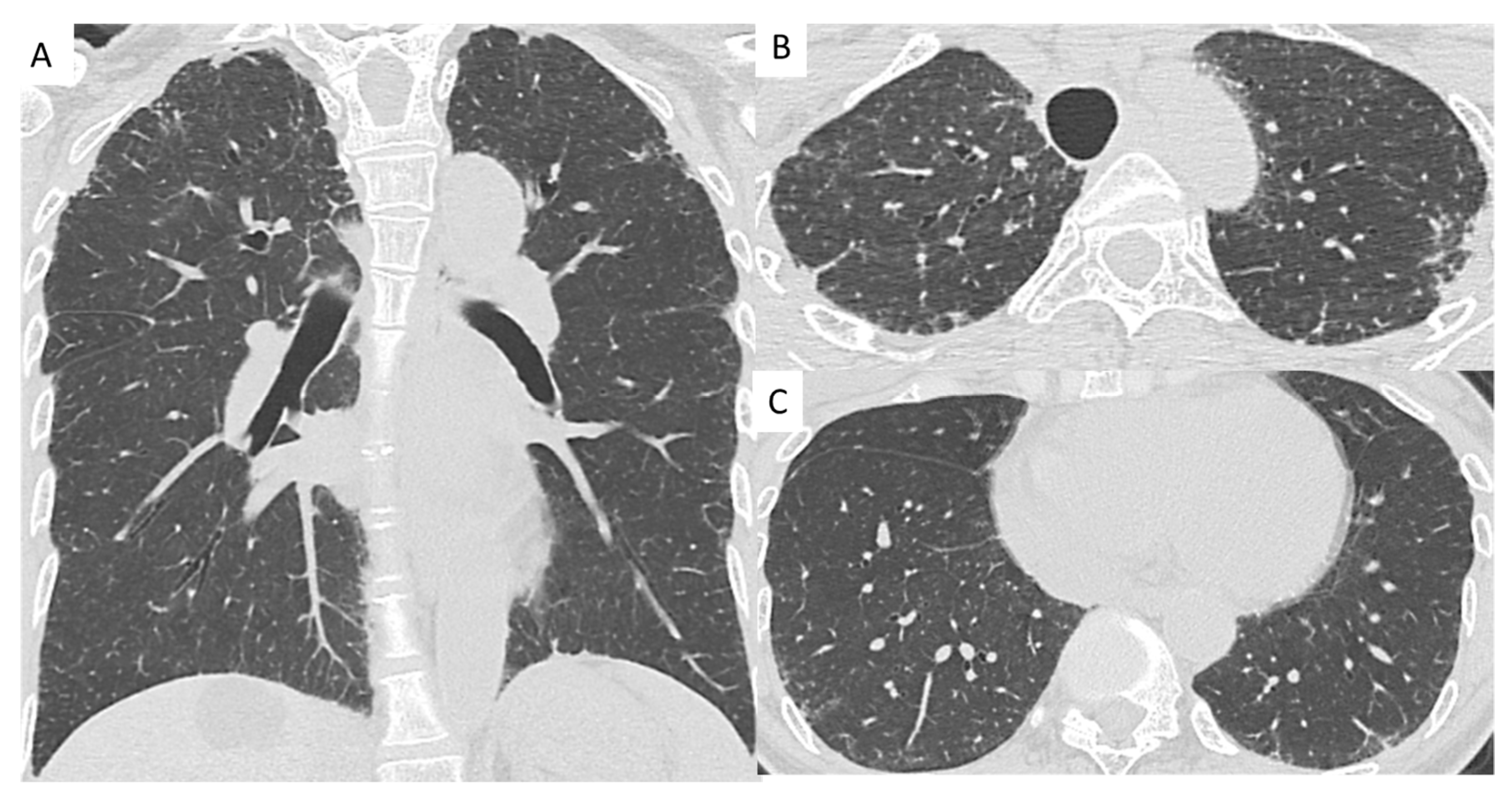
2.2. High-Resolution Computed Tomography Evaluation
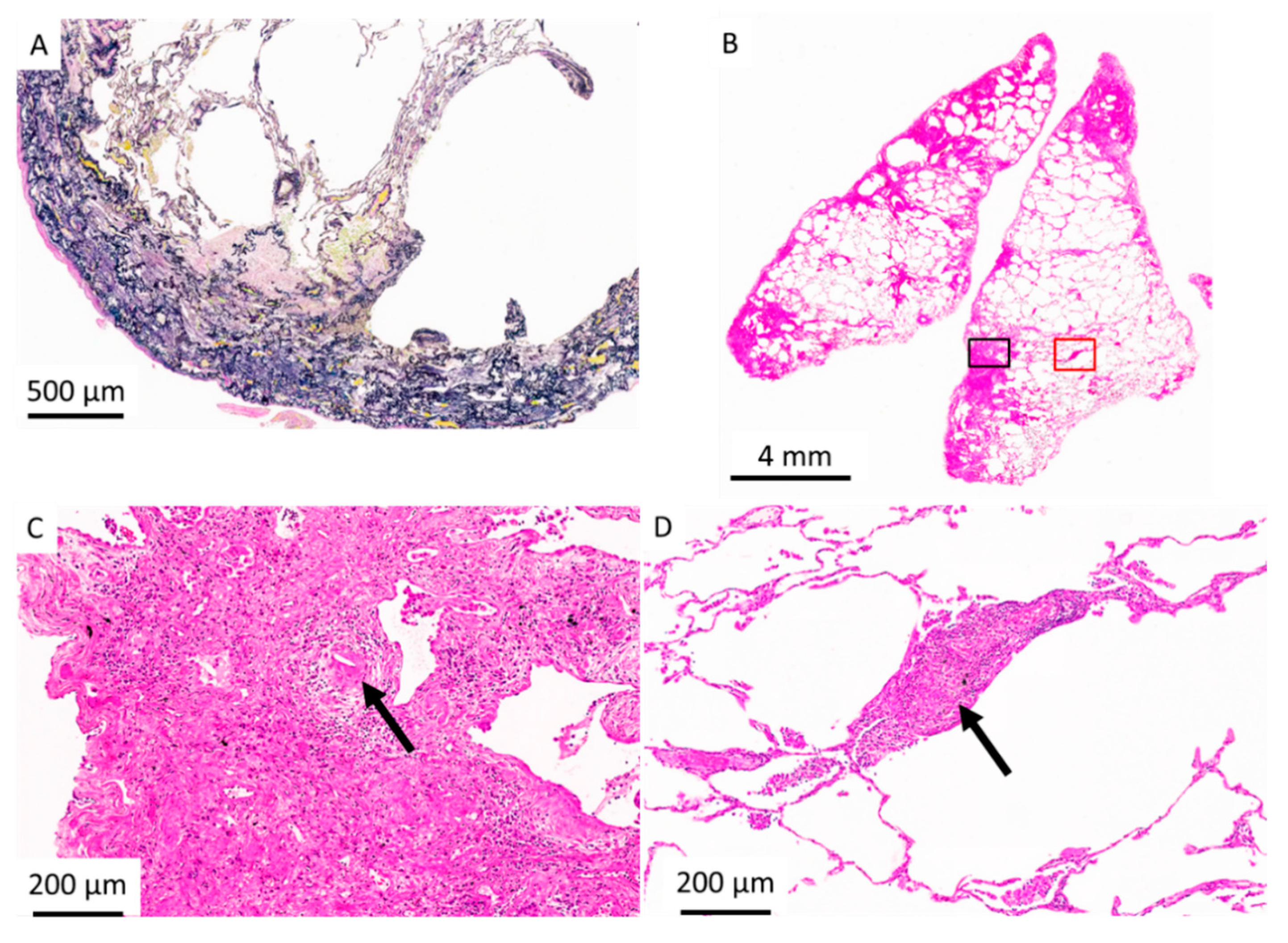
2.3. Histopathologic Criteria for PPFE
2.4. Statistical Analyses
3. Results
3.1. Clinical Characteristics, PFTs, and BAL Fluid Findings
3.2. Predictor of Mortality for Idiopathic and Secondary PPFE
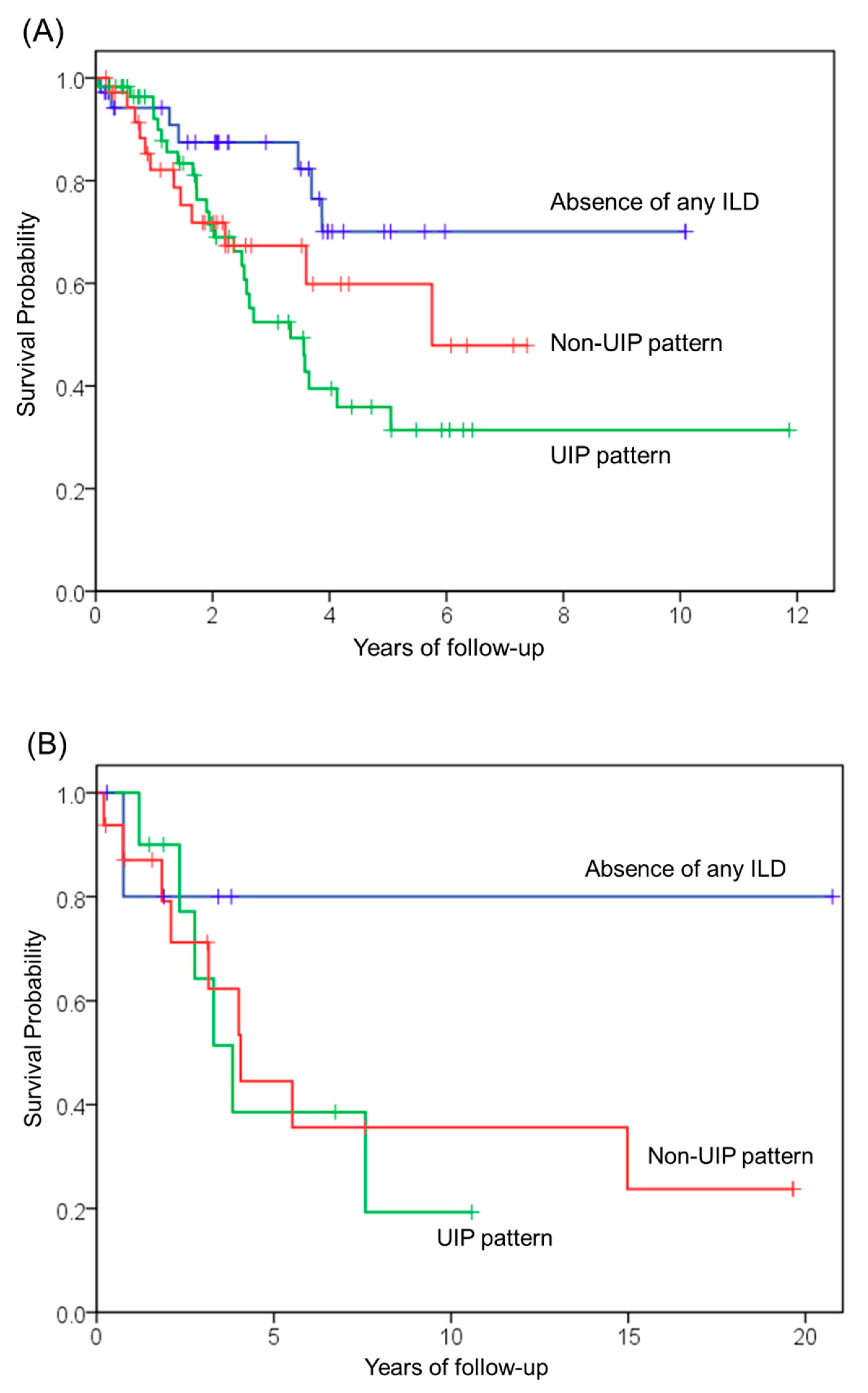
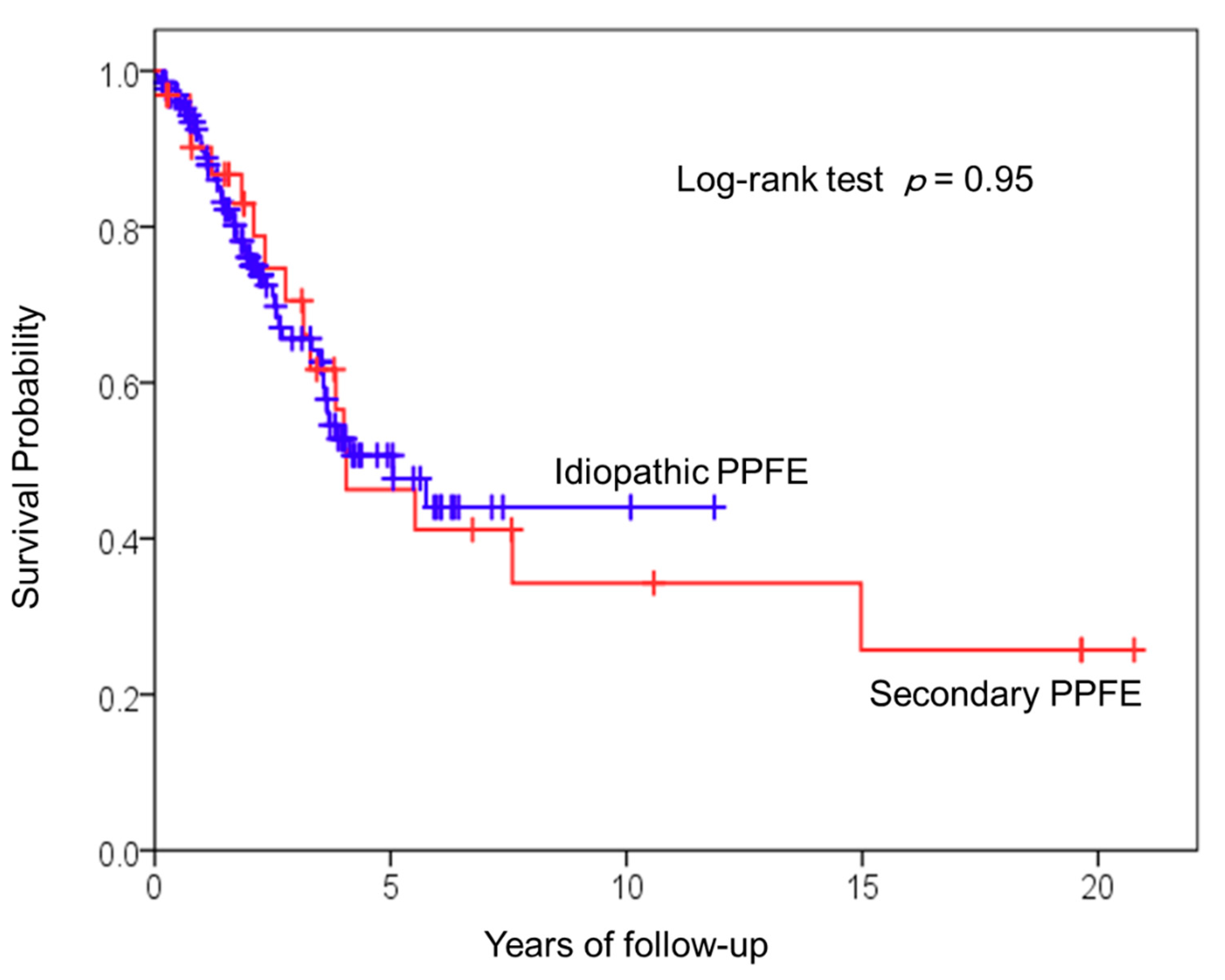
3.3. Survival Analysis of Patients with Idiopathic and Secondary PPFE
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frankel, S.K.; Cool, C.D.; Lynch, D.A.; Brown, K.K. Idiopathic pleuroparenchymal fibroelastosis: Description of a novel clinicopathologic entity. Chest 2004, 126, 2007–2013. [Google Scholar] [CrossRef]
- Mariani, F.; Gatti, B.; Rocca, A.; Bonifazi, F.; Cavazza, A.; Fanti, S.; Tomassetti, S.; Piciucchi, S.; Poletti, V.; Zompatori, M. Pleuroparenchymal fibroelastosis: The prevalence of secondary forms in hematopoietic stem cell and lung transplantation recipients. Diagn. Interv. Radiol. 2016, 22, 400–406. [Google Scholar] [CrossRef]
- von der Thusen, J.H.; Hansell, D.M.; Tominaga, M.; Veys, P.A.; Ashworth, M.T.; Owens, C.M.; Nicholson, A.G. Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod. Pathol. 2011, 24, 1633–1639. [Google Scholar] [CrossRef]
- Ofek, E.; Sato, M.; Saito, T.; Wagnetz, U.; Roberts, H.C.; Chaparro, C.; Waddell, T.K.; Singer, L.G.; Hutcheon, M.A.; Keshavjee, S.; et al. Restrictive allograft syndrome post lung transplantation is characterized by pleuroparenchymal fibroelastosis. Mod. Pathol. 2013, 26, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Nakamura, Y.; Colby, T.V.; Johkoh, T.; Sumikawa, H.; Nishimoto, K.; Yoshimura, K.; Matsushima, S.; Oyama, Y.; Hozumi, H.; et al. Radiologic pleuroparenchymal fibroelastosis-like lesion in connective tissue disease-related interstitial lung disease. PLoS ONE 2017, 12, e0180283. [Google Scholar] [CrossRef]
