
Primary Immune Thrombocytopenia: Novel Insights into Pathophysiology and Disease Management




Abstract
1. Introduction
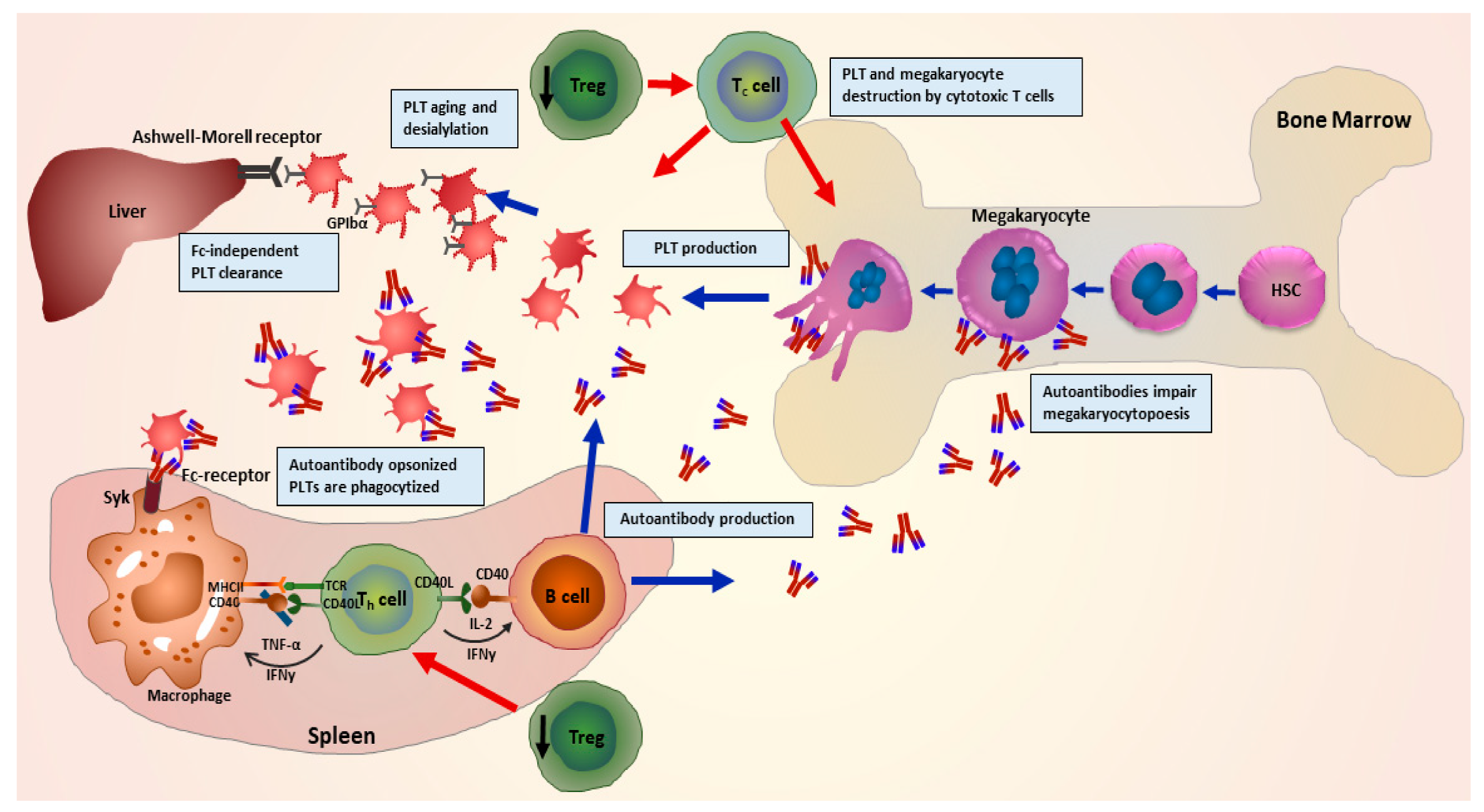
2. Pathophysiology of ITP
3. Clinical Manifestations
4. Diagnosis
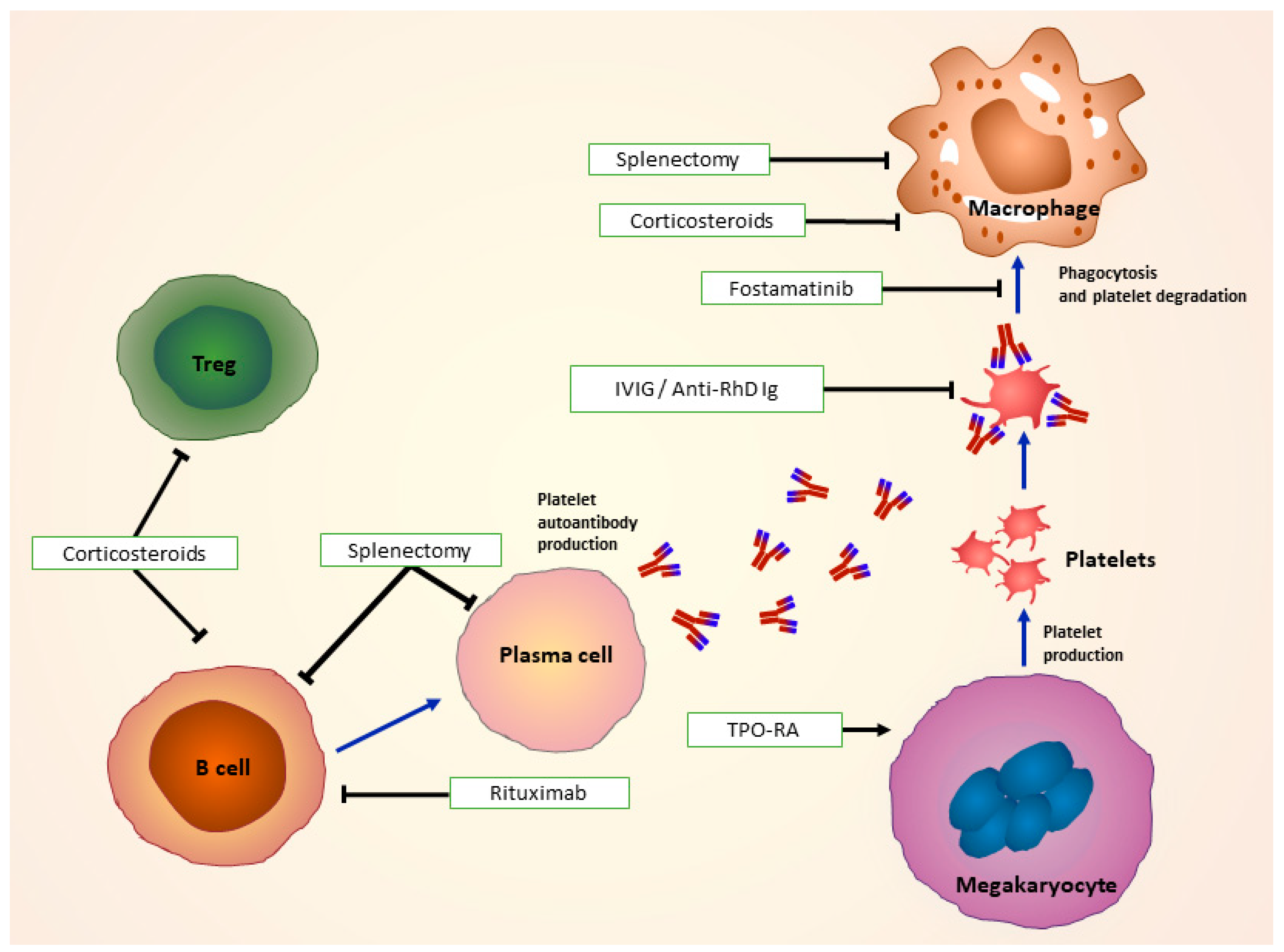
5. Treatment of ITP
5.1. First-Line Treatments and Treatment of Bleeding Emergencies
5.1.1. Glucocorticoids
5.1.2. Intravenous Immunoglobulin (IVIG)
5.1.3. Anti-RhD Immunoglobulin (Ig)
5.2. Treatment of Active Bleeding
5.3. Second-line treatments
5.3.1. Thrombopoietin-Receptor Agonists (TPO-RA)
5.3.2. Immunomodulators
5.3.3. Splenectomy
5.4. New Drugs under Investigation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Provan, D.; Stasi, R.; Newland, A.C.; Blanchette, V.S.; Bolton-Maggs, P.; Bussel, J.B.; Chong, B.H.; Cines, D.B.; Gernsheimer, T.B.; Godeau, B.; et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010, 115, 168–186. [Google Scholar] [CrossRef]
- Neunert, C.; Lim, W.; Crowther, M.; Cohen, A.; Solberg, L.; Crowther, M.A. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011, 117, 4190–4207. [Google Scholar] [CrossRef]
- Moulis, G.; Palmaro, A.; Montastruc, J.-L.; Godeau, B.; Lapeyre-Mestre, M.; Sailler, L. Epidemiology of incident immune thrombocytopenia: a nationwide population-based study in France. Blood 2014, 124, 3308–3315. [Google Scholar] [CrossRef] [PubMed]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef]
- McKenzie, C.G.J.; Guo, L.; Freedman, J.; Semple, J.W. Cellular immune dysfunction in immune thrombocytopenia (ITP). Br. J. Haematol. 2013, 163, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Shulman, N.R.; Marder, V.J.; Weinrach, R.S. Similarities between known antiplatelet antibodies and the factor responsible for thrombocytopenia in idiopathic purpura. Physiologic, serologic and isotopic studies. Ann. NY Acad. Sci. 1965, 124, 499–542. [Google Scholar] [CrossRef]
- Ku, F.-C.; Tsai, C.-R.; Der Wang, J.; Wang, C.H.; Chang, T.-K.; Hwang, W.-L. Stromal-derived factor-1 gene variations in pediatric patients with primary immune thrombocytopenia. Eur. J. Haematol. 2013, 90, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Rank, A.; Weigert, O.; Ostermann, H. Management of chronic immune thrombocytopenic purpura: targeting insufficient megakaryopoiesis as a novel therapeutic principle. Biologics 2010, 4, 139–145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cines, D.B.; Liebman, H.A. The Immune Thrombocytopenia Syndrome: A Disorder of Diverse Pathogenesis and Clinical Presentation. Hematol. Oncol. Clin. North Am. 2009, 23, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, J.A.; Neely, J.; Farhoudi, N. ITP in Children. J. Pediatric Hematol. Oncol. 2013, 35, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Provan, D.; Arnold, D.M.; Bussel, J.B.; Chong, B.H.; Cooper, N.; Gernsheimer, T.; Ghanima, W.; Godeau, B.; González-López, T.J.; Grainger, J.; et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019, 3, 3780–3817. [Google Scholar] [CrossRef] [PubMed]
- Bakchoul, T.; Sachs, U.J. Platelet destruction in immune thrombocytopenia. Understanding the mechanisms. Hamostaseologie 2016, 36, 187–194. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, L.; Yang, G.; Zhuang, Y.; Qian, X.; Zhou, X.; Xiao, D.; Shen, Y. Contributions of T Lymphocyte Abnormalities to Therapeutic Outcomes in Newly Diagnosed Patients with Immune Thrombocytopenia. PLoS ONE 2015, 10, e0126601. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Zhang, L.; Peng, J.; Hou, M. T cell immune abnormalities in immune thrombocytopenia. J. Hematol. Oncol. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chu, X.; Wang, L.; Zhu, Y.; Li, L.; Ma, D.; Peng, J.; Hou, M. Cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2006, 76, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Li, X.; Zhang, F.; Wang, L.; Peng, J.; Hou, M. Increased cytotoxic T-lymphocyte-mediated cytotoxicity predominant in patients with idiopathic thrombocytopenic purpura without platelet autoantibodies. Haematologica 2008, 93, 1428–1430. [Google Scholar] [CrossRef]
- Bakchoul, T.; Walek, K.; Krautwurst, A.; Rummel, M.; Bein, G.; Santoso, S.; Sachs, U.J. Glycosylation of autoantibodies: insights into the mechanisms of immune thrombocytopenia. Thromb. Haemost. 2013, 110, 1259–1266. [Google Scholar] [CrossRef]
- Kashiwagi, H.; Tomiyama, Y. Pathophysiology and management of primary immune thrombocytopenia. Int. J. Hematol. 2013, 98, 24–33. [Google Scholar] [CrossRef]
- Nieswandt, B.; Bergmeier, W.; Rackebrandt, K.; Gessner, J.E.; Zirngibl, H. Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood 2000, 96, 2520–2527. [Google Scholar] [CrossRef]
