
Osteoporosis Treatment with Anti-Sclerostin Antibodies—Mechanisms of Action and Clinical Application


Abstract
1. Introduction
2. Sclerostin
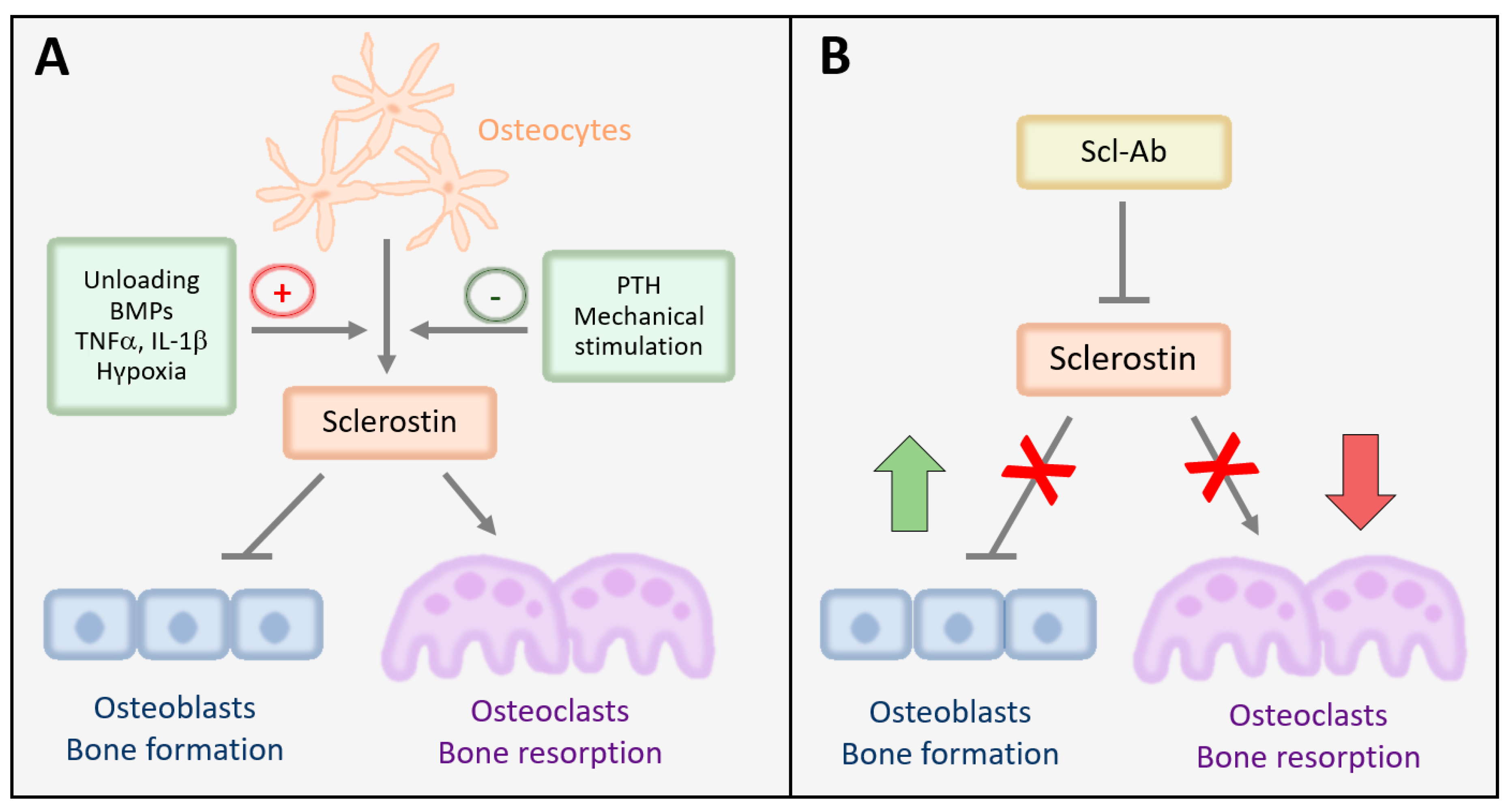
2.1. Expression, Mechanisms of Action, and Regulation of Sclerostin in Bone
2.2. Sclerostin Is a Critical Regulator of Bone Mass
3. Lessons Learnt about the Mode of Action of Sclerostin from Animal Models
3.1. Genetic Animal Models of Sclerostin Deficiency or Overexpression
3.2. Preclinical Models of Sclerostin Inhibition
4. Monoclonal Antibodies against Sclerostin in Human Osteoporosis Treatment
4.1. Blomosozumab
4.2. Romosozumab
4.2.1. Phase I and II Studies: Results on BMD and Bone Turnover Markers
4.2.2. Phase III Studies: Results on BMD and Anti-Fracture Activity

{kind=link}
| Type of Study | Aim | Study Population | Main Findings | Reference | ||
|---|---|---|---|---|---|---|
| (Patients) | BMD | Fx | BTMs | |||
| Phase II international, multicenter, placebo-controlled, parallel-group RCT | BMD changes after 12 months of Romo (5 doses) vs. placebo vs. ALN vs. TPTD | Postmenopausal women with T-score (FN/TH/LS) between −2.5 and −3.5 (419) | BMD LS: ↑11.3% (Romo 210 mg/month) ↑7.1% (TPTD) ↑4.1% (ALN) ↓0.1% (placebo) | NR | Romo: P1NP ↑ with maximum after 1 month, then gradual decrease CTX ↓ sustained through month 12 | [67] |
| Phase III international, multicenter, double-blind, placebo-controlled, parallel-group RCT (FRAME) | Primary: VFx at month 12 (Romo vs placebo) and month 24 (all Dmab) Secondary: (i) Clinical Fx, (ii) non-VFx | Postmenopausal women with T-score (FN/TH/LS) between −2.5 and −3.5 (7180) | BMD compared to placebo: ↑ at LS 13.3%; at TH 6.9%; at FN 5.9% | ↓ VFx at month 12 (73% lower RR with Romo) and sustained at month 24 ↓ Clinical VFx at month 12 (36% lower RR with Romo) No significant reduction of non-VFx | Romo: P1NP ↑ with maximum after 14 days, then gradual decrease CTX ↓ with minimum after 14 days, sustained decrease through month 12 | [69] |
| Phase III, international, multicenter, double-blind trial RCT vs. active comparator (ARCH) | Primary: (i) VFx at month 24 (0–12 month Romo vs ALN and 12–24 month all ALN) (ii) Clinical Fx at primary analysis Secondary: (i) BMD LS, FN, TH (ii) non-VFx at primary analysis | Postmenopausal women with T-score (FN/TH) ≤ −2.0 and two or more VFx or a hip Fx (4093) | Month 12 BMD LS:↑13.7% (Romo) ↑5.0% (ALN) Month 24 LS:↑15.2% (Romo→ALN) ↑7.1% (ALN→ALN) | ↓ VFx at month 24 (48% lower RR with Romo compared to ALN) ↓ Clinical VFx at primary analysis (27% lower RR with Romo compared to ALN) | Romo: P1NP ↑ with maximum after1 month, then gradual decrease and suppression after switch to ALN CTX ↓ sustained suppression through month 24 | [71] |
| Phase III international, multicenter, double-blind, placebo-controlled, parallel-group RCT in men (BRIDGE) | Primary: BMD LS changes after 12 months of Romo vs. placebo Secondary: (i) BMD FN/TH changes after 12 months of Romo vs. placebo (ii) BTMs iii) bone histology (subset of 20) | Men aged 55–90 years, with T-score (FN/TH/LS) between −1.5 and −2.5 and fragility VFx or non-VFx (245) | Month 12 BMD LS:↑12.1% (Romo) ↑1.2% (placebo) TH:↑2.5% (Romo) ↓0.5% (placebo) FN:↑2.2% (Romo) ↓0.2% (placebo) | NR | Romo: P1NP ↑ with maximum after 1 month, then gradual decrease CTX ↓ with minimum after 1 month, sustained decrease through month 12 * histology: sustained reduction in bone resorption | [72] |
| Phase III, international, multicenter, double-blind trial RCT vs. active comparator (STRUCTURE) | Primary: BMD TH change at 12 months Secondary: (i) BMD TH change at 12 months (ii) cortical BMD at TH at 6 and 12 months (iii) BMC at TH at 6 and 12 months iv) estimated bone strength at TH at 6 and 12 months | Postmenopausal women with T-score (FN/TH/LS) ≤ −2.5 and fragility VFx or non-VFx; ≥ 3 years of prior BP therapy (436) | Month 12 BMD TH:↑2.6% (Romo) ↓0.6% (TPTD) Month 12 cortical BMD TH:↑1.1% (Romo) ↓3.6% (TPTD) Month 12 estimated bone strength TH:↑2.5% (Romo) ↓0.7% (TPTD) Month 12 BMC TH:↑3.6% (Romo) ≈0.0% (TPTD) | NR | NR | [76] |
4.2.3. Post-Hoc Analyses and Additional Exploratory End-Points
4.2.4. Extension Studies and Sequential Treatment Effects
4.2.5. Meta-Analyses
5. Safety Profile of Romosozumab
6. Translational Applications of Sclerostin Antibodies in Diseases other than Osteoporosis
6.1. Osteogenesis Imperfecta (OI)
6.2. X-Linked Hypophosphatemia (XLH)
6.3. Malignant Disease
6.4. Other
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy, 7–29 March 2000: Highlights of the conference. South Med. J. 2001, 94, 569–573.
- Manolagas, S.C.; O’Brien, C.A.; Almeida, M. The role of estrogen and androgen receptors in bone health and disease. Nat. Rev. Endocrinol. 2013, 9, 699–712. [Google Scholar] [CrossRef]
