
Improving Familial Hypercholesterolemia Index Case Detection: Sequential Active Screening from Centralized Analytical Data

 , ,
, ,
Abstract
1. Introduction
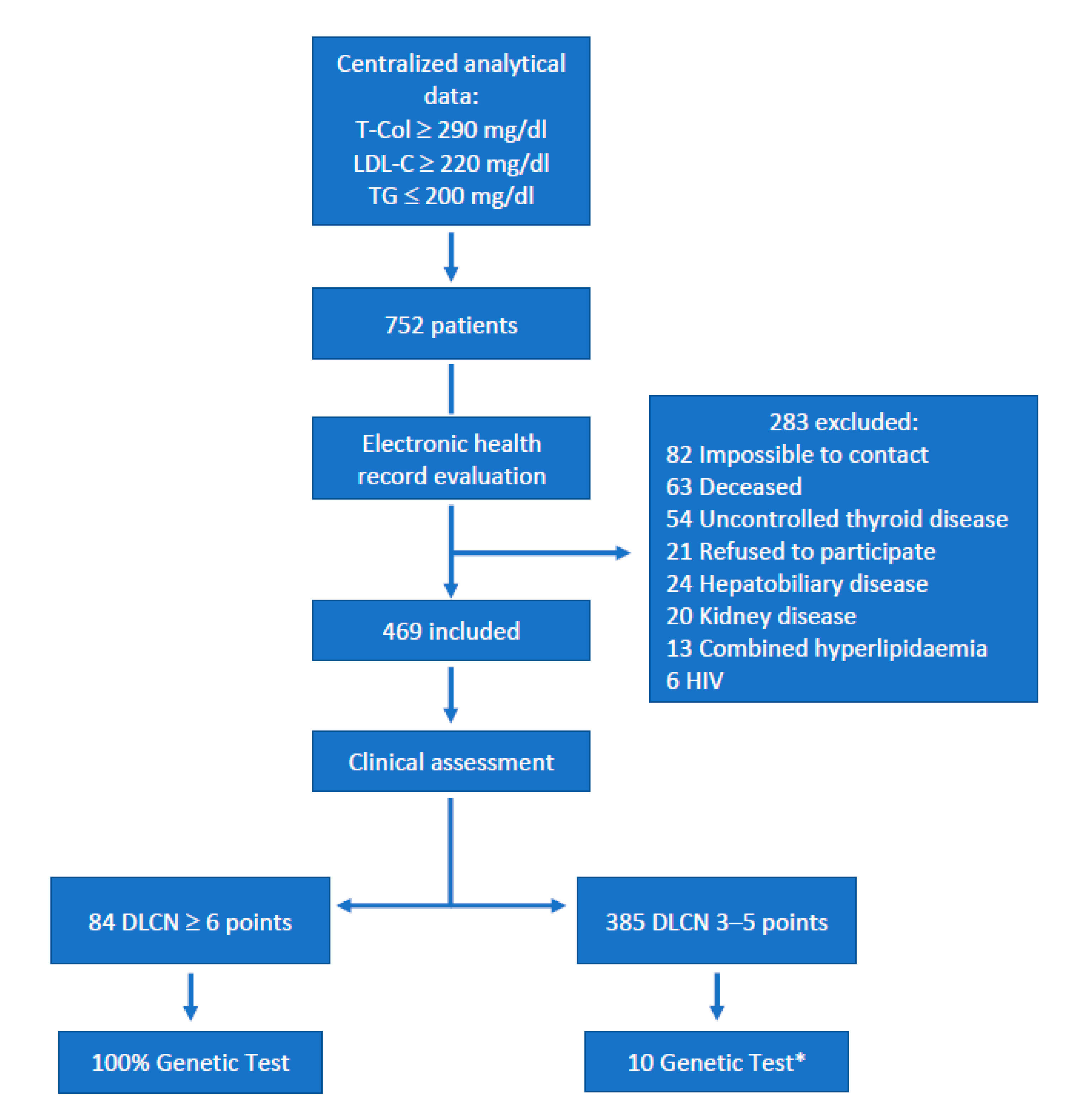
2. Materials and Methods
2.1. Population Study and Clinical Diagnostic Criteria for Familial Hypercholesterolemia
2.2. Genetic Testing
2.3. Statistical Analysis
3. Results
3.1. Clinical Characteristics of the Studied Population
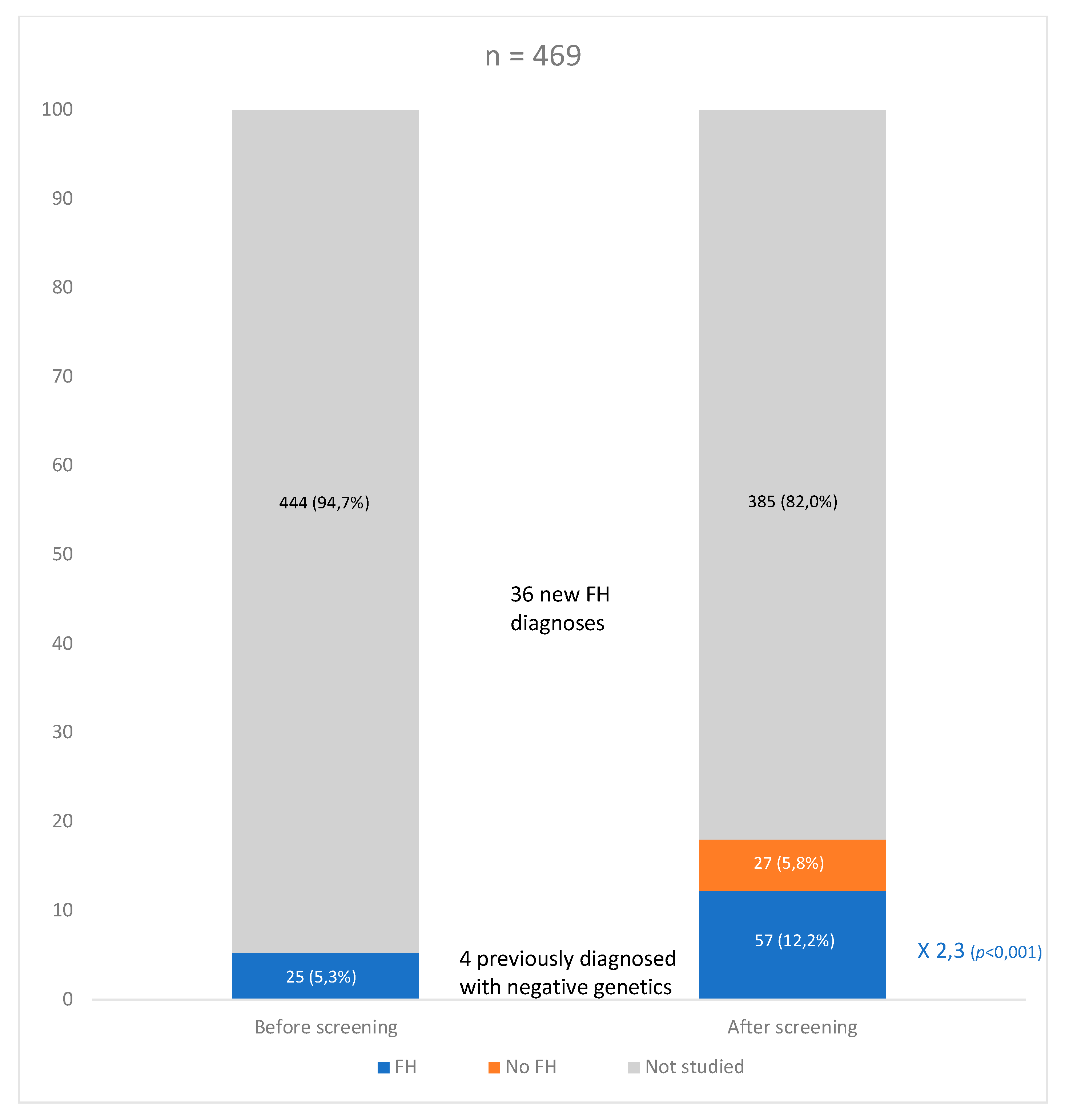
3.2. Genetic FH Diagnosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watts, G.F.; Gidding, S.S.; Mata, P.; Pang, J.; Sullivan, D.R.; Yamashita, S.; Raal, F.J.; Santos, R.D.; Ray, K.K. Familial hypercholesterolaemia: Evolving knowledge for designing adaptive models of care. Nat. Rev. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Benn, M.; Watts, G.F.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Mutations causative of familial hypercholesterolaemia: Screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur. Heart J. 2016, 37, 1384–1394. [Google Scholar] [CrossRef]
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef]
- Hu, P.; Dharmayat, K.I.; Stevens, C.A.T.; Sharabiani, M.T.A.; Jones, R.S.; Watts, G.F.; Genest, J.; Ray, K.K.; Vallejo-Vaz, A.J. Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease. Circulation 2020, 141, 1742–1759. [Google Scholar] [CrossRef]
- Akioyamen, L.E.; Genest, J.; Shan, S.D.; Reel, R.L.; Albaum, J.M.; Chu, A.; Tu, J.V. Estimating the prevalence of heterozygous familial hypercholesterolaemia: A systematic review and meta-analysis. BMJ Open 2017, 7, e016461. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2019, 41, 111–118. [Google Scholar] [CrossRef]