- Nieswandt, B.; Bergmeier, W.; Schulte, V.; Rackebrandt, K.; Gessner, J.E.; Zirngibl, H. Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcRgamma chain. J. Biol. Chem. 2000, 275, 23998–24002. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.L.; Sayeh, E.; Crow, M.; Chen, P.; Nieswandt, B.; Freedman, J.; Ni, H. Relative efficacy of intravenous immunoglobulin G in ameliorating thrombocytopenia induced by antiplatelet GPIIbIIIa versus GPIbalpha antibodies. Blood 2006, 108, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; van der Wal, D.E.; Zhu, G.; Xu, M.; Yougbare, I.; Ma, L.; Vadasz, B.; Carrim, N.; Grozovsky, R.; Ruan, M.; et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Liu, X.; Li, X.; Zhang, X.; Han, P.; Zhou, H.; Shao, L.; Hou, Y.; Min, Y.; Kong, Z.; et al. CD8(+) T cells induce platelet clearance in the liver via platelet desialylation in immune thrombocytopenia. Sci. Rep. 2016, 6, 27445. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zeng, Q.; Li, J.; Xu, M.; Wang, J.; Pan, Y.; Wang, H.; Tao, Q.; Chen, Y.; Peng, J.; et al. Platelet desialylation correlates with efficacy of first-line therapies for immune thrombocytopenia. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Quach, M.E.; Dragovich, M.A.; Chen, W.; Syed, A.K.; Cao, W.; Liang, X.; Deng, W.; de Meyer, S.F.; Zhu, G.; Peng, J.; et al. Fc-independent immune thrombocytopenia via mechanomolecular signaling in platelets. Blood 2018, 131, 787–796. [Google Scholar] [CrossRef]
- Marini, I.; Zlamal, J.; Faul, C.; Holzer, U.; Hammer, S.; Pelzl, L.; Bethge, W.; Althaus, K.; Bakchoul, T. Autoantibody-mediated desialylation impairs human thrombopoiesis and platelet lifespan. Haematologica 2020, 106, 196–207. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed Anuclear Cell Death Delimits Platelet Life Span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Van der Wal, D.E.; Gitz, E.; Du, V.X.; Lo, K.S.L.; Koekman, C.A.; Versteeg, S.; Akkerman, J.W.N. Arachidonic acid depletion extends survival of cold-stored platelets by interfering with the glycoprotein Ibα--14-3-3ζ association. Haematologica 2012, 97, 1514–1522. [Google Scholar] [CrossRef]
- Álvarez Román, M.; Bello, I.; Arias-Salgado, E.G.; Pollmar, M.I.; Yuste, V.; Salces, M.; Butta, N.V. Effects of thrombopoietin receptor agonists on procoagulant state in patients with immune thrombocytopenia. Thromb. Haemost. 2014, 112, 65–72. [Google Scholar] [CrossRef]
- Winkler, J.; Kroiss, S.; Rand, M.L.; Azzouzi, I.; Annie Bang, K.W.; Speer, O.; Schmugge, M. Platelet apoptosis in paediatric immune thrombocytopenia is ameliorated by intravenous immunoglobulin. Br. J. Haematol. 2012, 156, 508–515. [Google Scholar] [CrossRef]
- Goette, N.P.; Glembotsky, A.C.; Lev, P.R.; Grodzielski, M.; Contrufo, G.; Pierdominici, M.S.; Espasandin, Y.R.; Riveros, D.; García, A.J.; Molinas, F.C.; et al. Platelet Apoptosis in Adult Immune Thrombocytopenia: Insights into the Mechanism of Damage Triggered by Auto-Antibodies. PLoS ONE 2016, 11, e0160563. [Google Scholar] [CrossRef]
- Marini, I.; Bakchoul, T. Pathophysiology of Autoimmune Thrombocytopenia: Current Insight with a Focus on Thrombopoiesis. Hamostaseologie 2019, 39, 227–237. [Google Scholar] [CrossRef]
- McMillan, R.; Luiken, G.A.; Levy, R.; Yelenosky, R.; Longmire, R.L. Antibody against megakaryocytes in idiopathic thrombocytopenic purpura. JAMA 1978, 239, 2460–2462. [Google Scholar] [CrossRef]
- Takahashi, R.; Sekine, N.; Nakatake, T. Influence of monoclonal antiplatelet glycoprotein antibodies on in vitro human megakaryocyte colony formation and proplatelet formation. Blood 1999, 93, 1951–1958. [Google Scholar] [CrossRef]
- Chang, M.; Nakagawa, P.A.; Williams, S.A.; Schwartz, M.R.; Imfeld, K.L.; Buzby, J.S.; Nugent, D.J. Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood 2003, 102, 887–895. [Google Scholar] [CrossRef]
- McMillan, R.; Wang, L.; Tomer, A.; Nichol, J.; Pistillo, J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004, 103, 1364–1369. [Google Scholar] [CrossRef]
- Iraqi, M.; Perdomo, J.; Yan, F.; Choi, P.Y.-I.; Chong, B.H. Immune thrombocytopenia: antiplatelet autoantibodies inhibit proplatelet formation by megakaryocytes and impair platelet production in vitro. Haematologica 2015, 100, 623–632. [Google Scholar] [CrossRef]
- Yang, L.; Wang, L.; Zhao, C.; Zhu, X.; Hou, Y.; Jun, P.; Hou, M. Contributions of TRAIL-mediated megakaryocyte apoptosis to impaired megakaryocyte and platelet production in immune thrombocytopenia. Blood 2010, 116, 4307–4316. [Google Scholar] [CrossRef] [PubMed]
- Radley, J.M.; Haller, C.J. Fate of senescent megakaryocytes in the bone marrow. Br. J. Haematol. 1983, 53, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Houwerzijl, E.J.; Blom, N.R.; van der Want, J.J.L.; Esselink, M.T.; Koornstra, J.J.; Smit, J.W.; Louwes, H.; Vellenga, E.; de Wolf, J.T.M. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004, 103, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Vrbensky, J.R.; Nazy, I.; Toltl, L.J.; Ross, C.; Ivetic, N.; Smith, J.W.; Kelton, J.G.; Arnold, D.M. Megakaryocyte apoptosis in immune thrombocytopenia. Platelets 2018, 29, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Neylon, A.J.; Saunders, P.W.G.; Howard, M.R.; Proctor, S.J.; Taylor, P.R.A. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: A prospective study of a population-based cohort of 245 patients. Br. J. Haematol. 2003, 122, 966–974. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, J.-H.; Lee, H.; Kang, B.; Kim, J.-W.; Kim, S.H.; Lee, J.-O.; Kim, J.W.; Kim, Y.J.; Lee, K.-W.; et al. Epidemiology and management of primary immune thrombocytopenia: A nationwide population-based study in Korea. Thromb. Res. 2017, 155, 86–91. [Google Scholar] [CrossRef]
- Schoonen, W.M.; Kucera, G.; Coalson, J.; Li, L.; Rutstein, M.; Mowat, F.; Fryzek, J.; Kaye, J.A. Epidemiology of immune thrombocytopenic purpura in the General Practice Research Database. Br. J. Haematol. 2009, 145, 235–244. [Google Scholar] [CrossRef]
