
Genome Editing for β-Hemoglobinopathies: Advances and Challenges


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Globin Gene Regulation
3. Basics of Genome Editing Technologies
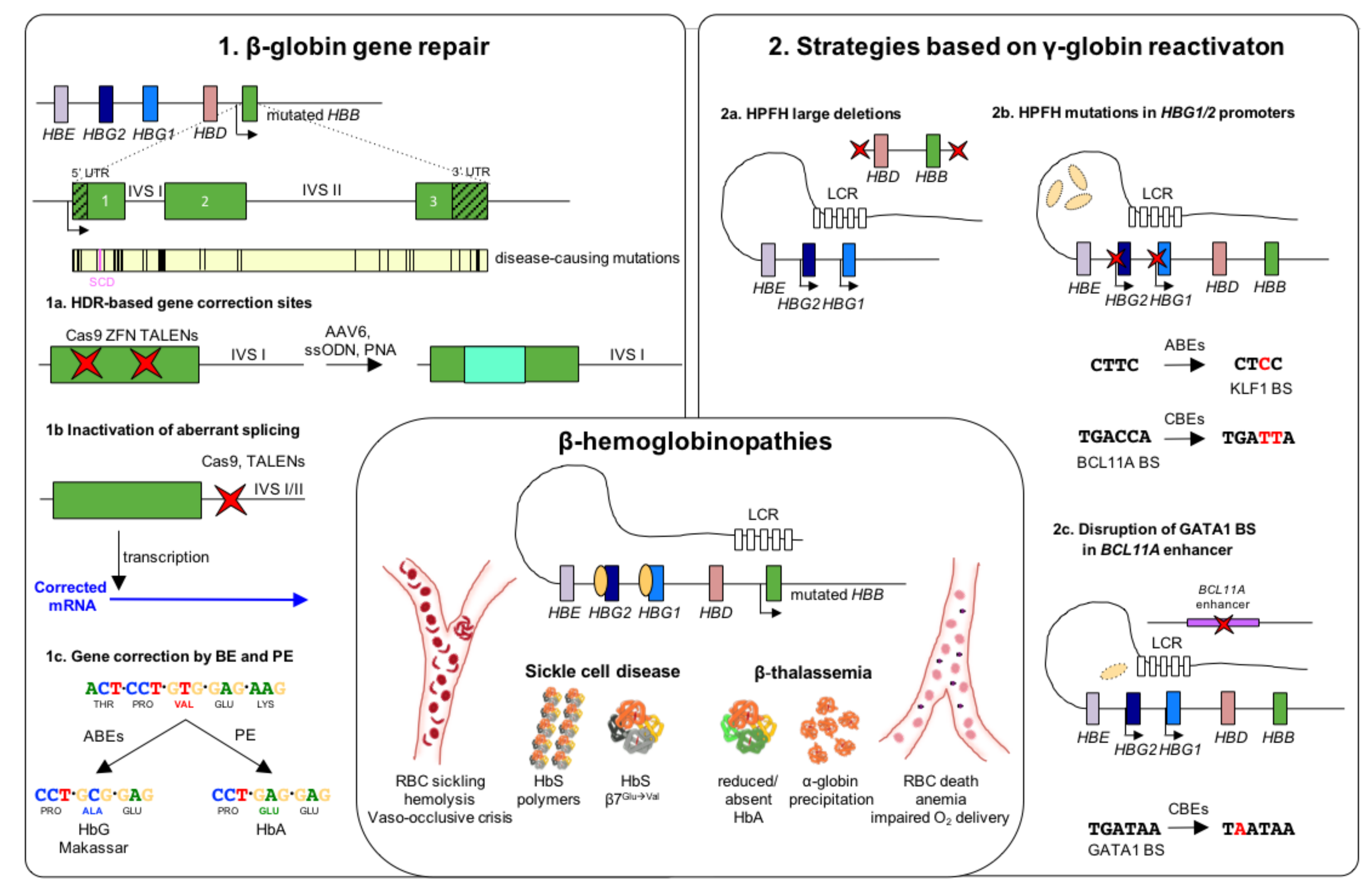
4. Nuclease-Mediated Strategies for β-Globin Gene Repair
4.1. HDR-Based Gene Correction Strategies
4.2. NHEJ-Based Gene Correction Strategies
5. Nuclease-Mediated Strategies for γ-Globin Reactivation
6. Base and Prime Editing Approaches for β-Hemoglobinopathies
7. Nuclease-Free Strategies for β-Hemoglobinopathies Based on Peptide Nucleic Acids
8. Off-Target Activity of Genome Editing Systems
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piel, F.B.; Patil, A.P.; Howes, E.R.; Nyangiri, A.O.; Gething, P.W.; Dewi, M.; Temperley, W.H.; Williams, T.N.; Weatherall, D.J.; Hay, I.S. Global epidemiology of sickle haemoglobin in neonates: A contemporary geostatistical model-based map and population estimates. Lancet 2013, 381, 142–151. [Google Scholar] [CrossRef]
- Angelucci, E.; Matthes-Martin, S.; Baronciani, D.; Bernaudin, F.; Bonanomi, S.; Cappellini, M.D.; Dalle, J.-H.; Di Bartolomeo, P.; De Heredia, C.D.; Dickerhoff, R.; et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: Indications and management recommendations from an international expert panel. Haematologica 2014, 99, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J.; Simões, B.P.; Ferster, A.; Dupont, S.; De La Fuente, J.; et al. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Andreani, M.; Angelucci, E. The cure of thalassemia by bone marrow transplantation. Blood Rev. 2002, 16, 81–85. [Google Scholar] [CrossRef]
- Locatelli, F.; Merli, P.; Strocchio, L. Transplantation for thalassemia major. Curr. Opin. Hematol. 2016, 23, 515–523. [Google Scholar] [CrossRef]
- Strocchio, L.; Locatelli, F. Hematopoietic Stem Cell Transplantation in Thalassemia. Hematol. Clin. N. Am. 2018, 32, 317–328. [Google Scholar] [CrossRef]
- May, C.; Rivella, S.; Callegari, J.; Heller, G.; Gaensler, K.M.L.; Luzzatto, L.; Sadelain, M. Therapeutic haemoglobin synthesis in β-thalassaemic mice expressing lentivirus-encoded human β-globin. Nature 2000, 406, 82–86. [Google Scholar] [CrossRef]
- Pawliuk, R.; Westerman, K.A.; Fabry, M.E.; Payen, E.; Tighe, R.; Bouhassira, E.E.; Acharya, S.A.; Ellis, J.; London, I.M.; Eaves, C.J.; et al. Correction of Sickle Cell Disease in Transgenic Mouse Models by Gene Therapy. Science 2001, 294, 2368–2371. [Google Scholar] [CrossRef]
- Rivella, S.; May, C.; Chadburn, A.; Riviere, I.; Sadelain, M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human β-globin gene transfer. Blood 2003, 101, 2932–2939. [Google Scholar] [CrossRef]
- Miccio, A.; Cesari, R.; Lotti, F.; Rossi, C.; Sanvito, F.; Ponzoni, M.; Routledge, S.J.E.; Chow, C.-M.; Antoniou, M.N.; Ferrari, G. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of -thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 10547–10552. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, A.K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Kanter, J.; Walters, M.C.; Hsieh, M.; Krishnamurti, L.; Kwiatkowski, J.L.; Kamble, R.; von Kalle, C.; Joseney-Antoine, M.; Pierciey, F.J.; Shi, W.; et al. Interim Results from a Phase 1/2 Clinical Study of Lentiglobin Gene Therapy for Severe Sickle Cell Disease. Blood 2017, 130, 527. [Google Scholar] [CrossRef]