- Hernlund, E.; Svedbom, A.; Ivergard, M.; Compston, J.; Cooper, C.; Stenmark, J.; McCloskey, E.V.; Jönsson, B.; Kanis, J.A. Osteoporosis in the European Union: Medical management, epidemiology and economic burden. A report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch. Osteoporos. 2013, 8, 136. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.; Silverman, S.L.; Cooper, C.; Hanley, D.A.; Barton, I.; Broy, S.B.; Licata, A.; Benhamou, L.; Geusens, P.; Flowers, K.; et al. Risk of new vertebral fracture in the year following a fracture. JAMA 2001, 285, 320–323. [Google Scholar] [CrossRef]
- Betella, N.; Biamonte, E.; Matarazzo, C.; Piccini, S.; Olivetti, R.; Cellini, M.; Lania, A.G.; Mazziotti, G. Suboptimal medication adherence may favor the progression of vertebral fractures in women with post-menopausal osteoporosis treated with denosumab. Minerva Endocrinol. 2020, 45, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, S.; Francomano, D.; Romagnoli, E.; Marocco, C.; Fornari, R.; Resmini, G.; Buffa, A.; Di Pietro, G.; Corvaglia, S.; Gimigliano, F.; et al. Persistence with denosumab therapy in women affected by osteoporosis with fragility fractures: A multicenter observational real practice study in Italy. J. Endocrinol. Investig. 2017, 40, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep. 2014, 3, 481. [Google Scholar] [CrossRef]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef]
- Colette, N.M.; Genetos, D.C.; Economides, A.N.; Xie, L.; Shahnazari, M.; Yao, W.; Lane, N.E.; Harland, R.M.; Loots, G.G. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc. Natl. Acad. Sci. USA 2012, 109, 14092–14097. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, E.; Wang, W.; Tam, C.; Hwang, J.; Zhang, Y.; Spittler, D.; Huang, O.W.; Gong, Y.; Estevez, A.; Zilberleyb, I.; et al. Wnt antagonists bind through a short peptide to the first beta-propeller domain of LRP5/6. Structure 2011, 19, 1433–1442. [Google Scholar] [CrossRef]
- Ellies, D.L.; Viviano, B.; McCarthy, J.; Rey, J.P.; Itasaki, N.; Saunders, S.; Krumlauf, R. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J. Bone Miner. Res. 2006, 21, 1738–1749. [Google Scholar] [CrossRef]
- Semenov, M.; Tamai, K.; He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 2005, 280, 26770–26775. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Fuentes, F.J.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.G.; Sutherland, M.S.; Ojala, E.; Turcott, E.; Geoghegan, J.C.; Shpektor, D.; Skonier, J.E.; Yu, C.; Latham, J.A. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J. Biol Chem 2005, 280, 2498–2502. [Google Scholar] [CrossRef]
- Krause, C.; Korchynskyi, O.; de Rooij, K.; Weidauer, S.E.; de Gorter, D.J.; van Bezooijen, R.L.; Hatsell, S.; Economides, A.N.; Mueller, T.D.; Löwik, C.W.; et al. Distinct modes of inhibition by sclerostin on bone morphogenetic protein and Wnt signaling pathways. J. Biol. Chem. 2010, 285, 41614–41626. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Turcott, E.; Skonier, J.E.; Winkler, D.G.; Latham, J.A. Sclerostin promotes the apoptosis of human osteoblastic cells: A novel regulation of bone formation. Bone 2004, 35, 828–835. [Google Scholar] [CrossRef]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef]
- Ryan, Z.C.; Ketha, H.; McNulty, M.S.; McGee-Lawrence, M.; Craig, T.A.; Grande, J.P.; Westendorf, J.J.; Singh, R.J.; Kumar, R. Sclerostin alters serum vitamin D metabolite and fibroblast growth factor 23 concentrations and the urinary excretion of calcium. Proc. Natl. Acad. Sci. USA 2013, 110, 6199–6204. [Google Scholar] [CrossRef]
- Florio, M.; Gunasekaran, K.; Stolina, M.; Li, X.; Liu, L.; Tipton, B.; Salimi-Moosavi, H.; Asuncion, F.J.; Li, C.; Sun, B.; et al. A bispecific antibody targeting sclerostin and DKK-1 promotes bone mass accrual and fracture repair. Nat. Commun. 2016, 7, 11505. [Google Scholar] [CrossRef]
- Taylor, S.; Ominsky, M.S.; Hu, R.; Pacheco, E.; He, Y.D.; Brown, D.L.; Aguirre, J.I.; Wronski, T.J.; Buntich, S.; Afshari, C.A.; et al. Time-dependent cellular and transcriptional changes in the osteoblast lineage associated with sclerostin antibody treatment in ovariectomized rats. Bone 2016, 84, 148–159. [Google Scholar] [CrossRef]
- Guañabens, N.; Ruiz-Gaspa, S.; Gifre, L.; Miquel, R.; Peris, P.; Monegal, A.; Dubrueil, M.; Arias, A.; Parés, A. Sclerostin expression in bile ducts of patients with chronic cholestasis may influence the bone disease in primary biliary cirrhosis. J. Bone Miner. Res. 2016, 31, 1725–1733. [Google Scholar] [CrossRef]
- Kim, S.P.; Frey, J.L.; Li, Z.; Kushwaha, P.; Zoch, M.L.; Tomlinson, R.E.; Da, H.; Aja, S.; Noh, H.L.; Kim, J.K.; et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E11238–E11247. [Google Scholar] [CrossRef] [PubMed]
- Roudier, M.; Li, X.; Niu, Q.T.; Pacheco, E.; Pretorius, J.K.; Graham, K.; Yoon, B.R.; Gong, J.; Warmington, K.; Ke, H.Z.; et al. Sclerostin is expressed in articular cartilage but loss or inhibition does not affect cartilage remodeling during aging or following mechanical injury. Arthritis Rheum. 2013, 65, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Van Bezooijen, R.L.; Deruiter, M.C.; Vilain, N.; Monteiro, R.M.; Visser, A.; van der Wee-Pals, L.; van Munsteren, C.J.; Hogendoorn, P.C.; Aguet, M.; Mummery, C.L.; et al. SOST expression is restricted to the great arteries during embryonic and neonatal cardiovascular development. Dev. Dyn. 2007, 236, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Mackenzie, N.C.; Millan, J.L.; Farquharson, C.; MacRae, V.E. The appearance and modulation of osteocyte marker expression during calcification of vascular smooth muscle cells. PLoS ONE 2011, 6, e19595. [Google Scholar] [CrossRef] [PubMed]