- Grimaldi-Bensouda, L.; Nordon, C.; Michel, M.; Viallard, J.-F.; Adoue, D.; Magy-Bertrand, N.; Durand, J.-M.; Quittet, P.; Fain, O.; Bonnotte, B.; et al. Immune thrombocytopenia in adults: A prospective cohort study of clinical features and predictors of outcome. Haematologica 2016, 101, 1039–1045. [Google Scholar] [CrossRef]
- Sailer, T.; Lechner, K.; Panzer, S.; Kyrle, P.A.; Pabinger, I. The course of severe autoimmune thrombocytopenia in patients not undergoing splenectomy. Haematologica 2006, 91, 1041–1045. [Google Scholar] [CrossRef]
- Kühne, T.; Buchanan, G.R.; Zimmerman, S.; Michaels, L.A.; Kohan, R.; Berchtold, W.; Imbach, P. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J. Pediatrics 2003, 143, 605–608. [Google Scholar] [CrossRef]
- Imbach, P.; Kühne, T.; Müller, D.; Berchtold, W.; Zimmerman, S.; Elalfy, M.; Buchanan, G.R. Childhood ITP: 12 months follow-up data from the prospective registry I of the Intercontinental Childhood ITP Study Group (ICIS). Pediatr. Blood Cancer 2006, 46, 351–356. [Google Scholar] [CrossRef]
- Jaime-Pérez, J.C.; Aguilar-Calderón, P.; Jiménez-Castillo, R.A.; Ramos-Dávila, E.M.; Salazar-Cavazos, L.; Gómez-Almaguer, D. Treatment outcomes and chronicity predictors for primary immune thrombocytopenia: 10-year data from an academic center. Ann. Hematol. 2020, 99, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Matzdorff, A.; Meyer, O.; Ostermann, H.; Kiefel, V.; Eberl, W.; Kühne, T.; Pabinger, I.; Rummel, M. Immune Thrombocytopenia—Current Diagnostics and Therapy: Recommendations of a Joint Working Group of DGHO, ÖGHO, SGH, GPOH, and DGTI. Oncol. Res. Treat. 2018, 41 (Suppl. 5), 1–30. [Google Scholar] [CrossRef] [PubMed]
- Adelborg, K.; Kristensen, N.R.; Nørgaard, M.; Bahmanyar, S.; Ghanima, W.; Kilpatrick, K.; Frederiksen, H.; Ekstrand, C.; Sørensen, H.T.; Fynbo Christiansen, C. Cardiovascular and bleeding outcomes in a population-based cohort of patients with chronic immune thrombocytopenia. J. Thromb. Haemost. 2019, 17, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Mithoowani, S.; Cervi, A.; Shah, N.; Ejaz, R.; Sirotich, E.; Barty, R.; Li, N.; Nazy, I.; Arnold, D.M. Management of major bleeds in patients with immune thrombocytopenia. J. Thromb. Haemost. 2020, 18, 1783–1790. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Djulbegovic, B.; Shamai-Lubovitz, O.; Mozes, B. The bleeding risk and natural history of idiopathic thrombocytopenic purpura in patients with persistent low platelet counts. Arch. Intern. Med. 2000, 160, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Neunert, C.; Noroozi, N.; Norman, G.; Buchanan, G.R.; Goy, J.; Nazi, I.; Kelton, J.G.; Arnold, D.M. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J. Thromb. Haemost. 2015, 13, 457–464. [Google Scholar] [CrossRef]
- Forsythe, A.; Schneider, J.; Pham, T.; Bhor, M.; Said, Q.; Allepuz, A.; Socorro O Portella, M.D.; Kwon, C.S.; Roy, A.N. Real-world evidence on clinical outcomes in immune thrombocytopenia treated with thrombopoietin receptor agonists. J. Comp. Eff. Res. 2020, 9, 447–457. [Google Scholar] [CrossRef]
- Arnold, D.M.; Nazy, I.; Clare, R.; Jaffer, A.M.; Aubie, B.; Li, N.; Kelton, J.G. Misdiagnosis of primary immune thrombocytopenia and frequency of bleeding: Lessons from the McMaster ITP Registry. Blood Adv. 2017, 1, 2414–2420. [Google Scholar] [CrossRef]
- Piel-Julian, M.-L.; Mahévas, M.; Germain, J.; Languille, L.; Comont, T.; Lapeyre-Mestre, M.; Payrastre, B.; Beyne-Rauzy, O.; Michel, M.; Godeau, B.; et al. Risk factors for bleeding, including platelet count threshold, in newly diagnosed immune thrombocytopenia adults. J. Thromb. Haemost. 2018, 16, 1830–1842. [Google Scholar] [CrossRef]
- Hato, T.; Shimada, N.; Kurata, Y.; Kuwana, M.; Fujimura, K.; Kashiwagi, H.; Takafuta, T.; Murata, M.; Tomiyama, Y. Risk factors for skin, mucosal, and organ bleeding in adults with primary ITP: A nationwide study in Japan. Blood Adv. 2020, 4, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.L.; Reese, J.A.; Watson, S.I.; Vesely, S.K.; Bolton-Maggs, P.H.B.; George, J.N.; Terrell, D.R. Fatigue in adult patients with primary immune thrombocytopenia. Eur. J. Haematol. 2011, 86, 420–429. [Google Scholar] [CrossRef]
- Kuter, D.J.; Mathias, S.D.; Rummel, M.; Mandanas, R.; Giagounidis, A.A.; Wang, X.; Deuson, R.R. Health-related quality of life in nonsplenectomized immune thrombocytopenia patients receiving romiplostim or medical standard of care. Am. J. Hematol. 2012, 87, 558–561. [Google Scholar] [CrossRef]
- Blatt, J.; Weston, B.; Gold, S. Fatigue as marker of thrombocytopenia in childhood idiopathic thrombocytopenic purpura. Pediatr. Hematol. Oncol. 2010, 27, 65–67. [Google Scholar] [CrossRef]
- Hill, Q.A.; Newland, A.C. Fatigue in immune thrombocytopenia. Br. J. Haematol. 2015, 170, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Severinsen, M.T.; Engebjerg, M.C.; Farkas, D.K.; Jensen, A.Ø.; Nørgaard, M.; Zhao, S.; Sørensen, H.T. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: A Danish population-based cohort study. Br. J. Haematol. 2011, 152, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Doobaree, I.U.; Nandigam, R.; Bennett, D.; Newland, A.; Provan, D. Thromboembolism in adults with primary immune thrombocytopenia: A systematic literature review and meta-analysis. Eur. J. Haematol. 2016, 97, 321–330. [Google Scholar] [CrossRef]
- Hollenhorst, M.A.; Al-Samkari, H.; Kuter, D.J. Markers of autoimmunity in immune thrombocytopenia: Prevalence and prognostic significance. Blood Adv. 2019, 3, 3515–3521. [Google Scholar] [CrossRef] [PubMed]