- Hassoun, D.; Dirou, S.; Arrigoni, P.P.; Durant, C.; Hamidou, M.; Neel, A.; Agard, C. Radiological pleuroparenchymal fibroelastosis associated to limited cutaneous systemic sclerosis: A case report. BMC Pulm. Med. 2018, 18, 73. [Google Scholar] [CrossRef]
- Bonifazi, M.; Sverzellati, N.; Negri, E.; Jacob, J.; Egashira, R.; Moser, J.; Piciucchi, S.; Mei, F.; De Lauretis, A.; Visca, D.; et al. Pleuroparenchymal fibroelastosis in systemic sclerosis: Prevalence and prognostic impact. Eur. Respir. J. 2020, 56, 1902135. [Google Scholar] [CrossRef]
- Xu, L.; Rassaei, N.; Caruso, C. Pleuroparenchymal Fibroelastosis with Long History of Asbestos and Silicon Exposure. Int. J. Surg. Pathol. 2018, 26, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.D.; Gil, J.; Padilla, M.L. Idiopathic pleuroparenchymal fibroelastosis: An unrecognized or misdiagnosed entity? Mod. Pathol. 2008, 21, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Kikuchi, N.; Ishii, Y.; Kawabata, Y.; Moriyama, H.; Terada, M.; Suzuki, E.; Kobayashi, M.; Watanabe, K.; Hizawa, N. Upper lobe-dominant pulmonary fibrosis showing deposits of hard metal component in the fibrotic lesions. Intern. Med. 2010, 49, 2143–2145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yabuuchi, Y.; Goto, H.; Nonaka, M.; Tachi, H.; Akiyama, T.; Arai, N.; Ishikawa, H.; Hyodo, K.; Nemoto, K.; Miura, Y.; et al. A case of airway aluminosis with likely secondary pleuroparenchymal fibroelastosis. Multidiscip. Respir. Med. 2019, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.L.; Tominaga, M.; Hansell, D.M.; von der Thusen, J.; Rassl, D.; Parfrey, H.; Guy, S.; Twentyman, O.; Rice, A.; Maher, T.M.; et al. Pleuroparenchymal fibroelastosis: A spectrum of histopathological and imaging phenotypes. Eur. Respir. J. 2012, 40, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Nagata, N.; Kitasato, Y.; Wakamatsu, K.; Nabeshima, K.; Harada, T.; Hirota, T.; Shiraishi, M.; Fujita, M. Rapid decrease in forced vital capacity in patients with idiopathic pulmonary upper lobe fibrosis. Respir. Investig. 2012, 50, 88–97. [Google Scholar] [CrossRef]