- Kwiatkowski, J.L.; Thompson, A.A.; Rasko, J.; Hongeng, S.; Schiller, G.J.; Anurathapan, U.; Cavazzana, M.; Ho, P.J.; von Kalle, C.; Kletzel, M.; et al. Clinical Outcomes up to 3 Years Following Lentiglobin Gene Therapy for Transfusion-Dependent β-Thalassemia in the Northstar Hgb-204 Study. Blood 2017, 130, 360. [Google Scholar] [CrossRef]
- Locatelli, F.; Walters, M.C.; Kwiatkowski, J.L.; Porter, J.; Sauer, M.G.; Thuret, I.; Hongeng, S.; Kulozik, A.E.; Lal, A.; Thrasher, A.J.; et al. Lentiglobin Gene Therapy for Patients with Transfusion-Dependent β-Thalassemia (TDT): Results from the Phase 3 Northstar-2 and Northstar-3 Studies. Blood 2018, 132, 1025. [Google Scholar] [CrossRef]
- Malik, P.; Grimley, M.; Quinn, C.T.; Shova, A.; Courtney, L.; Lutzko, C.; Kalfa, T.A.; Niss, O.; Mehta, A.P.; Chandra, S.; et al. Gene Therapy for Sickle Cell Anemia Using a Modified Gamma Globin Lentivirus Vector and Reduced Intensity Conditioning Transplant Shows Promising Correction of the Disease Phenotype. Blood 2018, 132, 1021. [Google Scholar] [CrossRef]
- Mansilla-Soto, J.; Riviere, I.; Boulad, F.; Sadelain, M. Cell and Gene Therapy for the Beta-Thalassemias: Advances and Prospects. Hum. Gene Ther. 2016, 27, 295–304. [Google Scholar] [CrossRef]
- Marktel, S.; Scaramuzza, S.; Cicalese, M.P.; Giglio, F.; Galimberti, S.; Lidonnici, M.R.; Calbi, V.; Assanelli, A.; Bernardo, M.E.; Rossi, C.; et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent β-thalassemia. Nat. Med. 2019, 25, 234–241. [Google Scholar] [CrossRef]
- Ribeil, J.-A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Sii-Felice, K.; Giorgi, M.; Leboulch, P.; Payen, E. Hemoglobin disorders: Lentiviral gene therapy in the starting blocks to enter clinical practice. Exp. Hematol. 2018, 64, 12–32. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef]
- Tisdale, J.F.; Kanter, J.; Mapara, M.Y.; Kwiatkowski, J.L.; Krishnamurti, L.; Schmidt, M.; Miller, A.L.; Pierciey, J.F.J.; Shi, W.; Ribeil, J.-A.; et al. Current Results of Lentiglobin Gene Therapy in Patients with Severe Sickle Cell Disease Treated Under a Refined Protocol in the Phase 1 Hgb-206 Study. Blood 2018, 132, 1026. [Google Scholar] [CrossRef]
- Walters, M.C.; Hongeng, S.; Kwiatkowski, J.L.; Locatelli, F.; Porter, J.B.; Sauer, M.G.; Thrasher, A.J.; Thuret, I.; Yannaki, E.; Elliot, H.; et al. Results from the Hgb-207 (Northstar-2) Trial: A Phase 3 Study to Evaluate Safety and Efficacy of Lentiglobin Gene Therapy for Transfusion-Dependent β-Thalassemia (TDT) in Patients with Non-β0/β0 Genotypes. Blood 2017, 130, 526. [Google Scholar] [CrossRef]
- Magrin, E.; Miccio, A.; Cavazzana, M. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood 2019, 134, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Ghiaccio, V.; Chappell, M.; Rivella, S.; Breda, L. Gene Therapy for Beta-Hemoglobinopathies: Milestones, New Therapies and Challenges. Mol. Diagn. Ther. 2019, 23, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Bungert, J.; Dave, U.; Lim, K.C.; Lieuw, K.H.; Shavit, A.J.; Liu, Q.; Engel, J.D. Synergistic regulation of human beta-globin gene switching by locus control region elements HS3 and HS4. Genes Dev. 1995, 9, 3083–3096. [Google Scholar] [CrossRef]
- Kim, A.; Dean, A. Chromatin loop formation in the β-globin locus and its role in globin gene transcription. Mol. Cells 2012, 34, 1–5. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene Editing ofCCR5in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef]