- Peerschke, E.I.; Yin, W.; Ghebrehiwet, B. Complement activation on platelets: Implications for vascular inflammation and thrombosis. Mol. Immunol. 2010, 47, 2170–2175. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J.; Bussel, J.B.; Newland, A.; Baker, R.I.; Lyons, R.M.; Wasser, J.; Viallard, J.-F.; Macik, G.; Rummel, M.; Nie, K.; et al. Long-term treatment with romiplostim in patients with chronic immune thrombocytopenia: Safety and efficacy. Br. J. Haematol. 2013, 161, 411–423. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Stasi, R.; Giagounidis, A.; Viallard, J.-F.; Godeau, B.; Pabinger, I.; Cines, D.; Liebman, H.; Wang, X.; Woodard, P. Long-term safety and tolerability of romiplostim in patients with primary immune thrombocytopenia: A pooled analysis of 13 clinical trials. Eur. J. Haematol. 2013, 91, 423–436. [Google Scholar] [CrossRef]
- Frederiksen, H.; Maegbaek, M.L.; Nørgaard, M. Twenty-year mortality of adult patients with primary immune thrombocytopenia: A Danish population-based cohort study. Br. J. Haematol. 2014, 166, 260–267. [Google Scholar] [CrossRef]
- Vollenberg, R.; Jouni, R.; Norris, P.A.A.; Burg-Roderfeld, M.; Cooper, N.; Rummel, M.J.; Bein, G.; Marini, I.; Bayat, B.; Burack, R.; et al. Glycoprotein V is a relevant immune target in patients with immune thrombocytopenia. Haematologica 2019, 104, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Kiefel, V.; Freitag, E.; Kroll, H.; Santoso, S.; Mueller-Eckhardt, C. Platelet autoantibodies (IgG, IgM, IgA) against glycoproteins IIb/IIIa and Ib/IX in patients with thrombocytopenia. Ann. Hematol. 1996, 72, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Newland, A.; Lee, E.-J.; McDonald, V.; Bussel, J.B. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy 2018, 10, 9–25. [Google Scholar] [CrossRef]
- Page, L.K.; Psaila, B.; Provan, D.; Michael Hamilton, J.; Jenkins, J.M.; Elish, A.S.; Lesser, M.L.; Bussel, J.B. The immune thrombocytopenic purpura (ITP) bleeding score: Assessment of bleeding in patients with ITP. Br. J. Haematol. 2007, 138, 245–248. [Google Scholar] [CrossRef]
- Palau, J.; Jarque, I.; Sanz, M.A. Long-term management of chronic immune thrombocytopenic purpura in adults. Int. J. Gen. Med. 2010, 3, 305–311. [Google Scholar] [CrossRef][Green Version]
- McGrath, L.J.; Kilpatrick, K.; Overman, R.A.; Reams, D.; Sharma, A.; Altomare, I.; Wasser, J.; Brookhart, M.A. Treatment Patterns Among Adults with Primary Immune Thrombocytopenia Diagnosed in Hematology Clinics in the United States. Clin. Epidemiol. 2020, 12, 435–445. [Google Scholar] [CrossRef]
- Branehög, I.; Weinfeld, A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP) before and during treatment with corticosteroids. Scand. J. Haematol. 1974, 12, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Gernsheimer, T.; Stratton, J.; Ballem, P.J.; Slichter, S.J. Mechanisms of response to treatment in autoimmune thrombocytopenic purpura. N. Engl. J. Med. 1989, 320, 974–980. [Google Scholar] [CrossRef]
- Wang, L.; Xu, L.; Hao, H.; Jansen, A.J.G.; Liu, G.; Li, H.; Liu, X.; Zhao, Y.; Peng, J.; Hou, M. First line treatment of adult patients with primary immune thrombocytopenia: A real-world study. Platelets 2020, 31, 55–61. [Google Scholar] [CrossRef]
- Mithoowani, S.; Gregory-Miller, K.; Goy, J.; Miller, M.C.; Wang, G.; Noroozi, N.; Kelton, J.G.; Arnold, D.M. High-dose dexamethasone compared with prednisone for previously untreated primary immune thrombocytopenia: A systematic review and meta-analysis. Lancet Haematol. 2016, 3, e489–e496. [Google Scholar] [CrossRef]
- Ma, J.; Fu, L.; Chen, Z.; Gu, H.; Ma, J.; Wu, R. High-dose dexamethasone as a replacement for traditional prednisone as the first-line treatment in children with previously untreated primary immune thrombocytopenia: A prospective, randomized single-center study. Int. J. Hematol. 2020, 112, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.; Ghanima, W. Response of first line treatment with corticosteroids in a population-based cohort of adults with primary immune thrombocytopenia. Eur. J. Intern. Med. 2017, 37, e23–e25. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wong, R.S.; Soo, Y.O.; Chui, C.H.; Lau, F.Y.; Chan, N.P.; Wong, W.S.; Cheng, G. Initial Treatment of Immune Thrombocytopenic Purpura with High-Dose Dexamethasone. N. Engl. J. Med. 2003, 349, 831–836. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Wang, C.; Zhang, Y.; Gao, C.; Lang, H.; Chen, X. Efficacy and Safety of the Combination Treatment of Rituximab and Dexamethasone for Adults with Primary Immune Thrombocytopenia (ITP): A Meta-Analysis. Biomed Res. Int. 2018, 2018, 1316096. [Google Scholar] [CrossRef] [PubMed]
- Imbach, P. Treatment of immune thrombocytopenia with intravenous immunoglobulin and insights for other diseases. A historical review. Swiss Med. Wkly. 2012, 142, w13593. [Google Scholar] [CrossRef]
- Lazarus, A.H.; Crow, A.R. Mechanism of action of IVIG and anti-D in ITP. Transfus. Apher. Sci. 2003, 28, 249–255. [Google Scholar] [CrossRef]
- Beck, C.E.; Nathan, P.C.; Parkin, P.C.; Blanchette, V.S.; Macarthur, C. Corticosteroids Versus Intravenous Immune Globulin for the Treatment of Acute Immune Thrombocytopenic Purpura in Children: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Pediatrics 2005, 147, 521–527. [Google Scholar] [CrossRef]