- Baroke, E.; Heussel, C.P.; Warth, A.; Eichinger, M.; Oltmanns, U.; Palmowski, K.; Herth, F.J.; Kreuter, M. Pleuroparenchymal fibroelastosis in association with carcinomas. Respirology 2016, 21, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Beynat-Mouterde, C.; Beltramo, G.; Lezmi, G.; Pernet, D.; Camus, C.; Fanton, A.; Foucher, P.; Cottin, V.; Bonniaud, P. Pleuroparenchymal fibroelastosis as a late complication of chemotherapy agents. Eur. Respir. J. 2014, 44, 523–527. [Google Scholar] [CrossRef]
- Khiroya, R.; Macaluso, C.; Montero, M.A.; Wells, A.U.; Chua, F.; Kokosi, M.; Maher, T.M.; Devaraj, A.; Rice, A.; Renzoni, E.A.; et al. Pleuroparenchymal Fibroelastosis: A Review of Histopathologic Features and the Relationship Between Histologic Parameters and Survival. Am. J. Surg. Pathol. 2017, 41, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Odink, A.; Brun, A.L.; Macaluso, C.; de Lauretis, A.; Kokosi, M.; Devaraj, A.; Desai, S.; Renzoni, E.; Wells, A.U. Functional associations of pleuroparenchymal fibroelastosis and emphysema with hypersensitivity pneumonitis. Respir. Med. 2018, 138, 95–101. [Google Scholar] [CrossRef]
- Camus, P.; von der Thusen, J.; Hansell, D.M.; Colby, T.V. Pleuroparenchymal fibroelastosis: One more walk on the wild side of drugs? Eur. Respir. J. 2014, 44, 289–296. [Google Scholar] [CrossRef]
- Bonifazi, M.; Montero, M.A.; Renzoni, E.A. Idiopathic Pleuroparenchymal Fibroelastosis. Curr. Pulmonol. Rep. 2017, 6, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Watanabe, K.; Kushima, H.; Baba, T.; Watanabe, S.; Yamada, Y.; Arai, T.; Tsushima, K.; Kondoh, Y.; Nakamura, Y.; et al. Pleuroparenchymal fibroelastosis diagnosed by multidisciplinary discussions in Japan. Respir. Med. 2018, 141, 190–197. [Google Scholar] [CrossRef]