- Nakade, S.; Tsubota, T.; Sakane, Y.; Kume, S.; Sakamoto, N.; Obara, M.; Daimon, T.; Sezutsu, H.; Yamamoto, T.; Sakuma, T.; et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014, 5, 5560. [Google Scholar] [CrossRef]
- Lieber, M.R. The Mechanism of Human Nonhomologous DNA End Joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef]
- Yanik, M.; Ponnam, S.P.G.; Wimmer, T.; Trimborn, L.; Müller, C.; Gambert, I.; Ginsberg, J.; Janise, A.; Domicke, J.; Wende, W.; et al. Development of a Reporter System to Explore MMEJ in the Context of Replacing Large Genomic Fragments. Mol. Ther. Nucleic Acids 2018, 11, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Crudele, J.M.; Chamberlain, J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018, 9, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, V.L.; McGill, J.; McMahon, S.; Wang, J.; Jiang, H.; Sauna, Z.E. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther. Methods Clin. Dev. 2018, 10, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Yao, Y.; Zhang, C.-Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR-Cas9 ge-nome editing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Turchiano, G.; Andrieux, G.; Blattner, G.; Pennucci, V.; Klermund, J.; Monaco, G.; Poddar, S.; Mussolino, C.; Cornu, T.I.; Boerries, M.; et al. Quantitative Evaluation of Chromosomal Rearrangements in Primary Gene-Edited Human Stem Cells by Preclinical CAST-Seq. Cell Stem Cell 2020. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Schiroli, G.; Conti, A.; Ferrari, S.; Della Volpe, L.; Jacob, A.; Albano, L.; Beretta, S.; Calabria, A.; Vavassori, V.; Gasparini, P.; et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell 2019, 24, 551–565. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Exline, C.M.; Declercq, J.J.; Llewellyn, G.N.; Hayward, S.B.; Li, P.W.-L.; Shivak, D.A.; Surosky, R.T.; Gregory, P.D.; Holmes, M.C.; et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015, 33, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Pattabhi, S.; Lotti, S.N.; Berger, M.P.; Singh, S.; Lux, C.; Jacoby, K.; Lee, C.; Negre, O.; Scharenberg, A.M.; Rawlings, D.J. In Vivo Outcome of Homology-Directed Repair at the HBB Gene in HSC Using Alternative Donor Template Delivery Methods. Mol. Ther. Nucleic Acids 2019, 17, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Romero, Z.; Lomova, A.; Said, S.; Miggelbrink, A.; Kuo, C.Y.; Campo-Fernandez, B.; Hoban, M.D.; Masiuk, K.E.; Clark, D.N.; Long, J.; et al. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 2019, 27, 1389–1406. [Google Scholar] [CrossRef]
- Martin, R.M.; Ikeda, K.; Cromer, M.K.; Uchida, N.; Nishimura, T.; Romano, R.; Tong, A.J.; Lemgart, V.T.; Camarena, J.; Pavel-Dinu, M.; et al. Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell 2019, 24, 821–828. [Google Scholar] [CrossRef]
- Park, S.; Gianotti-Sommer, A.; Molina-Estevez, F.J.; Vanuytsel, K.; Skvir, N.; Leung, A.; Rozelle, S.S.; Shaikho, E.M.; Weir, I.; Jiang, Z.; et al. A Comprehensive, Ethnically Diverse Library of Sickle Cell Disease-Specific Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 1076–1085. [Google Scholar] [CrossRef]
- Hoban, M.D.; Cost, G.J.; Mendel, M.C.; Romero, Z.; Kaufman, M.L.; Joglekar, A.V.; Ho, M.; Lumaquin, D.; Gray, D.; Lill, G.R.; et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 2015, 125, 2597–2604. [Google Scholar] [CrossRef]