- Colette, N.M.; Yee, C.S.; Murugesh, D.; Sebastian, A.; Taher, L.; Gale, N.W.; Economides, A.N.; Harland, R.M.; Loots, G.G. Sost and its paralog Sostdc1 coordinate digit number in a Gli3-dependent manner. Dev. Biol. 2013, 383, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Sevetson, B.; Taylor, S.; Pan, Y. Cbfa1/RUNX2 directs specific expression of the sclerosteosis gene (SOST). J. Biol. Chem. 2004, 279, 13849–13858. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tang, W.; So, S.; de Crombrugghe, B.; Zhang, C. Sclerostin is a direct target of osteoblast-specific transcription factor osterix. Biochem. Biophys. Res. Commun. 2010, 400, 684–688. [Google Scholar] [CrossRef][Green Version]
- Delgado-Calle, J.; Arozamena, J.; Pérez-López, J.; Bolado-Carrancio, A.; Sañudo, C.; Agudo, G.; de la Vega, R.; Alonso, M.A.; Rodríguez-Rey, J.C.; Riancho, J.A. Role of BMPs in the regulation of sclerostin as revealed by an epigenetic modifier of human bone cells. Mol. Cell Endocrinol. 2013, 369, 27–34. [Google Scholar] [CrossRef]
- Fujiwara, M.; Kubota, T.; Wang, W.; Ohata, Y.; Miura, K.; Kitaoka, T.; Okuzaki, D.; Namba, N.; Michigami, T.; Kitabatake, Y.; et al. Successful induction of sclerostin in human-derived fibroblasts by 4 transcription factors and its regulation by parathyroid hormone, hypoxia, and prostaglandin E2. Bone 2016, 85, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, N.; Kobayashi, T.; Mochida, Y.; Yu, P.B.; Yamauchi, M.; Kronenberg, H.M.; Mishina, Y. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J. Bone Miner. Res. 2010, 25, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, A.; Loots, G.G. Transcriptional control of Sost in bone. Bone 2017, 96, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Kramer, I.; Loots, G.G.; Studer, A.; Keller, H.; Kneissel, M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J. Bone Miner. Res. 2010, 25, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef]
- Lin, C.; Jiang, X.; Dai, Z.; Guo, X.; Weng, T.; Wang, J.; Li, Y.; Feng, G.; Gao, X.; He, L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J. Bone Miner. Res. 2009, 24, 1651–1661. [Google Scholar] [CrossRef]
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012, 50, 209–217. [Google Scholar] [CrossRef]
- Loots, G.G.; Keller, H.; Leupin, O.; Murugesh, D.; Collette, N.M.; Genetos, D.C. TGF-beta regulates sclerostin expression via the ECR5 enhancer. Bone 2012, 50, 663–669. [Google Scholar] [CrossRef]
- Nguyen, J.; Tang, S.Y.; Nguyen, D.; Alliston, T. Load regulates bone formation and sclerostin expression through a TGFbeta-dependent mechanism. PLoS ONE 2013, e53813. [Google Scholar] [CrossRef]
- Keller, H.; Kneissel, M. SOST is a target gene for PTH in bone. Bone 2005, 37, 148–158. [Google Scholar] [CrossRef]
- Chan, B.Y.; Fuller, E.S.; Russell, A.K.; Smith, S.M.; Smith, M.M.; Jackson, M.T.; Cake, M.A.; Read, R.A.; Bateman, J.F.; Sambrook, P.N.; et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartil. 2011, 19, 874–885. [Google Scholar] [CrossRef]
- Pathak, J.L.; Bakker, A.D.; Luyten, F.P.; Verschueren, P.; Lems, W.F.; Klein-Nulend, J.; Bravenboer, N. Systemic inflammation affects human osteocyte-specific protein and cytokine expression. Calcif. Tissue Int. 2016, 98, 596–608. [Google Scholar] [CrossRef]
- Kim, B.J.; Bae, S.J.; Lee, S.Y.; Lee, Y.S.; Baek, J.E.; Park, S.Y.; Lee, S.H.; Koh, J.M.; Kim, G.S. TNF-alpha mediates the stimulation of sclerostin expression in an estrogen-deficient condition. Biochem. Biophys. Res. Commun. 2012, 424, 170–175. [Google Scholar] [CrossRef]
- Vincent, C.; Findlay, D.M.; Welldon, K.J.; Wijenayaka, A.R.; Zheng, T.S.; Haynes, D.R.; Fazzalari, N.L.; Evdokiou, A.; Atkins, G.J. Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J. Bone Miner. Res. 2009, 24, 1434–1449. [Google Scholar] [CrossRef]
- Chen, X.X.; Baum, W.; Dwyer, D.; Stock, M.; Schwabe, K.; Ke, H.Z.; Stolina, M.; Schett, G.; Bozec, A. Sclerostin inhibition reverses systemic, periarticular and local bone loss in arthritis. Ann. Rheum. Dis. 2013, 72, 1732–1736. [Google Scholar] [CrossRef]
- Genetos, D.C.; Toupadakis, C.A.; Raheja, L.F.; Wong, A.; Papanicolaou, S.E.; Fyhrie, D.P.; Loots, G.G.; Yellowley, C.E. Hypoxia decreases sclerostin expression and increases Wnt signaling in osteoblasts. J. Cell Biochem. 2010, 110, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Stegen, S.; Stockmans, I.; Moermans, K.; Thienpont, B.; Maxwell, P.H.; Carmeliet, P.; Carmeliet, G. Osteocytic oxygen sensing controls bone mass through epigenetic regulation of sclerostin. Nat. Commun. 2018, 9, 2557. [Google Scholar] [CrossRef]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef]