- Shioya, M.; Otsuka, M.; Yamada, G.; Umeda, Y.; Ikeda, K.; Nishikiori, H.; Kuronuma, K.; Chiba, H.; Takahashi, H. Poorer Prognosis of Idiopathic Pleuroparenchymal Fibroelastosis Compared with Idiopathic Pulmonary Fibrosis in Advanced Stage. Can. Respir. J. 2018, 429, 6043053. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sasaki, S.; Kurokawa, K.; Nakamura, T.; Yamada, T.; Sasano, H.; Arano, N.; Komura, M.; Ihara, H.; Nagashima, O.; et al. Usual Interstitial Pneumonia Pattern in the Lower Lung Lobes as a Prognostic Factor in Idiopathic Pleuroparenchymal Fibroelastosis. Respiration 2019, 97, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Fujita, Y.; Takeda, K.; Miyashita, K.; Tsutsumi, A.; Kobayashi, T.; Miki, Y.; Hashimoto, D.; Enomoto, N.; Nakamura, Y.; et al. Clinical significance of lower-lobe interstitial lung disease on high-resolution computed tomography in patients with idiopathic pleuroparenchymal fibroelastosis. Respir. Med. 2019, 154, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Nakamura, Y.; Satake, Y.; Sumikawa, H.; Johkoh, T.; Colby, T.V.; Yasui, H.; Hozumi, H.; Karayama, M.; Suzuki, Y.; et al. Clinical diagnosis of idiopathic pleuroparenchymal fibroelastosis: A retrospective multicenter study. Respir. Med. 2017, 133, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tanizawa, K.; Handa, T.; Kubo, T.; Chen-Yoshikawa, T.F.; Aoyama, A.; Motoyama, H.; Hijiya, K.; Yoshizawa, A.; Oshima, Y.; Ikezoe, K.; et al. Clinical significance of radiological pleuroparenchymal fibroelastosis pattern in interstitial lung disease patients registered for lung transplantation: A retrospective cohort study. Respir. Res. 2018, 19, 162. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., III; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis. Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef]
- Vitali, C.; Bombardieri, S.; Jonsson, R.; Moutsopoulos, H.M.; Alexander, E.L.; Carsons, S.E.; Daniels, T.E.; Fox, P.C.; Fox, R.I.; Kassan, S.S.; et al. Classification criteria for Sjogren’s syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum. Dis. 2002, 61, 554–558. [Google Scholar] [CrossRef]