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.-J.; Mitros, T.; Muñoz, D.P.; Boffelli, D.; et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016, 8, 360ra134. [Google Scholar] [CrossRef]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, C.M.; Dever, D.P.; Davis, T.H.; Camarena, J.; Srifa, W.; Zhang, Y.; Paikari, A.; Chang, A.K.; Porteus, M.H.; et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019, 47, 7955–7972. [Google Scholar] [CrossRef] [PubMed]
- Magis, W.; DeWitt, M.A.; Wyman, S.K.; Vu, J.T.; Heo, S.-J.; Shao, S.J.; Hennig, F.; Romero, Z.G.; Campo-Fernandez, B.; McNeill, M.; et al. High-level correction of the sickle mutation amplified in vivo during erythroid differentiation. bioRxiv 2019. [Google Scholar] [CrossRef]
- Ferrari, S.; Jacob, A.; Beretta, S.; Unali, G.; Albano, L.; Vavassori, V.; Cittaro, D.; Lazarevic, D.; Brombin, C.; Cugnata, F.; et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat. Biotechnol. 2020, 38, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.J.; Schröder, M.S.; Caiado, F.; Wyman, S.K.; Bray, N.L.; Bordi, M.; DeWitt, M.A.; Vu, J.T.; Kim, W.-T.; Hockemeyer, D.; et al. Controlled Cycling and Quiescence Enables Efficient HDR in Engraftment-Enriched Adult Hematopoietic Stem and Progenitor Cells. Cell Rep. 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 andpiggyBac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef]
- Song, B.; Fan, Y.; He, W.; Zhu, D.; Niu, X.; Wang, D.; Ou, Z.; Luo, M.; Sun, X.-F. Improved Hematopoietic Differentiation Efficiency of Gene-Corrected Beta-Thalassemia Induced Pluripotent Stem Cells by CRISPR/Cas9 System. Stem Cells Dev. 2015, 24, 1053–1065. [Google Scholar] [CrossRef]
- Xu, P.; Tong, Y.; Liu, X.-Z.; Wang, T.-T.; Cheng, L.; Wang, B.-Y.; Lv, X.; Huang, Y.; Liu, D.-P. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2–654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Niu, X.; He, W.; Song, B.; Ou, Z.; Fan, D.; Chen, Y.; Fan, Y.; Sun, X. Combining Single Strand Oligodeoxynucleotides and CRISPR/Cas9 to Correct Gene Mutations in β-Thalassemia-induced Pluripotent Stem Cells. J. Biol. Chem. 2016, 291, 16576–16585. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Kang, X.; Lin, B.; Yu, Q.; Song, B.; Gao, G.; Chen, Y.; Sun, X.; Li, X.; et al. One-Step Biallelic and Scarless Correction of a β-Thalassemia Mutation in Patient-Specific iPSCs without Drug Selection. Mol. Ther. Nucleic Acids 2017, 6, 57–67. [Google Scholar] [CrossRef]
- Wattanapanitch, M.; Damkham, N.; Potirat, P.; Trakarnsanga, K.; Janan, M.; U-Pratya, Y.; Kheolamai, P.; Klincumhom, N.; Issaragrisil, S. One-step genetic correction of hemoglobin E/beta-thalassemia patient-derived iPSCs by the CRISPR/Cas9 system. Stem Cell Res. Ther. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Cai, L.; Bai, H.; Mahairaki, V.; Gao, Y.; He, C.; Wen, Y.; Jin, Y.-C.; Wang, Y.; Pan, R.L.; Qasba, A.; et al. A Universal Approach to Correct Various HBB Gene Mutations in Human Stem Cells for Gene Therapy of Beta-Thalassemia and Sickle Cell Disease. Stem Cells Transl. Med. 2017, 7, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Luk, K.; Yao, Q.; Shen, A.H.; Zeng, J.; Wu, Y.; Luo, H.-Y.; Brendel, C.; Pinello, L.; Chui, D.H.K.; et al. Editing aberrant splice sites efficiently restores β-globin expression in β-thalassemia. Blood 2019, 133, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Patsali, P.; Turchiano, G.; Papasavva, P.; Romito, M.; Loucari, C.C.; Stephanou, C.; Christou, S.; Sitarou, M.; Mussolino, C.; Cornu, T.I.; et al. Correction of IVS I-110(G > A) β-thalassemia by CRISPR/Cas-and TALEN-mediated disruption of aberrant regulatory elements in human hematopoietic stem and progenitor cells. Haematologica 2019, 104, e497–e501. [Google Scholar] [CrossRef] [PubMed]