- Van Lierop, A.H.; Appelman-Dijkstra, N.M.; Papapoulos, S.E. Sclerostin deficiency in humans. Bone 2017, 96, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ominsky, M.S.; Niu, Q.T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Hassler, N.; Roschger, A.; Gamsjaeger, S.; Kramer, I.; Lueger, S.; van Lierop, A.; Roschger, P.; Klaushofer, K.; Paschalis, E.P.; Kneissel, M.; et al. Sclerostin deficiency is linked to altered bone composition. J. Bone Miner. Res. 2014, 29, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Loots, G.G.; Kneissel, M.; Keller, H.; Baptist, M.; Chang, J.; Collette, N.M.; Ovcharenko, D.; Plajzer-Frick, I.; Rubin, E.M. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005, 15, 928–935. [Google Scholar] [CrossRef]
- Smith, S.Y.; Jolette, J.; Turner, C.H. Skeletal health: Primate model of postmenopausal osteoporosis. Am. J. Primatol. 2009, 71, 752–765. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ominsky, M.S.; Warmington, K.S.; Morony, S.; Gong, J.; Cao, J.; Gao, Y.; Shalhoub, V.; Tipton, B.; Haldankar, R.; et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J. Bone Miner. Res. 2009, 24, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Warmington, K.S.; Niu, Q.T.; Asuncion, F.J.; Barrero, M.; Grisanti, M.; Dwyer, D.; Stouch, B.; Thway, T.M.; Stolina, M.; et al. Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J. Bone Miner. Res. 2010, 25, 2647–2656. [Google Scholar] [CrossRef]
- Li, X.; Niu, Q.-T.; Warmington, K.S.; Asuncion, F.J.; Dwyer, D.; Grisanti, M.; Han, C.-Y.; Stolina, M.; Eschenberg, M.J.; Kostenuik, P.J.; et al. Progressive increases in bone mass and bone strength in an ovariectomized rat model of osteoporosis after 26 weeks of treatment with a sclerostin antibody. Endocrinology 2014, 155, 4785–4797. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Ominsky, M.S.; Warmington, K.S.; Niu, Q.-T.; Asuncion, F.J.; Barrero, M.; Dwyer, D.; Grisanti, M.; Stolina, M.; Kostenuik, P.J.; et al. Increased bone formation and bone mass induced by sclerostin antibody is not affected by pretreatment or cotreatment with alendronate in osteopenic, ovariectomized rats. Endocrinology 2011, 152, 3312–3322. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ominsky, M.S.; Vlasseros, F.; Jolette, J.; Smith, S.Y.; Stouch, B.; Doellgast, G.; Gong, J.; Gao, Y.; Cao, J.; Graham, K.; et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J. Bone Miner. Res. 2010, 25, 948–959. [Google Scholar] [CrossRef]
- Ominsky, M.S.; Boyd, S.K.; Varela, A.; Jolette, J.; Felx, M.; Doyle, N.; Mellal, N.; Smith, S.Y.; Locher, K.; Buntich, S.; et al. Romosozumab improves bone mass and strength while maintaining bone quality in ovariectomized cynomolgus monkeys. J. Bone Miner. Res. 2017, 32, 788–801. [Google Scholar] [CrossRef]
- Boyce, R.W.; Niu, Q.-T.; Ominsky, M.S. Kinetic reconstruction reveals time-dependent effects of romosozumab on bone formation and osteoblast function in vertebral cancellous and cortical bone in cynomolgus monkeys. Bone 2017, 101, 77–87. [Google Scholar] [CrossRef]
- Matheny, J.B.; Torres, A.M.; Ominsky, M.S.; Hernandez, C.J. Romosozumab treatment converts trabecular rods into trabecular plates in male cynomolgus monkeys. Calcif. Tissue Int. 2017, 101, 82–91. [Google Scholar] [CrossRef]
- McColm, J.; Hu, L.; Womack, T.; Tang, C.C.; Chiang, A.Y. Single- and multiple-dose randomized studies of blosozumab, a monoclonal antibody against sclerostin, in healthy postmenopausal women. J. Bone Miner. Res. 2014, 29, 935–943. [Google Scholar] [CrossRef]
- Recker, R.R.; Benson, C.T.; Matsumoto, T.; Bolognese, M.A.; Robins, D.A.; Alam, J.; Chiang, A.Y.; Hu, L.; Krege, J.H.; Sowa, H.; et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J. Bone Miner. Res. 2015, 30, 216–224. [Google Scholar] [CrossRef]
- Recknor, C.P.; Recker, R.R.; Benson, C.T.; Robins, D.A.; Chiang, A.Y.; Alam, J.; Hu, L.; Matsumoto, T.; Sowa, H.; Sloan, J.H.; et al. The effect of discontinuing treatment with blosozumab: Follow-up results of a phase 2 randomized clinical trial in postmenopausal women with low bone mineral density. J. Bone Miner. Res. 2015, 30, 1717–1725. [Google Scholar] [CrossRef]
- Padhi, D.; Jang, G.; Stouch, B.; Fang, L.; Posvar, E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J. Bone Miner. Res. 2011, 26, 19–26. [Google Scholar] [CrossRef]
- Padhi, D.; Allison, M.; Kivitz, A.J.; Gutierrez, M.J.; Stouch, B.; Wang, C.; Jang, G. Multiple doses of sclerostin antibody romosozumab in healthy men and postmenopausal women with low bone mass: A randomized, double-blind, placebo-controlled study. J. Clin. Pharmacol. 2014, 54, 168–178. [Google Scholar] [CrossRef]