- van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A., Jr.; Carreira, P.E.; et al. 2013 classification criteria for systemic sclerosis: An American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis. Rheum. 2013, 65, 2737–2747. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care. Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef]
- Oda, T.; Ogura, T.; Kitamura, H.; Hagiwara, E.; Baba, T.; Enomoto, Y.; Iwasawa, T.; Okudela, K.; Takemura, T.; Sakai, F.; et al. Distinct characteristics of pleuroparenchymal fibroelastosis with usual interstitial pneumonia compared with idiopathic pulmonary fibrosis. Chest 2014, 146, 1248–1255. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care. Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.I.; Chae, E.J.; Song, J.S.; Lee, J.H.; Song, J.W. Pleuroparenchymal fibroelastosis in patients with idiopathic pulmonary fibrosis. Respirology 2020, 25, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Watanabe, K.; Ishii, H.; Kushima, H.; Hamasaki, M.; Fujita, M.; Nabeshima, K. Pleuroparenchymal fibroelastosis as a histological background of autoimmune diseases. Virchows. Arch. 2019, 474, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Hunninghake, G.; King, T.E., Jr.; Lynch, D.A.; Colby, T.V.; Galvin, J.R.; Brown, K.K.; Chung, M.P.; Cordier, J.F.; du Bois, R.M.; et al. Idiopathic nonspecific interstitial pneumonia: Report of an American Thoracic Society project. Am. J. Respir. Crit. Care. Med. 2008, 177, 1338–1347. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cause of Secondary PPFE | n (%) |
|---|---|
| Chronic hypersensitivity pneumonitis | 10 (31.3) |
| Sjögren’s syndrome | 6 (18.8) |
| Pneumoconiosis | 4 (12.5) |
| Rheumatoid arthritis | 2 (6.3) |
| Metal lung | 2 (6.3) |
| Mycobacterium infection | 2 (6.3) |