- Alonso, R.; Perez de Isla, L.; Muñiz-Grijalvo, O.; Mata, P. Barriers to Early Diagnosis and Treatment of Familial Hypercholesterolemia: Current Perspectives on Improving Patient Care. Vasc. Health Risk Manag. 2020, 16, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, D.; Larsen, M.L.; Nissen, P.H.; Jensen, J.M.; Jensen, H.K.; Soerensen, V.R.; Jensen, L.G.; Faergeman, O. The relationship of molecular genetic to clinical diagnosis of familial hypercholesterolemia in a Danish population. Atherosclerosis 2005, 180, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Civeira, F.; Ros, E.; Jarauta, E.; Plana, N.; Zambon, D.; Puzo, J.; Martinez de Esteban, J.P.; Ferrando, J.; Zabala, S.; Almagro, F.; et al. Comparison of Genetic Versus Clinical Diagnosis in Familial Hypercholesterolemia. Am. J. Cardiol. 2008, 102, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef]
- Nanchen, D.; Gencer, B.; Muller, O.; Auer, R.; Aghlmandi, S.; Heg, D.; Klingenberg, R.; Räber, L.; Carballo, D.; Carballo, S.; et al. Prognosis of Patients with Familial Hypercholesterolemia after Acute Coronary Syndromes. Circulation 2016, 134, 698–709. [Google Scholar] [CrossRef]
- Pérez De Isla, L.; Alonso, R.; Mata, N.; Fernández-Pérez, C.; Muñiz, O.; Díaz-Díaz, J.L.; Saltijeral, A.; Fuentes-Jiménez, F.; De Andrés, R.; Zambón, D.; et al. Predicting cardiovascular events in familial hypercholesterolemia: The SAFEHEART registry (Spanish Familial Hypercholesterolemia Cohort Study). Circulation 2017, 135, 2133–2144. [Google Scholar] [CrossRef]
- De Isla, L.P.; Alonso, R.; Mata, N.; Saltijeral, A.; Muñiz, O.; Rubio-Marin, P.; Diaz-Diaz, J.L.; Fuentes, F.; De Andrs, R.; Zambn, D.; et al. Coronary heart disease, peripheral arterial disease, and stroke in familial hypercholesterolaemia: Insights from the SAFEHEART registry (Spanish familial hypercholesterolaemia cohort study). Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Perak, A.M.; Ning, H.; de Ferranti, S.D.; Gooding, H.C.; Wilkins, J.T.; Lloyd-Jones, D.M. Long-Term Risk of Atherosclerotic Cardiovascular Disease in US Adults With the Familial Hypercholesterolemia Phenotype. Circulation 2016, 134, 9–19. [Google Scholar] [CrossRef]
- Khera, A.V.; Won, H.H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients with Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef]
- Versmissen, J.; Oosterveer, D.M.; Yazdanpanah, M.; Defesche, J.C.; Basart, D.C.G.; Liem, A.H.; Heeringa, J.; Witteman, J.C.; Lansberg, P.J.; Kastelein, J.J.P.; et al. Efficacy of statins in familial hypercholesterolaemia: A long term cohort study. BMJ 2008, 337, a2423. [Google Scholar] [CrossRef] [PubMed]
- Besseling, J.; Hovingh, G.K.; Huijgen, R.; Kastelein, J.J.P.; Hutten, B.A. Statins in Familial Hypercholesterolemia: Consequences for Coronary Artery Disease and All-Cause Mortality. J. Am. Coll. Cardiol. 2016, 68, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.P.; Ginsberg, H.N.; Langslet, G.; Hovingh, G.K.; Ceska, R.; Dufour, R.; Blom, D.; Civeira, F.; Krempf, M.; Lorenzato, C.; et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur. Heart J. 2015, 36, 2996–3003. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Stein, E.A.; Dufour, R.; Turner, T.; Civeira, F.; Burgess, L.; Langslet, G.; Scott, R.; Olsson, A.G.; Sullivan, D.; et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 331–340. [Google Scholar] [CrossRef]
- Wilemon, K.A.; Patel, J.; Aguilar-Salinas, C.; Ahmed, C.D.; Alkhnifsawi, M.; Almahmeed, W.; Alonso, R.; Al-Rasadi, K.; Badimon, L.; Bernal, L.M.; et al. Reducing the Clinical and Public Health Burden of Familial Hypercholesterolemia: A Global Call to Action. JAMA Cardiol. 2020, 5, 217–229. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Masana, L.; Ibarretxe, D.; Plana, N. Maximum Low-density Lipoprotein Cholesterol Lowering Capacity Achievable With Drug Combinations. When 50 Plus 20 Equals 60. Rev. Española Cardiol. Engl. Ed. 2016, 69, 342–343. [Google Scholar] [CrossRef]
- Escobar, C.; Anguita, M.; Arrarte, V.; Barrios, V.; Cequier, Á.; Cosín-Sales, J.; Egocheaga, I.; López de Sa, E.; Masana, L.; Pallarés, V.; et al. Recommendations to improve lipid control. Consensus document of the Spanish Society of Cardiology. Rev. Esp. Cardiol. 2020, 73, 161–167. [Google Scholar] [CrossRef]
- Mata, P.; Alonso, R.; Ruiz, A.; Gonzalez-Juanatey, J.R.; Badimón, L.; Díaz-Díaz, J.L.; Muñoz, M.T.; Muñiz, O.; Galve, E.; Irigoyen, L.; et al. Diagnóstico y tratamiento de la hipercolesterolemia familiar en España: Documento de consenso. Aten Primaria 2015, 47, 56–65. [Google Scholar] [CrossRef]
- Amor-Salamanca, A.; Castillo, S.; Gonzalez-Vioque, E.; Dominguez, F.; Quintana, L.; Lluís-Ganella, C.; Escudier, J.M.; Ortega, J.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Genetically Confirmed Familial Hypercholesterolemia in Patients with Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2017, 70, 1732–1740. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; Mcgowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Loaiza, N.; Hartgers, M.L.; Reeskamp, L.F.; Balder, J.-W.; Rimbert, A.; Bazioti, V.; Wolters, J.C.; Winkelmeijer, M.; Jansen, H.P.G.; Dallinga-Thie, G.M.; et al. Taking One Step Back in Familial Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Ascaso, J.F.; Civeira, F.; Guijarro, C.; López Miranda, J.; Masana, L.; Mostaza, J.M.; Pedro-Botet, J.; Pintó, X.; Valdivielso, P. Indications of PCSK9 inhibitors in clinical practice. Recommendations of the Spanish Sociey of Arteriosclerosis (SEA), 2019. Clin. Investig. Arterioscler. 2019, 31, 128–139. [Google Scholar] [CrossRef]