- Zhou, Z.; Qiao, Z.; Li, H.; Luo, N.; Zhang, X.; Xue, F.; Yang, R. Different dosages of intravenous immunoglobulin (IVIg) in treating immune thrombocytopenia with long-term follow-up of three years: Results of a prospective study including 167 cases. Autoimmunity 2016, 49, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.-H.; Zhou, T.-B.; Su, L.-N.; Lei, F.-Y.; Zhao, Y.-J.; Huang, W.-F. The efficacy of different dose intravenous immunoglobulin in treating acute idiopathic thrombocytopenic purpura: a meta-analysis of 13 randomized controlled trials. Blood Coagul. Fibrinolysis 2010, 21, 713–721. [Google Scholar] [CrossRef]
- Jacobs, P.; Wood, L.; Novitzky, N. Intravenous gammaglobulin has no advantages over oral corticosteroids as primary therapy for adults with immune thrombocytopenia: A prospective randomized clinical trial. Am. J. Med. 1994, 97, 55–59. [Google Scholar] [CrossRef]
- Godeau, B.; Chevret, S.; Varet, B.; Lefrère, F.; Zini, J.-M.; Bassompierre, F.; Chèze, S.; Legouffe, E.; Hulin, C.; Grange, M.-J.; et al. Intravenous immunoglobulin or high-dose methylprednisolone, with or without oral prednisone, for adults with untreated severe autoimmune thrombocytopenic purpura: A randomised, multicentre trial. Lancet 2002, 359, 23–29. [Google Scholar] [CrossRef]
- Peng, J.; Ma, S.-H.; Liu, J.; Hou, Y.; Liu, X.-M.; Niu, T.; Xu, R.-R.; Guo, C.-S.; Wang, X.-M.; Cheng, Y.-F.; et al. Association of autoantibody specificity and response to intravenous immunoglobulin G therapy in immune thrombocytopenia: A multicenter cohort study. J. Thromb. Haemost. 2014, 12, 497–504. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Rosovsky, R.P.; Karp Leaf, R.S.; Smith, D.B.; Goodarzi, K.; Fogerty, A.E.; Sykes, D.B.; Kuter, D.J. A modern reassessment of glycoprotein-specific direct platelet autoantibody testing in immune thrombocytopenia. Blood Adv. 2020, 4, 9–18. [Google Scholar] [CrossRef]
- Dash, C.H.; Gillanders, K.R.; Stratford Bobbitt, M.E.; Gascoigne, E.W.; Leach, S.J. Safety and efficacy of Gammaplex® in idiopathic thrombocytopenic purpura (ClinicalTrials.gov--NCT00504075). PLoS ONE 2014, 9, e96600. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, F.A. Intravenous immunoglobulin: Adverse reactions and management. J. Allergy Clin. Immunol. 2008, 122, 1238–1239. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Graziano, J.N.; Kimberly, R.P.; Pahwa, S.; Aledort, L.M. Intravenous anti-D treatment of immune thrombocytopenic purpura: Analysis of efficacy, toxicity, and mechanism of effect. Blood 1991, 77, 1884–1893. [Google Scholar] [CrossRef]
- Cheung, E.; Liebman, H.A. Anti-RhD immunoglobulin in the treatment of immune thrombocytopenia. Biologics 2009, 3, 57–62. [Google Scholar] [PubMed]
- Meyer, O.; Kiesewetter, H.; Hermsen, M.; Petriedes, P.; Rose, M.; Seibt, H.; Salama, A. Replacement of intravenous administration of anti-D by subcutaneous administration in patients with autoimmune thrombocytopenia. Pediatr. Blood Cancer 2006, 47, 721–722. [Google Scholar] [CrossRef] [PubMed]
- Scaradavou, A.; Woo, B.; Woloski, B.M.; Cunningham-Rundles, S.; Ettinger, L.J.; Aledort, L.M.; Bussel, J.B. Intravenous anti-D treatment of immune thrombocytopenic purpura: Experience in 272 patients. Blood 1997, 89, 2689–2700. [Google Scholar] [CrossRef]
- Gaines, A.R. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following Rh(0)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005, 106, 1532–1537. [Google Scholar] [CrossRef]
- Tarantino, M.D.; Bussel, J.B.; Cines, D.B.; McCrae, K.R.; Gernsheimer, T.; Liebman, H.A.; Wong, W.-Y.; Kulkarni, R.; Grabowski, E.; McMillan, R. A closer look at intravascular hemolysis (IVH) following intravenous anti-D for immune thrombocytopenic purpura (ITP). Blood 2007, 109, 5527. [Google Scholar] [CrossRef]
- Roumier, M.; Le Burel, S.; Audia, S.; Chauchet, A.; Gousseff, M.; Hamidou, M.; Liferman, F.; Moulis, G.; Lioger, B.; Galicier, L.; et al. High dose romiplostim as a rescue therapy for adults with severe bleeding and refractory immune thrombocytopenia. Am. J. Hematol. 2020. [Google Scholar] [CrossRef]
- Mayer, B.; Salama, A. Successful treatment of bleeding with tranexamic acid in a series of 12 patients with immune thrombocytopenia. Vox Sang. 2017, 112, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, J.R.; Salgia, R.; Bell, W.R. Control of bleeding in patients with immune and nonimmune thrombocytopenia with aminocaproic acid. Arch. Intern. Med. 1989, 149, 1959–1961. [Google Scholar] [CrossRef] [PubMed]
- Randall, M.M.; Nurse, J.; Singh, K.P. Tranexamic Acid in a Case Report of Life-threatening Nontraumatic Hemorrhage in Immune Thrombocytopenic Purpura. Clin. Pract. Cases Emerg. Med. 2020, 4, 421–423. [Google Scholar] [CrossRef]
- Gurion, R.; Siu, A.; Weiss, A.R.; Masterson, M. Use of Recombinant Factor VIIa in a Pediatric Patient with Initial Presentation of Refractory Acute Immune Thrombocytopenic Purpura and Severe Bleeding. J. Pediatr. Pharmacol. Ther. 2012, 17, 274–280. [Google Scholar] [CrossRef]
- Gerotziafas, G.T.; Zervas, C.; Gavrielidis, G.; Tokmaktsis, A.; Hatjiharissi, E.; Papaioannou, M.; Lazaridou, A.; Constantinou, N.; Samama, M.M.; Christakis, J. Effective hemostasis with rFVIIa treatment in two patients with severe thrombocytopenia and life-threatening hemorrhage. Am. J. Hematol. 2002, 69, 219–222. [Google Scholar] [CrossRef]