| Chemotherapeutic agents | 2 (6.3) |
| Systemic sclerosis | 1 (3.1) |
| Crohn’s disease | 1 (3.1) |
| Bone marrow transplantation | 1 (3.1) |
| Radiation | 1 (3.1) |
| Parameters | Idiopathic PPFE (n = 132) | Secondary PPFE | |||
|---|---|---|---|---|---|
| All (n = 32) | Chronic HP (n = 10) | Sjögren’s Syndrome (n = 6) | Others (n = 16) | ||
| Demographics | |||||
| Age, years | 68.5 (14) | 59.5 (13) * | 63 (14) | 60 (13) | 55 (17) |
| Sex, male n (%) | 68 (51.5) | 13 (40.6) | 4 (40) | 2 (33.3) | 7 (43.7) |
| Smoker n (%) | 57 (43.2) | 9 (28.1) | 4 (40) | 1 (20) | 4 (25) |
| Body mass index, kg/m2 | 19.8 (4.1) | 19.7 (2.6) | 20.3 (3.5) | 17.2 (4.5) | 18.2 (3.7) |
| Follow-up periods, years | 2.1 (2.7) | 3.1 (4.9) | 4.8 (6.8) | 3.6 (6.9) | 2.2 (2.9) |
| Diagnosis | |||||
| Biopsy-proven/ No biopsy-proven, n | 45/87 | 23/9 * | 10/0 | 4/2 | 9/7 |
| Symptoms on initial examination | |||||
| No symptoms, n (%) | 29 (22.0) | 9 (28.1) | 3 (30) | 3 (50) | 3 (18.8) |
| Cough, n (%) | 55 (41.7) | 14 (43.8) | 4 (40) | 1 (16.7) | 9 (56.3) |
| Dyspnea on exertion, n (%) | 76 (57.6) | 13 (40.6) | 3 (30) | 2 (33.3) | 8 (50) |
| Pulmonary function test data | |||||
| FVC, % predicted | 73.0 (31.6) | 70.0 (21.7) | 73.4 (14.0) | 65.9 (27.9) | 52.5 (30.9) |
| DLCO, % predicted | 89.4 (33.1) | 78.8 (59.1) | 97.6 (63.0) | 88.3 (51.3) | 60.9 (31.8) |
| RV/TLC, % | 45.2 (16.9) | 44.9 (11.3) | 39.5 (16.6) | 44.3 (14.5) | 47.5 (12.0) |
| Laboratory data | |||||
| KL-6, IU/mL | 638.5 (447) | 557.5 (290) | 544 (299) | 529.5 (374) | 593.5 (552) |
| BAL, Macrophages, % | 83.8 (16.5) | 62.9 (22.4) † | 59.5 (13.8) | 80.0 (n = 3) | 69.8 (21.3) (n = 8) |
| BAL, Lymphocytes, % | 11.3 (11.7) | 23.9 (17.9) ‡ | 31.3 (19.5) | 17.2 (n = 3) | 19.4 (16.0) (n = 8) |
| Lower-lobe ILD pattern | |||||
| UIP pattern, n (%) | 59 (44.7) | 10 (31.3) § | 5 (50) | 3 (50) | 2 (12.5) |
| NSIP patten, n (%) | 5 (3.8) | 4 (12.5) | 1 (10) | 2 (33.3) | 1 (6.3) |