- Graham, C.A.; McIlhatton, B.P.; Kirk, C.W.; Beattie, E.D.; Lyttle, K.; Hart, P.; Neely, R.D.G.; Young, I.S.; Nicholls, D.P. Genetic screening protocol for familial hypercholesterolemia which includes splicing defects gives an improved mutation detection rate. Atherosclerosis 2005, 182, 331–340. [Google Scholar] [CrossRef]
- Taylor, A.; Wang, D.; Patel, K.; Whittall, R.; Wood, G.; Farrer, M.; Neely, R.; Fairgrieve, S.; Nair, D.; Barbir, M.; et al. Mutation detection rate and spectrum in familial hypercholesterolaemia patients in the UK pilot cascade project. Clin. Genet. 2010, 77, 572–580. [Google Scholar] [CrossRef]
- Palacios, L.; Grandoso, L.; Cuevas, N.; Olano-Martín, E.; Martinez, A.; Tejedor, D.; Stef, M. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis 2012, 221, 137–142. [Google Scholar] [CrossRef]
- Wald, D.S.; Bestwick, J.P.; Wald, N.J. Child-parent screening for familial hypercholesterolaemia: Screening strategy based on a meta-analysis. BMJ 2007, 335, 599. [Google Scholar] [CrossRef] [PubMed]
- Kirke, A.B.; Barbour, R.A.; Burrows, S.; Bell, D.A.; Vickery, A.W.; Emery, J.; Watts, G.F. Systematic Detection of Familial Hypercholesterolaemia in Primary Health Care: A Community Based Prospective Study of Three Methods. Hearth Lung Circ. 2015, 24, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Reeskamp, L.F.; Tromp, T.R.; Defesche, J.C.; Grefhorst, A.; Stroes, E.S.G.; Hovingh, G.K.; Zuurbier, L. Next-generation sequencing to confirm clinical familial hypercholesterolemia. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Wang, J.; Dron, J.S.; Ban, M.R.; Robinson, J.F.; McIntyre, A.D.; Alazzam, M.; Zhao, P.J.; Dilliott, A.A.; Cao, H.; Huff, M.W.; et al. Polygenic Versus Monogenic Causes of Hypercholesterolemia Ascertained Clinically. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2439–2445. [Google Scholar] [CrossRef]