- McClung, M.R.; Grauer, A.; Boonen, S.; Bolognese, M.A.; Brown, J.P.; Diez-Perez, A.; Langdahl, B.L.; Reginster, J.Y.; Zanchetta, J.R.; Wasserman, S.M.; et al. Romosozumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2014, 370, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, H.; Crittenden, D.B.; Miyauchi, A.; Libanati, C.; Maddox, J.; Fan, M.; Chen, L.; Grauer, A. Romosozumab increases bone mineral density in postmenopausal Japanese women with osteoporosis: A phase 2 study. Bone 2017, 103, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab treatment in postmenopausal women with osteoporosis. N. Engl. J. Med. 2017, 375, 1532–1543. [Google Scholar] [CrossRef]
- McClung, M.R. Denosumab for the treatment of osteoporosis. Osteoporos. Sarcopenia 2017, 3, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N. Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Lewiecki, E.M.; Blicharski, T.; Goemaere, S.; Lippuner, K.; Meisner, P.D.; Miller, P.D.; Miyauchi, A.; Maddox, J.; Chen, L.; Horlait, S. A Phase III randomized placebo-controlled trial to evaluate efficacy and safety of romosozumab in men with osteoporosis. J. Clin. Endocrinol. Metab. 2018, 103, 3183–3193. [Google Scholar] [CrossRef] [PubMed]
- Golds, G.; Houdek, D.; Arnason, T. Male hypogonadism and osteoporosis: The effects, clinical consequences, and treatment of testosterone deficiency in bone health. Int. J. Endocrinol. 2017, 4602129. [Google Scholar] [CrossRef]
- Liu, X.X.; Jiang, L.; Liu, Q.; Zhang, J.; Niu, W.; Liu, J.; Zhang, Q. Low bone turnover markers in young and middle-aged male patients with type 2 diabetes mellitus. J. Diabetes Res. 2020, 6191468. [Google Scholar] [CrossRef]
- Salcuni, A.S.; Carnevale, V.; Battista, C.; Palmieri, S.; Eller-Vainicher, C.; Guarnieri, V.; Pugliese, F.; Guglielmi, G.; Desina, G.; Minisola, S.; et al. Primary aldosteronism as a cause of secondary osteoporosis. Eur. J. Endocrinol. 2017, 177, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: A randomised, open-label, phase 3 trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- Nishiyama, K.K.; Cohen, A.; Young, P.; Wang, J.; Lappe, J.M.; Guo, X.E.; Dempster, D.W.; Recker, R.R.; Shane, E. Teriparatide increases strength of the peripheral skeleton in premenopausal women with idiopathic osteoporosis: A pilot HR-pQCT study. J. Clin. Endocrinol. Metab. 2014, 99, 2418–2425. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Ferrari, S.; Lewiecki, E.M.; Jaller-Raad, J.; Zerbini, C.; Milmont, C.E.; Meisner, P.D.; Libanati, C.; Grauer, A. Romosozumab FRAME study: A post hoc analysis of the role of regional background fracture risk on nonvertebral fracture outcome. J. Bone Miner. Res. 2018, 33, 1407–1416. [Google Scholar] [CrossRef]
- Miyauchi, A.; Dinavahi, R.V.; Crittenden, D.B.; Yang, W.; Maddox, J.C.; Hamaya, E.; Nakamura, Y.; Libanati, C.; Grauer, A.; Shimauchi, J. Increased bone mineral density for 1 year of romosozumab, vs placebo, followed by 2 years of denosumab in the Japanese subgroup of the pivotal FRAME trial and extension. Arch. Osteoporos. 2019, 14, 59. [Google Scholar] [CrossRef]
- Lau, E.M.C.; Dinavahi, R.; Woo, Y.C.; Wu, C.H.; Guan, J.; Maddox, J.; Tolman, C.; Yang, W.; Shin, C.S. Romosozumab or alendronate for fracture prevention in East Asian patients: A subanalysis of the phase III, randomized ARCH study. Osteoporos. Int. 2020, 31, 677–685. [Google Scholar] [CrossRef]
- Geusens, P.; Oates, M.; Miyauchi, A.; Adachi, J.D.; Lazaretti-Castro, M.; Ebeling, P.R.; Perez Niño, C.A.; Milmont, C.E.; Grauer, A.; Libanati, C. The effect of 1 year of romosozumab on the incidence of clinical vertebral fractures in postmenopausal women with osteoporosis: Results from the FRAME study. JBMR Plus 2019, 3, e10211. [Google Scholar] [CrossRef] [PubMed]
- Bone, H.G.; Wagman, R.B.; Brandi, M.L.; Brown, J.P.; Chapurlat, R.; Cummings, S.R.; Czerwiński, E.; Fahrleitner-Pammer, A.; Kendler, D.L.; Lippuner, K.; et al. 10 years of denosumab treatment in postmenopausal women with osteoporosis: Results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol. 2017, 5, 513–523. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Ferrari, S.; Khan, A.; Lane, N.E.; Lippuner, K.; Matsumoto, T.; Milmont, C.E.; Libanati, C.; Grauer, A. FRAME Study: The foundation effect of building bone with 1 year of romosozumab leads to continued lower fracture risk after transition to denosumab. J. Bone Miner. Res. 2018, 33, 1219–1226. [Google Scholar] [CrossRef]
- Cosman, F.; Lewiecki, E.M.; Ebeling, P.R.; Hesse, E.; Napoli, N.; Matsumoto, T.; Crittenden, D.B.; Rojeski, M.; Yang, W.; Libanati, C.; et al. T-Score as an indicator of fracture risk during treatment with romosozumab or alendronate in the ARCH trial. J. Bone Miner. Res. 2020, 35, 1333–1342. [Google Scholar] [CrossRef]
- Graeff, C.; Campbell, G.M.; Peña, J.; Borggrefe, J.; Padhi, D.; Kaufman, A.; Chang, S.; Libanati, C.; Glüer, C.C. Administration of romosozumab improves vertebral trabecular and cortical bone as assessed with quantitative computed tomography and finite element analysis. Bone 2015, 81, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Keaveny, T.M.; Crittenden, D.B.; Bolognese, M.A.; Genant, H.K.; Engelke, K.; Oliveri, B.; Brown, J.P.; Langdahl, B.L.; Yan, C.; Grauer, A.; et al. Greater gains in spine and hip strength for romosozumab compared with teriparatide in postmenopausal women with low bone mass. J. Bone Miner. Res. 2017, 32, 1956–1962. [Google Scholar] [CrossRef]