- Forget, B.G. Molecular Basis of Hereditary Persistence of Fetal Hemoglobin. Ann. N. Y. Acad. Sci. 1998, 850, 38–44. [Google Scholar] [CrossRef]
- Ye, L.; Wang, J.; Tan, Y.; Beyer, A.I.; Xie, F.; Muench, M.O.; Kan, Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proc. Natl. Acad. Sci. USA 2016, 113, 10661–10665. [Google Scholar] [CrossRef]
- Antoniani, C.; Meneghini, V.; Lattanzi, A.; Felix, T.; Romano, O.; Magrin, E.; Weber, L.; Pavani, G.; El Hoss, S.; Kurita, R.; et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood 2018, 131, 1960–1973. [Google Scholar] [CrossRef]
- Traxler, E.A.; Yao, Y.; Wang, Y.-D.; Woodard, K.J.; Kurita, R.; Nakamura, Y.; Hughes, J.R.; Hardison, R.C.; Blobel, G.A.; Li, C.; et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016, 22, 987–990. [Google Scholar] [CrossRef]
- Martyn, G.E.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.M.; Funnell, A.P.W.; et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018, 50, 498–503. [Google Scholar] [CrossRef]
- Métais, J.-Y.; Doerfler, P.A.; Mayuranathan, T.; Bauer, D.E.; Fowler, S.C.; Hsieh, M.; Katta, V.; Keriwala, S.; Lazzarotto, C.R.; Luk, K.; et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019, 3, 3379–3392. [Google Scholar] [CrossRef]
- Humbert, O.; Radtke, S.; Samuelson, C.; Carrillo, R.R.; Perez, A.M.; Reddy, S.S.; Lux, C.; Pattabhi, S.; Schefter, L.E.; Negre, O.; et al. Therapeutically relevant engraftment of a CRISPR-Cas9–edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Li, C.; Psatha, N.; Sova, P.; Gil, S.; Wang, H.; Kim, J.; Kulkarni, C.; Valensisi, C.; Hawkins, R.D.; Stamatoyannopoulos, G.; et al. Reactivation of γ-globin in adult β-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood 2018, 131, 2915–2928. [Google Scholar] [CrossRef]
- Peterson, K.R.; Clegg, C.H.; Huxley, C.; Josephson, B.M.; Haugen, H.S.; Furukawa, T.; Stamatoyannopoulos, G. Transgenic mice containing a 248-kb yeast artificial chromosome carrying the human beta-globin locus display proper developmental control of human globin genes. Proc. Natl. Acad. Sci. USA 1993, 90, 7593–7597. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.; Frati, G.; Felix, T.; Hardouin, G.; Casini, A.; Wollenschlaeger, C.; Meneghini, V.; Masson, C.; De Cian, A.; Chalumeau, A.; et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Xu, J.; Ragoczy, T.; Ippolito, G.C.; Walkley, C.R.; Maika, S.D.; Fujiwara, Y.; Ito, M.; Groudine, M.; Bender, M.A.; et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature 2009, 460, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Liu, K.; Sun, C.-W.; Pawlik, K.M.; Townes, T.M. KLF1 regulates BCL11A expression and γ- to β-globin gene switching. Nat. Genet. 2010, 42, 742–744. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Xu, J.; Orkin, S.H. Transcriptional silencing of fetal hemoglobin by BCL11A. Ann. N. Y. Acad. Sci. 2010, 1202, 64–68. [Google Scholar] [CrossRef]