- Trinder, M.; Li, X.; DeCastro, M.L.; Cermakova, L.; Sadananda, S.; Jackson, L.M.; Azizi, H.; Mancini, G.B.J.; Francis, G.A.; Frohlich, J.; et al. Risk of Premature Atherosclerotic Disease in Patients With Monogenic Versus Polygenic Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2019, 74, 512–522. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Manickam, K.; Jones, L.K.; Wright, E.A.; Hartzel, D.N.; Gonzaga-Jauregui, C.; O’Dushlaine, C.; Leader, J.B.; Kirchner, H.L.; Lindbuchler, D.M.; et al. Genetic identification of familial hypercholesterolemia within a single U.S. Health care system. Science 2016, 354, aaf7000. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, P.; Perez de Isla, L.; Watts, G.F.; Alonso, R.; Norman, R.; Muniz, O.; Fuentes, F.; Mata, N.; Lopez-Miranda, J.; Gonzalez-Juanatey, J.R.; et al. Cost-effectiveness of a cascade screening program for the early detection of familial hypercholesterolemia. J. Clin. Lipidol. 2017, 11, 260–271. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
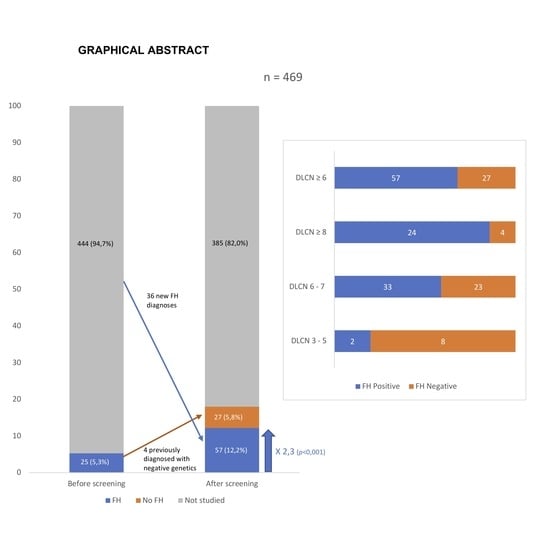
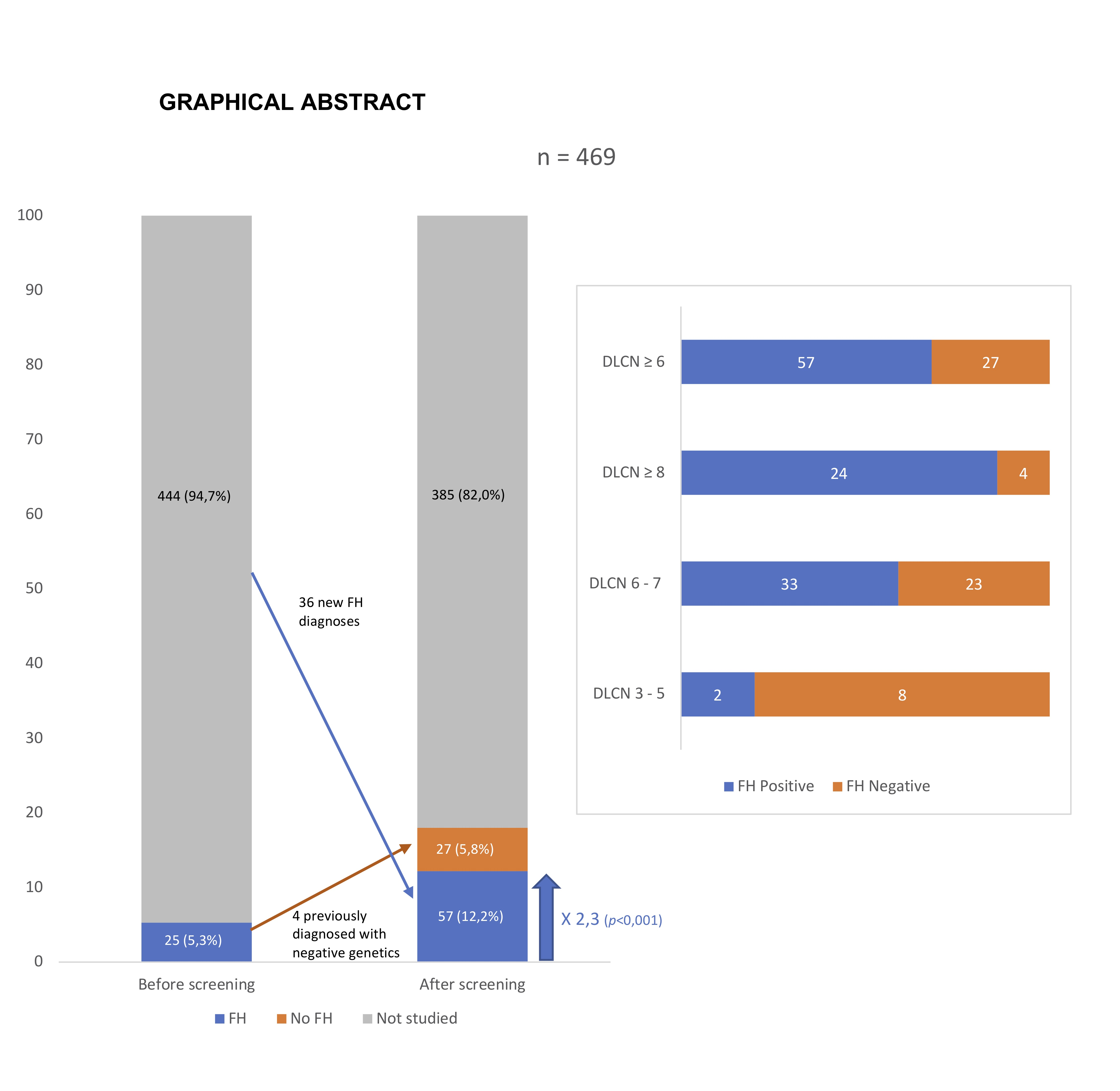
| Variable | Overall (n = 469) | DLCN 3–5 (n = 385) | DLCN ≥ 6 (n = 84) | Standardized Effect Size (95% CI) |
|---|---|---|---|---|
| Male | 191 (40.7) | 162 (42.1) | 29 (34.5) | 0,73 (0.44 to 1.19) |
| Mean age, years | 53.2 ± 12.8 | 54.6 ± 12.3 | 47.1 ± 12.9 | 0.59 (0.36 to 0.82) * |
| Hypertension | 148 (31.6) | 130 (33.8) | 18 (21.4) | 0.54 (0.31 to 0.94) * |
| Diabetes | 47 (10.0) | 43 (11.2) | 4 (4.8) | 0.39 (0.14 to 1.14) |