- Genant, H.K.; Engelke, K.; Bolognese, M.A.; Mautalen, C.; Brown, J.P.; Recknor, C.; Goemaere, S.; Fuerst, T.; Yang, Y.C.; Grauer, A.; et al. Effects of romosozumab compared with teriparatide on bone density and mass at the spine and hip in postmenopausal women with low bone mass. J. Bone Miner. Res. 2017, 32, 181–187. [Google Scholar] [CrossRef]
- Chavassieux, P.; Chapurlat, R.; Portero-Muzy, N.; Roux, J.P.; Garcia, P.; Brown, J.P.; Libanati, C.; Boyce, R.W.; Wang, A.; Grauer, A. Bone-forming and antiresorptive effects of romosozumab in postmenopausal women with osteoporosis: Bone histomorphometry and microcomputed tomography analysis after 2 and 12 months of treatment. J. Bone Miner. Res. 2019, 34, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Takada, J.; Dinavahi, R.; Miyauchi, A.; Hamaya, E.; Hirama, T.; Libanati, C.; Nakamura, Y.; Milmont, C.E.; Grauer, A. Relationship between P1NP, a biochemical marker of bone turnover, and bone mineral density in patients transitioned from alendronate to romosozumab or teriparatide: A post hoc analysis of the STRUCTURE trial. J. Bone Miner. Metab. 2020, 38, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Lewiecki, E.M.; Dinavahi, R.V.; Lazaretti-Castro, M.; Ebeling, P.R.; Adachi, J.D.; Miyauchi, A.; Gielen, E.; Milmont, C.E.; Libanati, C.; Grauer, A. One year of romosozumab followed by two years of denosumab maintains fracture risk reductions: Results of the FRAME extension study. J. Bone Miner. Res. 2019, 34, 419–428. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.R.; Brown, J.P.; Diez-Perez, A.; Resch, H.; Caminis, J.; Meisner, P.; Bolognese, M.A.; Goemaere, S.; Bone, H.G.; Zanchetta, J.R.; et al. Effects of 24 months of treatment with romosozumab followed by 12 months of denosumab or placebo in postmenopausal women with low bone mineral density: A randomized, double-blind, Phase 2, parallel group study. J. Bone Miner. Res. 2018, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Kendler, D.L.; Bone, H.G.; Massari, F.; Gielen, E.; Palacios, S.; Maddox, J.; Yan, C.; Yue, S.; Dinavahi, R.V.; Libanati, C.; et al. Bone mineral density gains with a second 12-month course of romosozumab therapy following placebo or denosumab. Osteoporos. Int. 2019, 30, 2437–2448. [Google Scholar] [CrossRef]
- McClung, M.R.; Bolognese, M.A.; Brown, J.P.; Reginster, J.Y.; Langdahl, B.L.; Maddox, J.; Shi, Y.; Rojeski, M.; Meisner, P.D.; Grauer, A. A single dose of zoledronate preserves bone mineral density for up to 2 years after a second course of romosozumab. Osteoporos. Int. 2020, 31, 2231–2241. [Google Scholar] [CrossRef]
- Migliorini, F.; Colarossi, G.; Baroncini, A.; Eschweiler, J.; Tingart, M.; Maffulli, N. Pharmacological management of postmenopausal osteoporosis: A level I evidence based expert opinion. Exp. Rev. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Wen, F.; Du, H.; Ding, L.; Hu, J.; Huang, Z.; Huang, H.; Li, K.; Mo, Y.; Kuang, A. Clinical efficacy and safety of drug interventions for primary and secondary prevention of osteoporotic fractures in postmenopausal women: Network meta-analysis followed by factor and cluster analysis. PLoS ONE 2020, 15, e0234123. [Google Scholar] [CrossRef]
- Ding, L.L.; Wen, F.; Wang, H.; Wang, D.H.; Liu, Q.; Mo, Y.X.; Tan, X.; Qiu, M.; Hu, J.X. Osteoporosis drugs for prevention of clinical fracture in white postmenopausal women: A network meta-analysis of survival data. Osteoporos. Int. 2020, 31, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.V.; Pérez-López, F.R.; Piscoya, A.; Pasupuleti, V.; Roman, Y.M.; Thota, P.; Herrera, A. Comparative efficacy of bone anabolic therapies in women with postmenopausal osteoporosis: A systematic review and network meta-analysis of randomized controlled trials. Maturitas 2019, 129, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Möckel, L.; Bartneck, M.; Möckel, C. Risk of falls in postmenopausal women treated with romosozumab: Preliminary indices from a meta-analysis of randomized, controlled trials. Osteoporos Sarcopenia 2020, 6, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Kaveh, S.; Hosseinifard, H.; Ghadimi, N.; Vojdanian, M.; Aryankhesal, A. Efficacy and safety of Romosozumab in treatment for low bone mineral density: A systematic review and meta-analysis. Clin. Rheumatol. 2020, 39, 3261–3276. [Google Scholar] [CrossRef]
- Lv, F.; Cai, X.; Yang, W.; Gao, L.; Chen, L.; Wu, J.; Ji, L. Denosumab or romosozumab therapy and risk of cardiovascular events in patients with primary osteoporosis: Systematic review and meta- analysis. Bone 2020, 130, 115121. [Google Scholar] [CrossRef]
- Cummings, S.R.; McCulloch, C. Explanations for the difference in rates of cardiovascular events in a trial of alendronate and romosozumab. Osteoporos. Int. 2020, 31, 1019–1021. [Google Scholar] [CrossRef]