| Absence of any ILD in the lower lobes, n (%) | 36 (27.3) | 6 (18.8) | 0 | 0 | 6 (37.5) |
| Respiratory complications | |||||
| Pneumothorax or pneumomediastinum, n (%) | 63 (47.7) | 16 (50) | 5 (50) | 5 (50) | 6 (37.5) |
| Acute exacerbation, n (%) | 5 (3.8) | 2 (6.3) | 1 (10) | 0 | 1 (6.3) |
| Dead, n (%) | 45 (34.1) | 16 (50) | 3 (30) | 5 (83.3) | 8 (50) |
| Parameters | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|
| Hazard Ratio (95% CI) | p Value | Hazard Ratio (95% CI) | p Value | |
| Age | 1.04 (1.01–1.07) | 0.02 | 1.04 (0.99–1.08) | 0.09 |
| Sex, male | 1.58 (0.87–2.87) | 0.14 | ||
| Body mass index | 0.93 (0.83–1.04) | 0.21 | ||
| FVC, % predicted | 0.94 (0.93–0.96) | <0.01 | 0.93 (0.90–0.96) | <0.01 |
| DLCO, % predicted | 0.96 (0.94–0.97) | <0.01 | 0.98 (0.960–0.999) | 0.04 |
| RV/TLC | 1.04 (1.00–1.07) | 0.03 | 0.93 (0.94–1.03) | 0.51 |
| KL-6 | 1.001 (1.000–1.001) | <0.01 | 1.001 (1.000–1.002) | 0.18 |
| Lymphocyte % in the BAL fluid | 0.99 (0.94–1.03) | 0.50 | ||
| Presence of UIP pattern in the lower lobes | 1.86 (1.03–3.37) | 0.04 | 7.56 (3.39–16.9) | <0.01 |
| Absence of any ILD in the lower lobes | 0.40 (0.18–0.89) | 0.03 | 0.93 (0.27–3.22) | 0.93 |
| Parameters | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|
| Hazard Ratio (95% CI) | p Value | Hazard Ratio (95% CI) | p Value | |
| Age | 1.02 (0.97–1.08) | 0.41 | ||
| Sex, male | 1.34 (0.48–3.76) | 0.59 | ||
| Body mass index | 0.92 (0.75–1.14) | 0.44 | ||
| FVC, % predicted | 0.98 (0.95–1.01) | 0.28 | ||
| DLCO, % predicted | 0.95 (0.92–0.98) | <0.01 | 0.95 (0.92–0.99) | <0.01 |
| RV/TLC | 0.98 (0.93–1.04) | 0.57 | ||
| KL-6 | 1.002 (1.000–1.003) | 0.01 | 1.000 (0.999–1.002) | 0.56 |
| Lymphocyte % in the BAL fluid | 0.95 (0.89–1.01) | 0.09 | ||
| Presence of UIP pattern in the lower lobes | 1.33 (0.47–3.74) | 0.59 | ||
| Absence of any ILD in the lower lobes | 0.35 (0.05–2.68) | 0.31 | ||
| Parameters | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|