| Current or past smoking | 233 (49.7) | 181 (47.0) | 52 (61.9) | 1.83 (1.13 to 2.97) * |
| Body Mass Index | 27.7 ± 4.4 | 27.8 ± 4.3 | 27.0 ± 4.7 | 0.19 (−0.04 to 0.43) |
| ASCVD | 26 (5.5) | 13 (3.4) | 13 (15.5) | 5.24 (2.33 to 11.77) * |
| Premature ASCVD | 11 (2.3) | 3 (0.8) | 8 (9.5) | 13.4 (3.48 to 51.68) * |
| Family history of premature ASCVD | 34 (7.2) | 15 (3.9) | 19 (22.6) | 7.21 (3.49 to 14.91) * |
| Family history of HCL | 131 (27.9) | 61 (15.9) | 70 (83.3) | 26.48 (14.02 to 49.99) * |
| Corneal arcus (<45 years) | 7 (6.2) | 0 | 7 (20.6) | - |
| Tendon Xanthoma | 1 (0.2) | 0 | 1 (1.1) | - |
| Total cholesterol (mg/dL) | 331.7 ± 48.3 | 317.7 ± 21.03 | 396.2 ± 77.5 | −1.62 (−1.97 to −1.27) |
| LDL-C (mg/dL) | 246.8 ± 38.2 | 234.9 ± 15.6 | 301.5 ± 58.5 | −1.74 (−2.08 to −1.41) |
| HDL-C (mg/dL) | 56.2 ± 14.0 | 56.4 ± 13.6 | 55.2 ± 15.9 | 0.89 (−1.47 to 0.33) |
| Triglycerides (mg/dl) | 128.3 ± 36.3 | 129.5 ± 35.8 | 122.8 ± 38.4 | 0.18 (−0.05 to 0.42) |
| TSH (mLU/L) | 2.1 ± 2.12 | 2.1 ± 2.3 | 2.2 ± 1.30 | −0.08 (−0.32 to 0.17) |
| Lipid-lowering treatment | 344 (73.4) | 269 (69.9) | 75 (89.3) | 3.59 (1.74 to 7.42) * |
| Maximal lipid-lowering therapy | 109 (23.2) | 64 (16.6) | 45 (53.6) | 5.79 (3.49 to 9.59) * |
| DLCN ≥ 6 no FH Mutation (n = 27) | DLCN ≥ 6 FH Mutation (n = 57) | p-Value | |
|---|---|---|---|
| ASCVD | 2 (7.4) | 11 (19.3) | 0.33 |
| ASCVD without previous FH diagnosis | 2 (100) | 7 (63.6) | 0.47 |
| Premature ASCVD | 1 (50) | 7 (63,6) | 0.43 |
| Premature ASCVD without previous FH diagnosis | 1 (100) | 3 (42.9) | 1 |
| Coronary artery disease | 2 (100) | 7 (63.6) | 0.72 |
| Cerebral vascular disease | 0 | 4 (36.4) | - |
| Peripheral vascular disease | 1 (50) | 1 (9.1) | 0.51 |
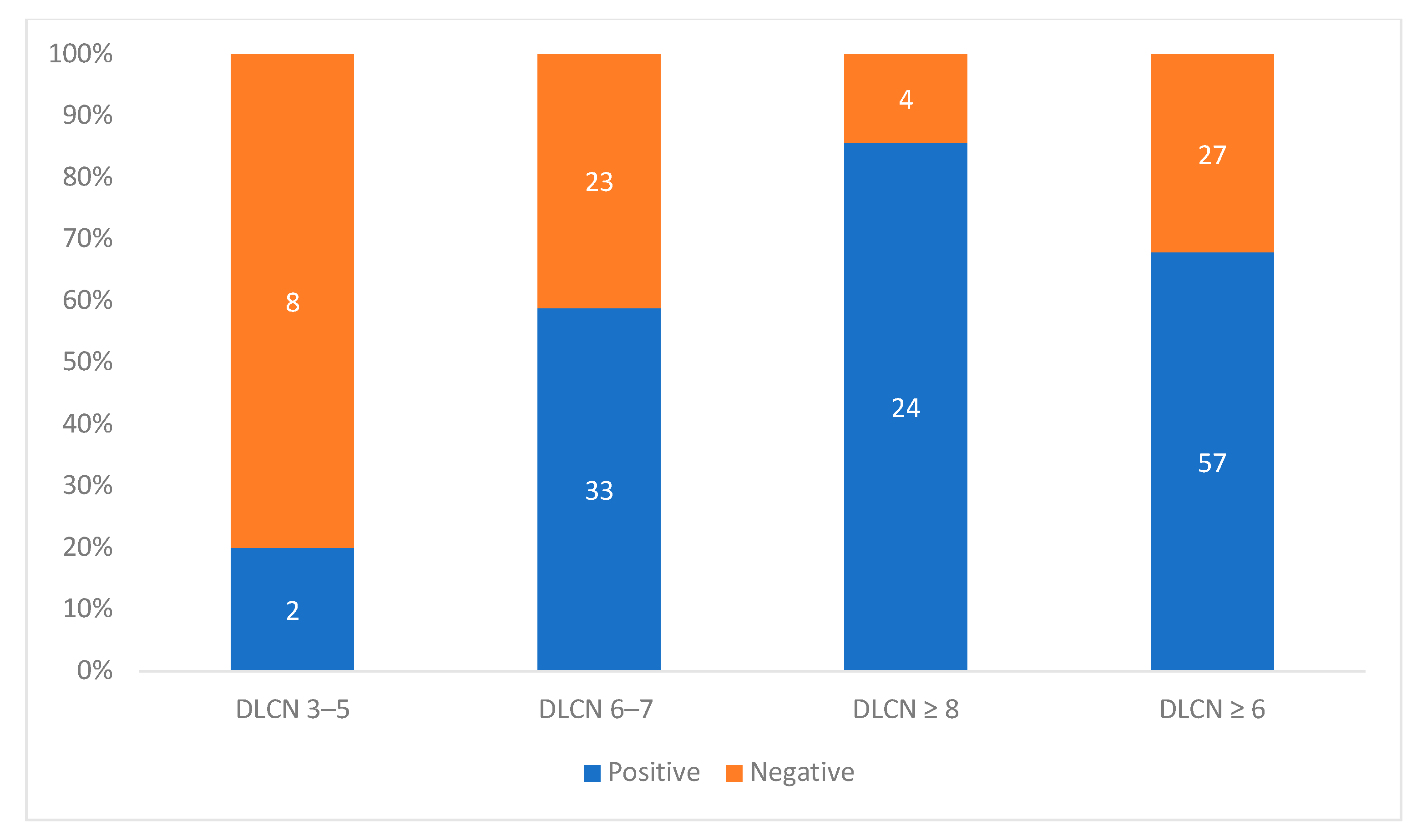
| All | FH Mutation | No FH Mutation | Odds Ratio (95% CI) | p-Value * | |