- Funnell, A.P.W.; Prontera, P.; Ottaviani, V.; Piccione, M.; Giambona, A.; Maggio, A.; Ciaffoni, F.; Stehling-Sun, S.; Marra, M.; Masiello, F.; et al. 2p15-p16.1 microdeletions encompassing and proximal to BCL11A are associated with elevated HbF in addition to neurologic impairment. Blood 2015, 126, 89–93. [Google Scholar] [CrossRef]
- Dias, C.; Estruch, S.B.; Graham, S.A.; McRae, J.; Sawiak, S.J.; Hurst, J.A.; Joss, S.K.; Holder, S.E.; Morton, J.E.; Turner, C.; et al. BCL11A Haploinsufficiency Causes an Intellectual Disability Syndrome and Dysregulates Transcription. Am. J. Hum. Genet. 2016, 99, 253–274. [Google Scholar] [CrossRef]
- Liu, P.; Keller, J.R.; Ortiz, M.; Tessarollo, L.; Rachel, R.A.; Nakamura, T.; Jenkins, N.A.; Copeland, N.G. Bcl11a is essential for normal lymphoid development. Nat. Immunol. 2003, 4, 525–532. [Google Scholar] [CrossRef]
- Luc, S.; Huang, J.; McEldoon, J.L.; Somuncular, E.; Li, D.; Rhodes, C.; Mamoor, S.; Hou, S.; Xu, J.; Orkin, S.H. Bcl11a Deficiency Leads to Hematopoietic Stem Cell Defects with an Aging-like Phenotype. Cell Rep. 2016, 16, 3181–3194. [Google Scholar] [CrossRef]
- Humbert, O.; Peterson, C.W.; Norgaard, Z.K.; Radtke, S.; Kiem, H.-P. A Nonhuman Primate Transplantation Model to Evaluate Hematopoietic Stem Cell Gene Editing Strategies for β-Hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2018, 8, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Peng, C.; Sankaran, V.G.; Shao, Z.; Esrick, E.B.; Chong, B.G.; Ippolito, G.C.; Fujiwara, Y.; Ebert, B.L.; Tucker, P.W.; et al. Correction of Sickle Cell Disease in Adult Mice by Interference with Fetal Hemoglobin Silencing. Science 2011, 334, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N. Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.E.; Kamran, S.C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M.C.; Smith, E.C.; Pinello, L.; et al. An Erythroid Enhancer of BCL11A Subject to Genetic Variation Determines Fetal Hemoglobin Level. Science 2013, 342, 253–257. [Google Scholar] [CrossRef]
- Canver, M.C.; Smith, J.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef]
- Vierstra, J.; Reik, A.; Chang, K.-H.; Stehling-Sun, S.; Zhou, Y.-Y.; Hinkley, S.J.; Paschon, D.E.; Zhang, L.; Psatha, N.; Bendana, Y.R.; et al. Functional footprinting of regulatory DNA. Nat. Methods 2015, 12, 927–930. [Google Scholar] [CrossRef]
- Chang, K.-H.; Smith, S.E.; Sullivan, T.; Chen, K.; Zhou, Q.; West, J.A.; Liu, M.; Liu, Y.; Vieira, B.F.; Sun, C.; et al. Long-Term Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34 + Hematopoietic Stem and Progenitor Cells. Mol. Ther. Methods Clin. Dev. 2017, 4, 137–148. [Google Scholar] [CrossRef]
- Psatha, N.; Reik, A.; Phelps, S.; Zhou, Y.; Dalas, D.; Yannaki, E.; Levasseur, D.N.; Urnov, F.D.; Holmes, M.C.; Papayannopoulou, T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with β-Thalassemia Major. Mol. Ther. Methods Clin. Dev. 2018, 10, 313–326. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, M.K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef]