| Hazard Ratio (95% CI) | p Value | Hazard Ratio (95% CI) | p Value | |
| Age | 1.03 (1.01–1.06) | 0.02 | 1.04 (1.01–1.07) | 0.01 |
| Sex, male | 1.46 (0.88–2.43) | 0.14 | ||
| Body mass index | 0.93 (0.84–1.03) | 0.93 | ||
| FVC, % predicted | 0.95 (0.94–0.97) | <0.01 | 0.96 (0.94–0.98) | <0.01 |
| DLCO, % predicted | 0.96 (0.95–0.97) | <0.01 | 0.97 (0.95–0.99) | <0.01 |
| RV/TLC | 1.03 (1.00–1.05) | 0.08 | ||
| KL-6 | 1.001 (1.000–1.001) | <0.01 | 1.001 (1.000–1.002) | 0.047 |
| Lymphocyte % in the BAL fluid | 0.98 (0.95–1.01) | 0.23 | ||
| Presence of UIP pattern in the lower lobes | 1.70 (1.03–2.83) | 0.04 | 7.08 (3.27–15.3) | <0.01 |
| Absence of any ILD in the lower lobes | 0.39 (0.19–0.82) | 0.01 | 1.72 (0.61–4.84) | 0.30 |
| Secondary PPFE | 1.02 (0.56–1.84) | 0.95 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oda, T.; Sekine, A.; Tabata, E.; Iwasawa, T.; Takemura, T.; Ogura, T. Comparison of Clinical Characteristics and Outcomes between Idiopathic and Secondary Pleuroparenchymal Fibroelastosis. J. Clin. Med. 2021, 10, 846. https://doi.org/10.3390/jcm10040846
Oda T, Sekine A, Tabata E, Iwasawa T, Takemura T, Ogura T. Comparison of Clinical Characteristics and Outcomes between Idiopathic and Secondary Pleuroparenchymal Fibroelastosis. Journal of Clinical Medicine. 2021; 10(4):846. https://doi.org/10.3390/jcm10040846
Chicago/Turabian StyleOda, Tsuneyuki, Akimasa Sekine, Erina Tabata, Tae Iwasawa, Tamiko Takemura, and Takashi Ogura. 2021. "Comparison of Clinical Characteristics and Outcomes between Idiopathic and Secondary Pleuroparenchymal Fibroelastosis" Journal of Clinical Medicine 10, no. 4: 846. https://doi.org/10.3390/jcm10040846
APA StyleOda, T., Sekine, A., Tabata, E., Iwasawa, T., Takemura, T., & Ogura, T. (2021). Comparison of Clinical Characteristics and Outcomes between Idiopathic and Secondary Pleuroparenchymal Fibroelastosis. Journal of Clinical Medicine, 10(4), 846. https://doi.org/10.3390/jcm10040846