|---|---|---|---|---|---|
| Previous FH diagnosis | 26 | 21 (80.8) | 5 (19.2) | 2,94 (0.99–8.73) | 0.08 |
| Possible FH | 10 | 2 (20) | 8 (80) | 1 (Reference) | - |
| Probable FH | 56 | 33 (58.9) | 23 (41.1) | 5.74 (1.12–29.54) | 0.037 |
| Definite FH | 28 | 24 (85.7) | 4 (14.3) | 24.0 (3.68–156.7) | 0.001 |
| DLCN ≥ 6 | 84 | 57 (67.9) | 27 (32.1) | 8.44 (1.68–42.49) | 0.005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabatel-Pérez, F.; Sánchez-Prieto, J.; Becerra-Muñoz, V.M.; Alonso-Briales, J.H.; Mata, P.; Rodríguez-Padial, L. Improving Familial Hypercholesterolemia Index Case Detection: Sequential Active Screening from Centralized Analytical Data. J. Clin. Med. 2021, 10, 749. https://doi.org/10.3390/jcm10040749
Sabatel-Pérez F, Sánchez-Prieto J, Becerra-Muñoz VM, Alonso-Briales JH, Mata P, Rodríguez-Padial L. Improving Familial Hypercholesterolemia Index Case Detection: Sequential Active Screening from Centralized Analytical Data. Journal of Clinical Medicine. 2021; 10(4):749. https://doi.org/10.3390/jcm10040749
Chicago/Turabian StyleSabatel-Pérez, Fernando, Joaquín Sánchez-Prieto, Víctor Manuel Becerra-Muñoz, Juan Horacio Alonso-Briales, Pedro Mata, and Luis Rodríguez-Padial. 2021. "Improving Familial Hypercholesterolemia Index Case Detection: Sequential Active Screening from Centralized Analytical Data" Journal of Clinical Medicine 10, no. 4: 749. https://doi.org/10.3390/jcm10040749
APA StyleSabatel-Pérez, F., Sánchez-Prieto, J., Becerra-Muñoz, V. M., Alonso-Briales, J. H., Mata, P., & Rodríguez-Padial, L. (2021). Improving Familial Hypercholesterolemia Index Case Detection: Sequential Active Screening from Centralized Analytical Data. Journal of Clinical Medicine, 10(4), 749. https://doi.org/10.3390/jcm10040749