- Demirci, S.; Zeng, J.; Wu, Y.; Uchida, N.; Shen, A.H.; Pellin, D.; Gamer, J.; Yapundich, M.; Drysdale, C.; Bonanno, J.; et al. BCL11A enhancer–edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J. Clin. Investig. 2020, 130, 6677–6687. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; De La Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Bobruff, Y.; Cappellini, M.D.; Corbacioglu, S.; Fernandez, R.C.M.; De La Fuente, J.; Grupp, A.S.; Handgretinger, R.; Ho, T.W.; Imren, S.; et al. Safety and Efficacy of CTX001 in Patients with Transfusion-Dependent β-Thalassemia and Sickle Cell Disease: Early Results from the Climb THAL-111 and Climb SCD-121 Studies of Autologous CRISPR-CAS9-Modified CD34+ Hematopoietic Stem and Progenitor Cells. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Grevet, J.D.; Lan, X.; Hamagami, N.; Edwards, C.R.; Sankaranarayanan, L.; Ji, X.; Bhardwaj, S.K.; Face, C.J.; Posocco, D.F.; Abdulmalik, O.; et al. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 2018, 361, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Peslak, S.A.; Lan, X.; Khandros, E.; Yano, J.A.; Sharma, M.; Keller, C.A.; Giardine, B.; Qin, K.; Abdulmalik, O.; et al. The HRI-regulated transcription factor ATF4 activates BCL11A transcription to silence fetal hemoglobin expression. Blood 2020, 135, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Peslak, S.A.; Khandros, E.; Huang, P.; Lan, X.; Geronimo, C.L.; Grevet, J.D.; Abdulmalik, O.; Zhang, Z.; Giardine, B.; Keller, C.A.; et al. HRI depletion cooperates with pharmacologic inducers to elevate fetal hemoglobin and reduce sickle cell formation. Blood Adv. 2020, 4, 4560–4572. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Bauer, D.E.; Kerenyi, M.A.; Vo, T.D.; Hou, S.; Hsu, Y.-J.; Yao, H.; Trowbridge, J.J.; Mandel, G.; Orkin, S.H. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc. Natl. Acad. Sci. USA 2013, 110, 6518–6523. [Google Scholar] [CrossRef]
- Amaya, M.; Desai, M.; Gnanapragasam, M.N.; Wang, S.Z.; Zu Zhu, S.; Williams, D.C.; Ginder, G.D. Mi2β-mediated silencing of the fetal γ-globin gene in adult erythroid cells. Blood 2013, 121, 3493–3501. [Google Scholar] [CrossRef]
- Sher, F.; Hossain, M.; Seruggia, D.; Schoonenberg, V.A.C.; Yao, Q.; Cifani, P.; Dassama, L.M.K.; Cole, M.A.; Ren, C.; Vinjamur, D.S.; et al. Rational targeting of a NuRD subcomplex guided by comprehensive in situ mutagenesis. Nat. Genet. 2019, 51, 1149–1159. [Google Scholar] [CrossRef]
- Lan, X.; Ren, R.; Feng, R.; Ly, L.C.; Lan, Y.; Zhang, Z.; Aboreden, N.; Qin, K.; Horton, J.R.; Grevet, J.D.; et al. ZNF410 Uniquely Activates the NuRD Component CHD4 to Silence Fetal Hemoglobin Expression. Mol. Cell 2021, 81, 239–254. [Google Scholar] [CrossRef]
- Antoniou, P.; Miccio, A.; Brusson, M. Base and prime editing technologies for blood disorders. Front. Genome 2021, 3. [Google Scholar] [CrossRef]
- Yeh, W.-H.; Chiang, H.; Rees, H.A.; Edge, A.S.B.; Liu, D.R. In vivo base editing of post-mitotic sensory cells. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Ding, C.; Sun, H.; Xie, X.; Xu, Y.; Zhang, X.; Sun, Y.; Xiong, Y.; Ma, W.; Liu, Y.; et al. Correction of β-thalassemia mutant by base editor in human embryos. Protein Cell 2017, 8, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, J.M.; Cervantes, O.; Clement, K.; Wu, Y.; Zeng, J.; Bauer, D.E.; Pinello, L.; Joung, J.K. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol. 2018, 36, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wu, Y.; Ren, C.; Bonanno, J.; Shen, A.H.; Shea, D.; Gehrke, J.M.; Clement, K.; Luk, K.; Yao, Q.; et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 2020, 26, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Viprakasit, V.; Wiriyasateinkul, A.; Sattayasevana, B.; Miles, K.L.; Laosombat, V. Hb G-Makassar (β6(A3)Glu→Ala; Codon 6 (GAG→GCG)): Molecular Characterization, Clinical, and Hematological Effects. Hemoglobin 2002, 26, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, A.S.; Hamzah, R.; Selvaratnam, V.; Yegapan, S.; Sathar, J. Human hemoglobin G-Makassar variant masquerading as sickle cell anemia. Hematol. Rep. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.M.; Wang, T.; Randolph, P.B.; Arbab, M.; Shen, M.W.; Huang, T.P.; Matuszek, Z.; Newby, G.A.; Rees, H.A.; Liu, D.R. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol. 2020, 38, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Zhu, B.; Wang, L.; Chen, C.; Hong, M.; Huang, Y.; Li, H.; Han, H.; Cai, B.; et al. Increasing the efficiency and targeting range of cytidine base editors through fusion of a single-stranded DNA-binding protein domain. Nat. Cell Biol. 2020, 22, 740–750. [Google Scholar] [CrossRef]
- Richter, M.F.; Zhao, K.T.; Eton, E.O.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.-J.; Liquori, A.J.; et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, L.; Ma, Y.; Hu, H.; Li, Q.; Yang, Y.; Liu, W.; Yin, S.; Li, W.; Fu, B.; et al. Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies. Cell Res. 2020, 30, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Bahal, R.; McNeer, N.A.; Quijano, E.; Liu, Y.; Sulkowski, P.; Turchick, A.; Lu, Y.-C.; Bhunia, D.C.; Manna, A.; Greiner, D.L.; et al. In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; López-Giráldez, F.; Coşkun, S.; Song, E.; et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Blattner, G.; Cavazza, A.; Thrasher, A.J.; Turchiano, G. Gene Editing and Genotoxicity: Targeting the Off-Targets. Front. Genome Ed. 2020, 2. [Google Scholar] [CrossRef]
- Bao, X.R.; Pan, Y.; Lee, C.M.; Davis, T.H.; Bao, G. Tools for experimental and computational analyses of off-target editing by programmable nucleases. Nat. Protoc. 2021, 16, 10–26. [Google Scholar] [CrossRef]
- Chaudhari, H.G.; Penterman, J.; Whitton, H.J.; Spencer, S.J.; Flanagan, N.; Zhang, M.C.L.; Huang, E.; Khedkar, A.S.; Toomey, J.M.; Shearer, C.A.; et al. Evaluation of Homology-Independent CRISPR-Cas9 Off-Target Assessment Methods. CRISPR J. 2020, 3, 440–453. [Google Scholar] [CrossRef]
- Coquerelle, S.; Ghardallou, M.; Rais, S.; Taupin, P.; Touzot, F.; Boquet, L.; Blanche, S.; Benaouadi, S.; Brice, T.; Tuchmann-Durand, C.; et al. Innovative Curative Treatment of Beta Thalassemia: Cost-Efficacy Analysis of Gene Therapy Versus Allogenic Hematopoietic Stem-Cell Transplantation. Hum. Gene Ther. 2019, 30, 753–761. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frati, G.; Miccio, A. Genome Editing for β-Hemoglobinopathies: Advances and Challenges. J. Clin. Med. 2021, 10, 482. https://doi.org/10.3390/jcm10030482
Frati G, Miccio A. Genome Editing for β-Hemoglobinopathies: Advances and Challenges. Journal of Clinical Medicine. 2021; 10(3):482. https://doi.org/10.3390/jcm10030482
Chicago/Turabian StyleFrati, Giacomo, and Annarita Miccio. 2021. "Genome Editing for β-Hemoglobinopathies: Advances and Challenges" Journal of Clinical Medicine 10, no. 3: 482. https://doi.org/10.3390/jcm10030482
APA StyleFrati, G., & Miccio, A. (2021). Genome Editing for β-Hemoglobinopathies: Advances and Challenges. Journal of Clinical Medicine, 10(3), 482. https://doi.org/10.3390/jcm10030482