- Waddington, D.P.; McAuley, F.T.; Hanley, J.P.; Summerfield, G.P. The use of recombinant factor viia in a jehovah’s witness with auto-immune thrombocytopenia and post-splenectomy haemorrhage. Br. J. Haematol. 2002, 119, 286–288. [Google Scholar] [CrossRef]
- Kuter, D.J.; Arnold, D.M.; Rodeghiero, F.; Janssens, A.; Selleslag, D.; Bird, R.; Newland, A.; Mayer, J.; Wang, K.; Olie, R. Safety and efficacy of self-administered romiplostim in patients with immune thrombocytopenia: Results of an integrated database of five clinical trials. Am. J. Hematol. 2020, 95, 643–651. [Google Scholar] [CrossRef]
- Kuter, D.J.; Newland, A.; Chong, B.H.; Rodeghiero, F.; Romero, M.T.; Pabinger, I.; Chen, Y.; Wang, K.; Mehta, B.; Eisen, M. Romiplostim in adult patients with newly diagnosed or persistent immune thrombocytopenia (ITP) for up to 1 year and in those with chronic ITP for more than 1 year: A subgroup analysis of integrated data from completed romiplostim studies. Br. J. Haematol. 2019, 185, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Murali, M.; Michel, M.; Viallard, J.-F.; Giagounidis, A.; Janssens, A.; Legg, J.; Deuson, R.; Danese, M.D. Evaluation of bleeding-related episodes in patients with immune thrombocytopenia (ITP) receiving romiplostim or medical standard of care. Int. J. Hematol. 2012, 96, 26–33. [Google Scholar] [CrossRef]
- Gernsheimer, T.B.; George, J.N.; Aledort, L.M.; Tarantino, M.D.; Sunkara, U.; Matthew Guo, D.; Nichol, J.L. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J. Thromb. Haemost. 2010, 8, 1372–1382. [Google Scholar] [CrossRef]
- Kuter, D.J.; Rummel, M.; Boccia, R.; Macik, B.G.; Pabinger, I.; Selleslag, D.; Rodeghiero, F.; Chong, B.H.; Wang, X.; Berger, D.P. Romiplostim or standard of care in patients with immune thrombocytopenia. N. Engl. J. Med. 2010, 363, 1889–1899. [Google Scholar] [CrossRef]
- Janssens, A.; Rodeghiero, F.; Anderson, D.; Chong, B.H.; Boda, Z.; Pabinger, I.; Červinek, L.; Terrell, D.R.; Wang, X.; Franklin, J. Changes in bone marrow morphology in adults receiving romiplostim for the treatment of thrombocytopenia associated with primary immune thrombocytopenia. Ann. Hematol. 2016, 95, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P. TPO-mimetics and myelofibrosis? A reticulin question! Blood 2009, 114, 3722–3723. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J.; Mufti, G.J.; Bain, B.J.; Hasserjian, R.P.; Davis, W.; Rutstein, M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009, 114, 3748–3756. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P. Spotlight on eltrombopag in treatment-refractory chronic primary immune thrombocytopenia. BioDrugs 2011, 25, 401–404. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Kuter, D.J. An alternative intermittent eltrombopag dosing protocol for the treatment of chronic immune thrombocytopenia. Br. J. Clin. Pharmacol. 2018, 84, 2673–2677. [Google Scholar] [CrossRef]
- Promacta® Prescription Information. Available online: www.accessdata.fda.gov/drugsatfda_docs/label/2015/207027s000lbl.pdf (accessed on 2 January 2021).
- Bussel, J.B.; Provan, D.; Shamsi, T.; Cheng, G.; Psaila, B.; Kovaleva, L.; Salama, A.; Jenkins, J.M.; Roychowdhury, D.; Mayer, B.; et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 641–648. [Google Scholar] [CrossRef]
- Tomiyama, Y.; Miyakawa, Y.; Okamoto, S.; Katsutani, S.; Kimura, A.; Okoshi, Y.; Ninomiya, H.; Kosugi, H.; Nomura, S.; Ozaki, K.; et al. A lower starting dose of eltrombopag is efficacious in Japanese patients with previously treated chronic immune thrombocytopenia. J. Thromb. Haemost. 2012, 10, 799–806. [Google Scholar] [CrossRef]
- Yang, R.; Hou, M.; Li, J. Effect of eltrombopag on platelet response and safety results in Chinese adults with chronic ITP-primary result of a phase III study (abstract). Blood 2014, 124, 1464. [Google Scholar] [CrossRef]
- Cheng, G.; Saleh, M.N.; Marcher, C.; Vasey, S.; Mayer, B.; Aivado, M.; Arning, M.; Stone, N.L.; Bussel, J.B. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): A 6-month, randomised, phase 3 study. Lancet 2011, 377, 393–402. [Google Scholar] [CrossRef]
- Wong, R.S.M.; Saleh, M.N.; Khelif, A.; Salama, A.; Portella, M.S.O.; Burgess, P.; Bussel, J.B. Safety and efficacy of long-term treatment of chronic/persistent ITP with eltrombopag: Final results of the EXTEND study. Blood 2017, 130, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- González-López, T.J.; Pascual, C.; Álvarez-Román, M.T.; Fernández-Fuertes, F.; Sánchez-González, B.; Caparrós, I.; Jarque, I.; Mingot-Castellano, M.E.; Hernández-Rivas, J.A.; Martín-Salces, M.; et al. Successful discontinuation of eltrombopag after complete remission in patients with primary immune thrombocytopenia. Am. J. Hematol. 2015, 90, E40–E43. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Kuter, D.J. Thrombopoietin level predicts response to treatment with eltrombopag and romiplostim in immune thrombocytopenia. Am. J. Hematol. 2018, 93, 1501–1508. [Google Scholar] [CrossRef]
- Mishra, K.; Pramanik, S.; Jandial, A.; Sahu, K.K.; Sandal, R.; Ahuja, A.; Yanamandra, U.; Kumar, R.; Kapoor, R.; Verma, T.; et al. Real-world experience of eltrombopag in immune thrombocytopenia. Am. J. Blood Res. 2020, 10, 240–251. [Google Scholar] [PubMed]
- Doptelet® Prescribtion Information. Available online: www.accessdata.fda.gov/drugsatfda_docs/label/2018/210238s000lbl.pdf (accessed on 8 January 2021).