- Hadaya, D.; Gkouveris, I.; Soundia, A.; Bezouglaia, O.; Boyce, R.W.; Stolina, M.; Dwyer, D.; Dry, S.M.; Pirih, F.Q.; Aghaloo, T.L.; et al. Clinically relevant doses of sclerostin antibody do not induce osteonecrosis of the jaw (ONJ) in rats with experimental periodontitis. J. Bone Miner. Res. 2019, 34, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H. Serious adverse events with romosozumab use in Japanese patients: Need for clear formulation of contraindications worldwide. J. Bone Miner. Res. 2020, 35, 994–995. [Google Scholar] [CrossRef] [PubMed]
- Mariscal, G.; Nuñez, J.H.; Bhatia, S.; Barrios, C.; Domenech-Fernández, P. Safety of romosozumab in osteoporotic men and postmenopausal women: A meta-analysis and systematic review. Monoclon. Antib. Immunodiagn. Immunother. 2020, 39, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Cardinal, M.; Tys, J.; Roels, T.; Lafont, S.; Ominsky, M.S.; Devogelaer, J.P.; Chappard, D.; Mabilleau, G.; Ammann, P.; Nyssen-Behets, C.; et al. Sclerostin antibody reduces long bone fractures in the oim/oim model of osteogenesis imperfecta. Bone 2019, 124, 137–147. [Google Scholar] [CrossRef]
- Sinder, B.P.; Salemi, J.D.; Ominsky, M.S.; Caird, M.S.; Marini, J.C.; Kozloff, K.M. Rapidly growing Brtl/+ mouse model of osteogenesis imperfecta improves bone mass and strength with sclerostin antibody treatment. Bone 2015, 71, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Sinder, B.P.; White, L.E.; Salemi, J.D.; Ominsky, M.S.; Caird, M.S.; Marini, J.C.; Kozloff, K.M. Adult Brtl/+ mouse model of osteogenesis imperfecta demonstrates anabolic response to sclerostin antibody treatment with increased bone mass and strength. Osteoporos. Int. 2014, 25, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Cardinal, M.; Dessain, A.; Roels, T.; Lafont, S.; Ominsky, M.S.; Devogelaer, J.P.; Chappard, D.; Mabilleau, G.; Ammann, P.; Nyssen-Behets, C.; et al. Sclerostin-antibody treatment decreases fracture rates in axial skeleton and improves the skeletal phenotype in growing oim/oim mice. Calcif. Tissue Int. 2020, 106, 494–508. [Google Scholar] [CrossRef]
- Roschger, A.; Roschger, P.; Keplingter, P.; Klaushofer, K.; Abdullah, S.; Kneissel, M.; Rauch, F. Effect of sclerostin antibody treatment in a mouse model of severe osteogenesis imperfecta. Bone 2014, 66, 182–188. [Google Scholar] [CrossRef]
- Glorieux, F.H.; Devogelaer, J.P.; Durigova, M.M.; Goemaere, S.; Hemsley, S.; Jakob, F.; Junker, U.; Ruckle, J.; Seefried, L.; Winkle, P.J. BPS804 anti-sclerostin antibody in adults with moderate osteogenesis imperfecta: Results of a randomized Phase 2a trial. J. Bone Miner. Res. 2017, 32, 1496–1504. [Google Scholar] [CrossRef]
- Perosky, J.E.; Khoury, B.M.; Jenks, T.N.; Ward, F.S.; Cortright, K.; Meyer, B.; Barton, D.K.; Sinder, B.P.; Marini, J.C.; Caird, M.S.; et al. Single dose of bisphosphonate preserves gains in bone mass following cessation of sclerostin antibody in Brtl/+ osteogenesis imperfecta model. Bone 2016, 93, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Olvera, D.; Stolzenfeld, R.; Marini, J.C.; Caird, M.S.; Kozloff, K.M. Low dose of bisphosphonate enhances sclerostin antibody-induced trabecular bone mass gains in Brtl/+ osteogenesis imperfecta mouse model. J. Bone Miner. Res. 2018, 33, 1272–1282. [Google Scholar] [CrossRef]
- Little, D.G.; Peacock, L.; Mikulec, K.; Kneissel, M.; Kramer, I.; Cheng, T.L.; Schindeler, A.; Munns, C. Combination sclerostin antibody and zoledronic acid treatment outperforms either treatment alone in a mouse model of osteogenesis imperfecta. Bone 2017, 101, 96–103. [Google Scholar] [CrossRef]
- Carpenter, K.A.; Ross, R.D. Sclerostin antibody treatment increases bone mass and normalizes circulating phosphate levels in growing Hyp mice. J. Bone Miner. Res. 2020, 35, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Han, X.; Jing, Y.; Yuan, B.; Ke, H.; Liu, M.; Feng, J.Q. Sclerostin antibody (Scl-Ab) improves osteomalacia phenotype in dentin matrix protein 1(Dmp1) knockout mice with little impact on serum levels of phosphorus and FGF23. Matrix Biol. 2016, 52–54, 151–161. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.M.; Reagan, M.R.; Youlten, S.E.; Mohanty, S.T.; Seckinger, A.; Terry, R.L.; Pettitt, J.A.; Simic, M.K.; Cheng, T.L.; Morse, A.; et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 2017, 129, 3452–3464. [Google Scholar] [CrossRef]
- Hesse, E.; Schröder, S.; Brandt, D.; Pamperin, J.; Saito, H.; Taipaleenmäki, H. Sclerostin inhibition alleviates breast cancer-induced bone metastases and muscle weakness. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Frost, C.; Hansen, R.R.; Heegaard, A.M. Bone pain: Current and future treatments. Curr. Opin. Pharmacol. 2016, 28, 31–37. [Google Scholar] [CrossRef]
- Harris, S.E.; Rediske, M.; Neitzke, R.; Rakian, A. Periodontal biology: Stem cells, Bmp2 gene, transcriptional enhancers, and use of sclerostin antibody and PTH for treatment of periodontal disease and bone loss. Cell Stem Cells Regen. Med. 2017, 3. [Google Scholar] [CrossRef]