- Jurczak, W.; Chojnowski, K.; Mayer, J.; Krawczyk, K.; Jamieson, B.D.; Tian, W.; Allen, L.F. Phase 3 randomised study of avatrombopag, a novel thrombopoietin receptor agonist for the treatment of chronic immune thrombocytopenia. Br. J. Haematol. 2018, 183, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Godeau, B. B-cell depletion in immune thrombocytopenia. Semin. Hematol. 2013, 50 (Suppl. 1), S75–S82. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.; Darvish-Kazem, S.; Lim, W.; Crowther, M.A.; Ghanima, W.; Wang, G.; Heddle, N.M.; Kelton, J.G.; Arnold, D.M. Rituximab plus standard of care for treatment of primary immune thrombocytopenia: A systematic review and meta-analysis. Lancet Haematol. 2015, 2, e75–e81. [Google Scholar] [CrossRef]
- Patel, V.L.; Mahévas, M.; Lee, S.Y.; Stasi, R.; Cunningham-Rundles, S.; Godeau, B.; Kanter, J.; Neufeld, E.; Taube, T.; Ramenghi, U.; et al. Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood 2012, 119, 5989–5995. [Google Scholar] [CrossRef]
- Ghanima, W.; Khelif, A.; Waage, A.; Michel, M.; Tjønnfjord, G.E.; Romdhan, N.B.; Kahrs, J.; Darne, B.; Holme, P.A. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 1653–1661. [Google Scholar] [CrossRef]
- Li, Y.; Shi, Y.; He, Z.; Chen, Q.; Liu, Z.; Yu, L.; Wang, C. The efficacy and safety of low-dose rituximab in immune thrombocytopenia: a systematic review and meta-analysis. Platelets 2019, 30, 690–697. [Google Scholar] [CrossRef]
- Tjønnfjord, E.; Holme, P.A.; Darne, B.; Khelif, A.; Waage, A.; Michel, M.; Ben Romdhan, N.; Ghanima, W. Long-term outcomes of patients treated with rituximab as second-line treatment for adult immune thrombocytopenia - Follow-up of the RITP study. Br. J. Haematol. 2020, 191, 460–465. [Google Scholar] [CrossRef]
- Hammond, W.A.; Vishnu, P.; Rodriguez, E.M.; Li, Z.; Dholaria, B.; Shreders, A.J.; Rivera, C.E. Sequence of Splenectomy and Rituximab for the Treatment of Steroid-Refractory Immune Thrombocytopenia: Does It Matter? Mayo Clin. Proc. 2019, 94, 2199–2208. [Google Scholar] [CrossRef]
- Wang, Y.-M.; Yu, Y.-F.; Liu, Y.; Liu, S.; Hou, M.; Liu, X.-G. The association between antinuclear antibody and response to rituximab treatment in adult patients with primary immune thrombocytopenia. Hematology 2020, 25, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Khellaf, M.; Charles-Nelson, A.; Fain, O.; Terriou, L.; Viallard, J.-F.; Cheze, S.; Graveleau, J.; Slama, B.; Audia, S.; Ebbo, M.; et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: Results from a prospective registry including 248 patients. Blood 2014, 124, 3228–3236. [Google Scholar] [CrossRef] [PubMed]
- Puavilai, T.; Thadanipon, K.; Rattanasiri, S.; Ingsathit, A.; McEvoy, M.; Attia, J.; Thakkinstian, A. Treatment efficacy for adult persistent immune thrombocytopenia: A systematic review and network meta-analysis. Br. J. Haematol. 2020, 188, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Podolanczuk, A.; Lazarus, A.H.; Crow, A.R.; Grossbard, E.; Bussel, J.B. Of mice and men: an open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood 2009, 113, 3154–3160. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.; Arnold, D.M.; Grossbard, E.; Mayer, J.; Treliński, J.; Homenda, W.; Hellmann, A.; Windyga, J.; Sivcheva, L.; Khalafallah, A.A.; et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am. J. Hematol. 2018, 93, 921–930. [Google Scholar] [CrossRef]
- Bussel, J.B.; Arnold, D.M.; Boxer, M.A.; Cooper, N.; Mayer, J.; Zayed, H.; Tong, S.; Duliege, A.-M. Long-term fostamatinib treatment of adults with immune thrombocytopenia during the phase 3 clinical trial program. Am. J. Hematol. 2019, 94, 546–553. [Google Scholar] [CrossRef]
- Boccia, R.; Cooper, N.; Ghanima, W.; Boxer, M.A.; Hill, Q.A.; Sholzberg, M.; Tarantino, M.D.; Todd, L.K.; Tong, S.; Bussel, J.B. Fostamatinib is an effective second-line therapy in patients with immune thrombocytopenia. Br. J. Haematol. 2020, 190, 933–938. [Google Scholar] [CrossRef]
- Tavalisse® Prescription Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/209299lbl.pdf (accessed on 2 January 2021).
- Chaturvedi, S.; Arnold, D.M.; McCrae, K.R. Splenectomy for immune thrombocytopenia: Down but not out. Blood 2018, 131, 1172–1182. [Google Scholar] [CrossRef]
- Palandri, F.; Polverelli, N.; Sollazzo, D.; Romano, M.; Catani, L.; Cavo, M.; Vianelli, N. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am. J. Hematol. 2016, 91, E267–E272. [Google Scholar] [CrossRef]
- Kojouri, K.; Vesely, S.K.; Terrell, D.R.; George, J.N. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004, 104, 2623–2634. [Google Scholar] [CrossRef]
- Lal, L.S.; Said, Q.; Andrade, K.; Cuker, A. Second-line treatments and outcomes for immune thrombocytopenia: A retrospective study with electronic health records. Res. Pract. Thromb. Haemost. 2020, 4, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Vianelli, N.; Palandri, F.; Polverelli, N.; Stasi, R.; Joelsson, J.; Johansson, E.; Ruggeri, M.; Zaja, F.; Cantoni, S.; Catucci, A.E.; et al. Splenectomy as a curative treatment for immune thrombocytopenia: A retrospective analysis of 233 patients with a minimum follow up of 10 years. Haematologica 2013, 98, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Amini, S.N.; Nelson, V.S.; Sobels, A.; Schoones, J.W.; Zwaginga, J.J.; Schipperus, M.R. Autologous platelet scintigraphy and clinical outcome of splenectomy in immune thrombocytopenia: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2020, 153, 103040. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Xu, J.; Jiao, C.; Cheng, Z.; Ren, S. Long-Term Outcomes of Laparoscopic Splenectomy Versus Open Splenectomy for Idiopathic Thrombocytopenic Purpura. Int. Surg. 2014, 99, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Nørgaard, M.; Cetin, K.; Maegbaek, M.L.; Kristensen, N.R.; Ghanima, W.; Bahmanyar, S.; Stryker, S.; Christiansen, C.F. Risk of arterial thrombotic and venous thromboembolic events in patients with primary chronic immune thrombocytopenia: A Scandinavian population-based cohort study. Br. J. Haematol. 2016, 174, 639–642. [Google Scholar] [CrossRef]
- Boyle, S.; White, R.H.; Brunson, A.; Wun, T. Splenectomy and the incidence of venous thromboembolism and sepsis in patients with immune thrombocytopenia. Blood 2013, 121, 4782–4790. [Google Scholar] [CrossRef]
- Maria, L.A.; Nour, A.; Eleanor, P.; Victor, B.; Paul, I.; Thomas, K.; Intercontinental, C.I.S.G. Long-term outcomes after splenectomy in children with immune thrombocytopenia: An update on the registry data from the Intercontinental Cooperative ITP Study Group. Haematologica 2019, 105, 2682–2685. [Google Scholar] [CrossRef]
- Kwiatkowska, A.; Radkowiak, D.; Wysocki, M.; Torbicz, G.; Gajewska, N.; Lasek, A.; Kulawik, J.; Budzyński, A.; Pędziwiatr, M. Prognostic Factors for Immune Thrombocytopenic Purpura Remission after Laparoscopic Splenectomy: A Cohort Study. Medicina (Kaunas) 2019, 55. [Google Scholar] [CrossRef]