- Hassan, M.G.; Zaher, A.R.; Palomo, J.M.; Palomo, L. Sclerostin modulation holds promise for dental indications. Healthcare 2018, 6, 134. [Google Scholar] [CrossRef]
- Taut, A.D.; Jin, Q.; Chung, J.H.; Galindo-Moreno, P.; Yi, E.S.; Sugai, J.V.; Ke, H.Z.; Liu, M.; Giannobile, W.V. Sclerostin antibody stimulates bone regeneration after experimental periodontitis. J. Bone Miner. Res. 2013, 28, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Seefried, L.; Baumann, J.; Hemsley, S.; Hofmann, C.; Kunstmann, E.; Kiese, B.; Huang, Y.; Chivers, S.; Valentin, M.A.; Borah, B.; et al. Efficacy of anti-sclerostin monoclonal antibody BPS804 in adult patients with hypophosphatasia. J. Clin. Investig. 2017, 127, 2148–2158. [Google Scholar] [CrossRef] [PubMed]
- Kedlaya, R.; Veera, S.; Horan, D.J.; Moss, R.E.; Ayturk, U.M.; Jacobsen, C.M.; Bowen, M.E.; Paszty, C.; Warman, M.L.; Robling, A.G. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci. Transl. Med. 2013, 5, 211ra158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moe, S.M.; Chen, N.X.; Newman, C.L.; Organ, J.M.; Kneissel, M.; Kramer, I.; Gattone, V.H., 2nd; Allen., M.R. Anti-sclerostin antibody treatment in a rat model of progressive renal osteodystrophy. J. Bone Miner. Res. 2015, 30, 499–509. [Google Scholar] [CrossRef]
- Newman, C.L.; Chen, N.X.; Smith, E.; Smith, M.; Brown, D.; Moe, S.M.; Allen, M.R. Compromised vertebral structural and mechanical properties associated with progressive kidney disease and the effects of traditional pharmacological interventions. Bone 2015, 77, 50–56. [Google Scholar] [CrossRef][Green Version]
- Marenzana, M.; Greenslade, K.; Eddleston, A.; Okoye, R.; Marshall, D.; Moore, A.; Robinson, M.K. Sclerostin antibody treatment enhances bone strength but does not prevent growth retardation in young mice treated with dexamethasone. Arthritis Rheum. 2011, 63, 2385–2395. [Google Scholar] [CrossRef] [PubMed]
- Marenzana, M.; Vugler, A.; Moore, A.; Robinson, M. Effect of sclerostin-neutralising antibody on periarticular and systemic bone in a murine model of rheumatoid arthritis: A microCT study. Arthritis Res. Ther. 2013, 15, R125. [Google Scholar] [CrossRef]
- Wehmeyer, C.; Frank, S.; Beckmann, D.; Böttcher, M.; Cromme, C.; König, U.; Fennen, M.; Held, A.; Paruzel, P.; Hartmann, C.; et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci. Transl. Med. 2016, 8, 330ra35. [Google Scholar] [CrossRef]
- Spatz, J.M.; Ellman, R.; Cloutier, A.M.; Louis, L.; van Vliet, M.; Dwyer, D.; Stolina, M.; Ke, H.Z.; Bouxsein, M.L. Sclerostin antibody inhibits skeletal deterioration in mice exposed to partial weight-bearing. Life Sci. Space Res. 2017, 12, 32–38. [Google Scholar] [CrossRef]
- Phillips, E.G.; Beggs, L.A.; Ye, F.; Conover, C.F.; Beck, D.T.; Otzel, D.M.; Ghosh, P.; Bassit, A.C.F.; Borst, S.E.; Yarrow, J.F. Effects of pharmacologic sclerostin inhibition or testosterone administration on soleus muscle atrophy in rodents after spinal cord injury. PLoS ONE 2018, 13, e0194440. [Google Scholar] [CrossRef]
- Shin, Y.K.; Yoon, Y.K.; Chung, K.B.; Rhee, Y.; Cho, S.R. Patients with non-ambulatory cerebral palsy have higher sclerostin levels and lower bone mineral density than patients with ambulatory cerebral palsy. Bone 2017, 103, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Schemitsch, E.H.; Miclau, T.; Karachalios, T.; Nowak, L.L.; Sancheti, P.; Poolman, R.W.; Caminis, J.; Daizadeh, N.; Dent-Acosta, R.E.; Egbuna, O.; et al. A randomized, placebo-controlled study of romosozumab for the treatment of hip fractures. J. Bone Jt. Surg. Am. 2020, 15, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, M.; Schemitsch, E.H.; Karachalios, T.; Sancheti, P.; Poolman, R.W.; Caminis, J.; Daizadeh, N.; Dent-Acosta, R.E.; Egbuna, O.; Chines, A.; et al. Romosozumab in skeletally mature adults with a fresh unilateral tibial diaphyseal fracture: A randomized phase-2 study. J. Bone Jt. Surg. Am. 2020, 102, 1416–1426. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rauner, M.; Taipaleenmäki, H.; Tsourdi, E.; Winter, E.M. Osteoporosis Treatment with Anti-Sclerostin Antibodies—Mechanisms of Action and Clinical Application. J. Clin. Med. 2021, 10, 787. https://doi.org/10.3390/jcm10040787
Rauner M, Taipaleenmäki H, Tsourdi E, Winter EM. Osteoporosis Treatment with Anti-Sclerostin Antibodies—Mechanisms of Action and Clinical Application. Journal of Clinical Medicine. 2021; 10(4):787. https://doi.org/10.3390/jcm10040787
Chicago/Turabian StyleRauner, Martina, Hanna Taipaleenmäki, Elena Tsourdi, and Elizabeth M. Winter. 2021. "Osteoporosis Treatment with Anti-Sclerostin Antibodies—Mechanisms of Action and Clinical Application" Journal of Clinical Medicine 10, no. 4: 787. https://doi.org/10.3390/jcm10040787
APA StyleRauner, M., Taipaleenmäki, H., Tsourdi, E., & Winter, E. M. (2021). Osteoporosis Treatment with Anti-Sclerostin Antibodies—Mechanisms of Action and Clinical Application. Journal of Clinical Medicine, 10(4), 787. https://doi.org/10.3390/jcm10040787