- Gonzalez-Porras, J.R.; Escalante, F.; Pardal, E.; Sierra, M.; Garcia-Frade, L.J.; Redondo, S.; Arefi, M.; Aguilar, C.; Ortega, F.; de Cabo, E.; et al. Safety and efficacy of splenectomy in over 65-yrs-old patients with immune thrombocytopenia. Eur. J. Haematol. 2013, 91, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Yi, H.G.; Kim, C.S.; Hong, J.; Park, J.; Lee, J.H.; Kim, H.Y.; Kim, H.J.; Zang, D.Y.; Kim, S.H.; et al. Clinical Outcome and Predictive Factors in the Response to Splenectomy in Elderly Patients with Primary Immune Thrombocytopenia: A Multicenter Retrospective Study. Acta Haematol. 2016, 135, 162–171. [Google Scholar] [CrossRef]
- Robak, T.; Kaźmierczak, M.; Jarque, I.; Musteata, V.; Treliński, J.; Cooper, N.; Kiessling, P.; Massow, U.; Woltering, F.; Snipes, R.; et al. Phase 2 multiple-dose study of an FcRn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood Adv. 2020, 4, 4136–4146. [Google Scholar] [CrossRef]
- Li, G.; Wang, S.; Li, N.; Liu, Y.; Feng, Q.; Zuo, X.; Li, X.; Hou, Y.; Shao, L.; Ma, C.; et al. Proteasome Inhibition with Bortezomib Induces Apoptosis of Long-Lived Plasma Cells in Steroid-Resistant or Relapsed Immune Thrombocytopaenia. Thromb. Haemost. 2018, 118, 1752–1764. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.D.; Rollins-Raval, M.A.; Raval, J.S.; Park, Y.A.; Mazepa, M.; Ma, A. Bortezomib for Refractory Immune-Mediated Thrombocytopenia Purpura. Am. J. Ther. 2018, 25, e270–e272. [Google Scholar] [CrossRef]
- Newland, A.C.; Sánchez-González, B.; Rejtő, L.; Egyed, M.; Romanyuk, N.; Godar, M.; Verschueren, K.; Gandini, D.; Ulrichts, P.; Beauchamp, J.; et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am. J. Hematol. 2020, 95, 178–187. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, Z.; Ma, J.; Ma, J.; Liu, F.; Wu, R. Foxp3 methylation status in children with primary immune thrombocytopenia. Hum. Immunol. 2014, 75, 1115–1119. [Google Scholar] [CrossRef]
- Ding, K.; Fu, R.; Liu, H.; Nachnani, D.A.; Shao, Z.-H. Effects of decitabine on megakaryocyte maturation in patients with myelodysplastic syndromes. Oncol. Lett. 2016, 11, 2347–2352. [Google Scholar] [CrossRef][Green Version]
- Zhou, H.; Hou, Y.; Liu, X.; Qiu, J.; Feng, Q.; Wang, Y.; Zhang, X.; Min, Y.; Shao, L.; Liu, X.; et al. Low-dose decitabine promotes megakaryocyte maturation and platelet production in healthy controls and immune thrombocytopenia. Thromb. Haemost. 2015, 113, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Qin, P.; Liu, Q.; Yuan, C.; Hao, Y.; Zhang, H.; Wang, Z.; Ran, X.; Chu, X.; Yu, W.; et al. A prospective, multicenter study of low dose decitabine in adult patients with refractory immune thrombocytopenia. Am. J. Hematol. 2019, 94, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Thachil, J. Coronavirus Disease 2019 Coagulopathy: Disseminated Intravascular Coagulation and Thrombotic Microangiopathy-Either, Neither, or Both. Semin. Thromb. Hemost. 2020, 46, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Pavord, S.; Thachil, J.; Hunt, B.J.; Murphy, M.; Lowe, G.; Laffan, M.; Makris, M.; Newland, A.C.; Provan, D.; Grainger, J.D.; et al. Practical guidance for the management of adults with immune thrombocytopenia during the COVID-19 pandemic. Br. J. Haematol. 2020, 189, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Mahévas, M.; Moulis, G.; Andres, E.; Riviere, E.; Garzaro, M.; Crickx, E.; Guillotin, V.; Malphettes, M.; Galicier, L.; Noel, N.; et al. Clinical characteristics, management and outcome of COVID-19-associated immune thrombocytopenia: a French multicentre series. Br. J. Haematol. 2020, 190, e224–e229. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Agent | Application Route and Dosage | Advantages | Disadvantages and Complications |
|---|---|---|---|
| First-line Therapies | |||
| Glucocorticoids | |||
| Predniso(lo)ne | Oral 1 mg/kg of body weight for 2–3 weeks (maximum 80 mg/d), gradual tapering | Response within 1–2 weeks Early response rate 60–80% | Low durable response rate after discontinuation (30–50%) Complications: hypertension, hyperglycaemia, sleep and mood disturbances, gastric ulceration, myopathy, glaucoma and osteoporosis |
| Dexamethasone | Oral 40 mg for 4 days Maximum 3 cycles | ||
| Immunoglobulin | Intravenous 0.4–1 gr/kg of body weight, total maximal dose of 2 gr/kg of body weight | Response within 1–4 days Early response rate 70–80% | Only a transient rise of platelet count Complications: headache, pyrexia, vomiting, acute kidney injury, aseptic meningitis and thrombotic events |
| Anti-Rhesus D Ig | Intravenous 50 to 75 µg/kg | Early response rate 65% | Only effective in Rh-positive patients Not approved for ITP in Europe Complications: headache, nausea, chills, fever and mild to moderate haemolysis Severe intravascular haemolysis and disseminated intravascular coagulation |
| Second-line Therapies | |||
| Thrombopoietin-receptor Agonists | |||
| Romiplostim | Subcutaneous 1–10 microg/kg/week | Response rate 70–80% Remission rate 10–30% | Cost Headache, arthralgia, myalgia, dizziness and insomnia Thromboembolism and bone marrow fibrosis |
| Eltrombobag | Oral 25–75 mg/day | Response rate 70–80% Remission rate 10–30% | Cost Dietary restrictions Gastrointestinal symptoms (nausea, vomiting, diarrhoea), mild transaminase elevations and headache Thromboembolism and bone marrow fibrosis |
| Avatrombopag | Oral 20–40 mg/day | Response rate 60% No dietary restrictions | Cost Headache, arthralgia, fatigue and diarrhoea |
| Immunomodulators | |||
| Rituximab | Intravenous 375 mg/m2 per week for 4 weeks 100 mg/m2 per week for 4 weeks | Response rate 60% at 6 months No need for chronic treatment | High relapse rate Contraindicated by patients with evidence of an active or previous HBV infection Increased tendency to minor infections; progressive multifocal leukoencephalopathy |
| Fostamatinib | Oral 100–150 mg twice daily | Response rate 43% within 12 weeks after treatment | Diarrhoea, hypertension and nausea Monthly follow up for hypertension, hepatotoxicity and neutropenia. |
| Splenectomy | Open or laparoscopic surgery | Durable remission rate 60 to 70% No need for chronic treatment | Surgical complications, thromboembolic events, infection with encapsulated bacteria, Sepsis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.; Uzun, G.; Bakchoul, T. Primary Immune Thrombocytopenia: Novel Insights into Pathophysiology and Disease Management. J. Clin. Med. 2021, 10, 789. https://doi.org/10.3390/jcm10040789
Singh A, Uzun G, Bakchoul T. Primary Immune Thrombocytopenia: Novel Insights into Pathophysiology and Disease Management. Journal of Clinical Medicine. 2021; 10(4):789. https://doi.org/10.3390/jcm10040789
Chicago/Turabian StyleSingh, Anurag, Günalp Uzun, and Tamam Bakchoul. 2021. "Primary Immune Thrombocytopenia: Novel Insights into Pathophysiology and Disease Management" Journal of Clinical Medicine 10, no. 4: 789. https://doi.org/10.3390/jcm10040789
APA StyleSingh, A., Uzun, G., & Bakchoul, T. (2021). Primary Immune Thrombocytopenia: Novel Insights into Pathophysiology and Disease Management. Journal of Clinical Medicine, 10(4), 789. https://doi.org/10.3390/jcm10040789