
The Antidiabetic Agent Metformin Inhibits IL-23 Production in Murine Bone-Marrow-Derived Dendritic Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Generation of BMDCs and Cell Culture
2.3. Transfection of Small Interfering RNAs (siRNAs) against Nfkbiz
2.4. qRT-PCR
2.5. Western Blotting
2.6. ELISA
2.7. Statistical Analysis
3. Results
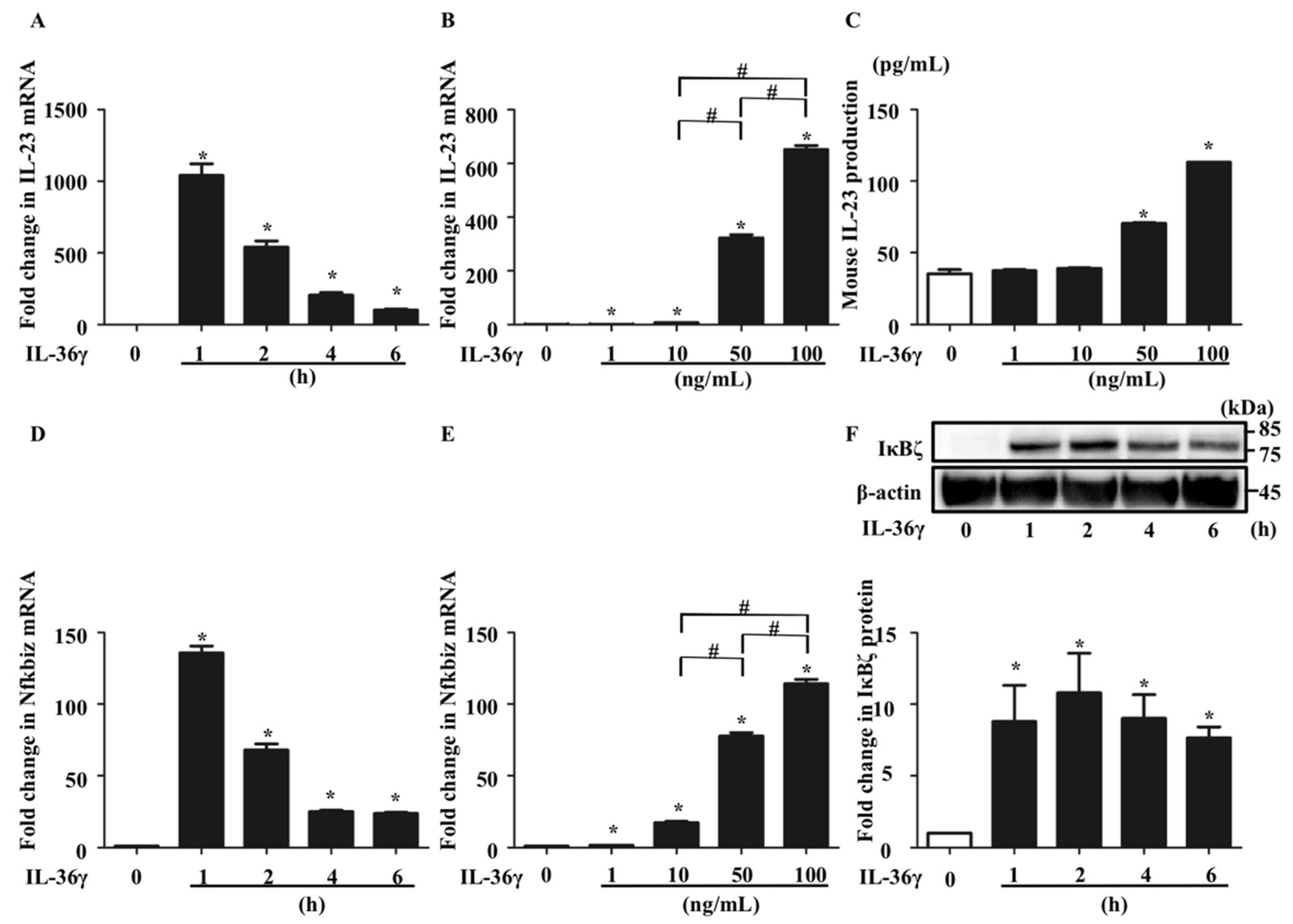
3.1. IL-36γ Stimulation Upregulated IL-23 and Nfkbiz Expression in BMDCs
3.2. IL-36γ Stimulation Upregulated IL-23 via Nfkbiz in BMDCs
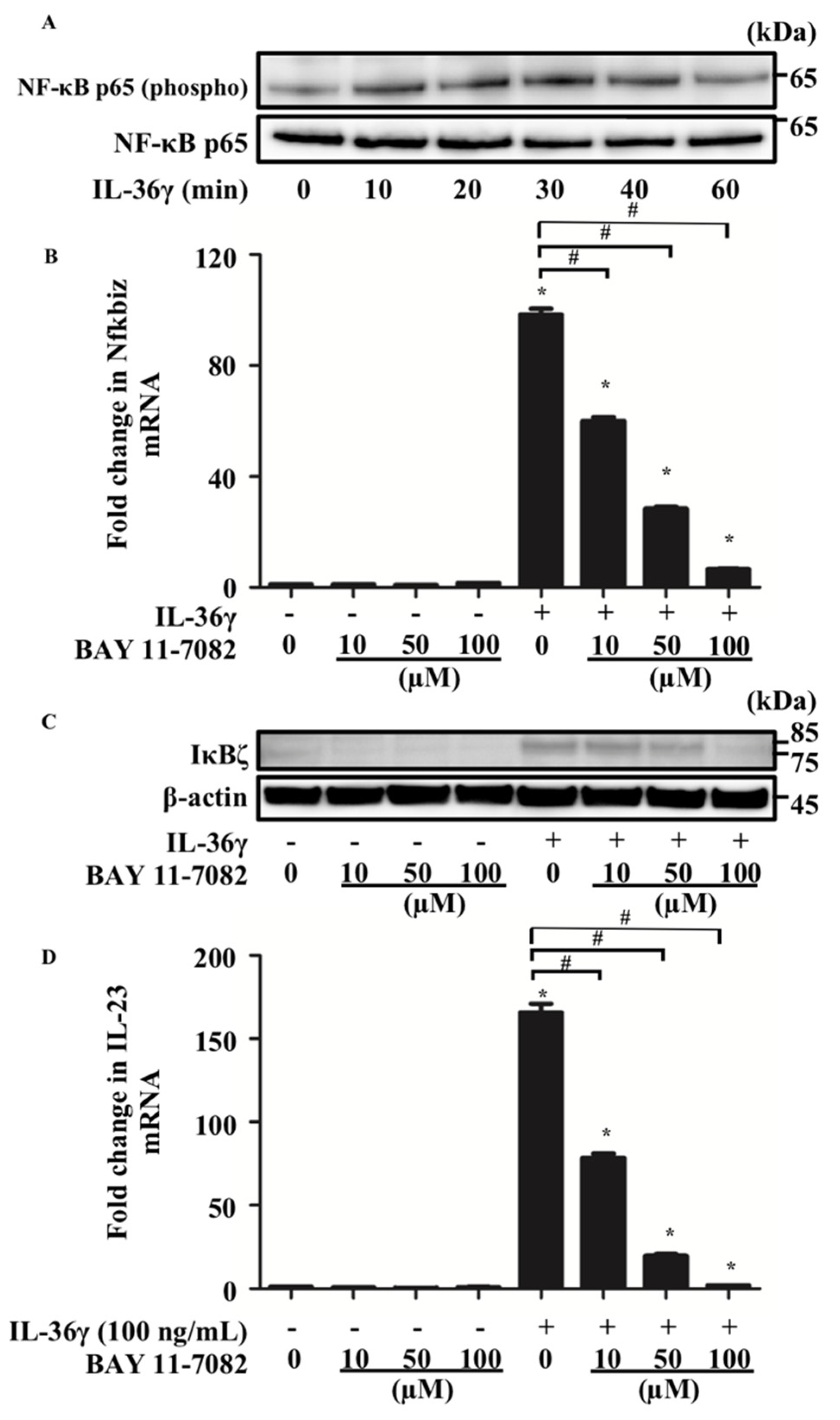
3.3. IL-36γ Upregulates Nfkbiz and IL-23 via the Activation of NF-κB Signaling
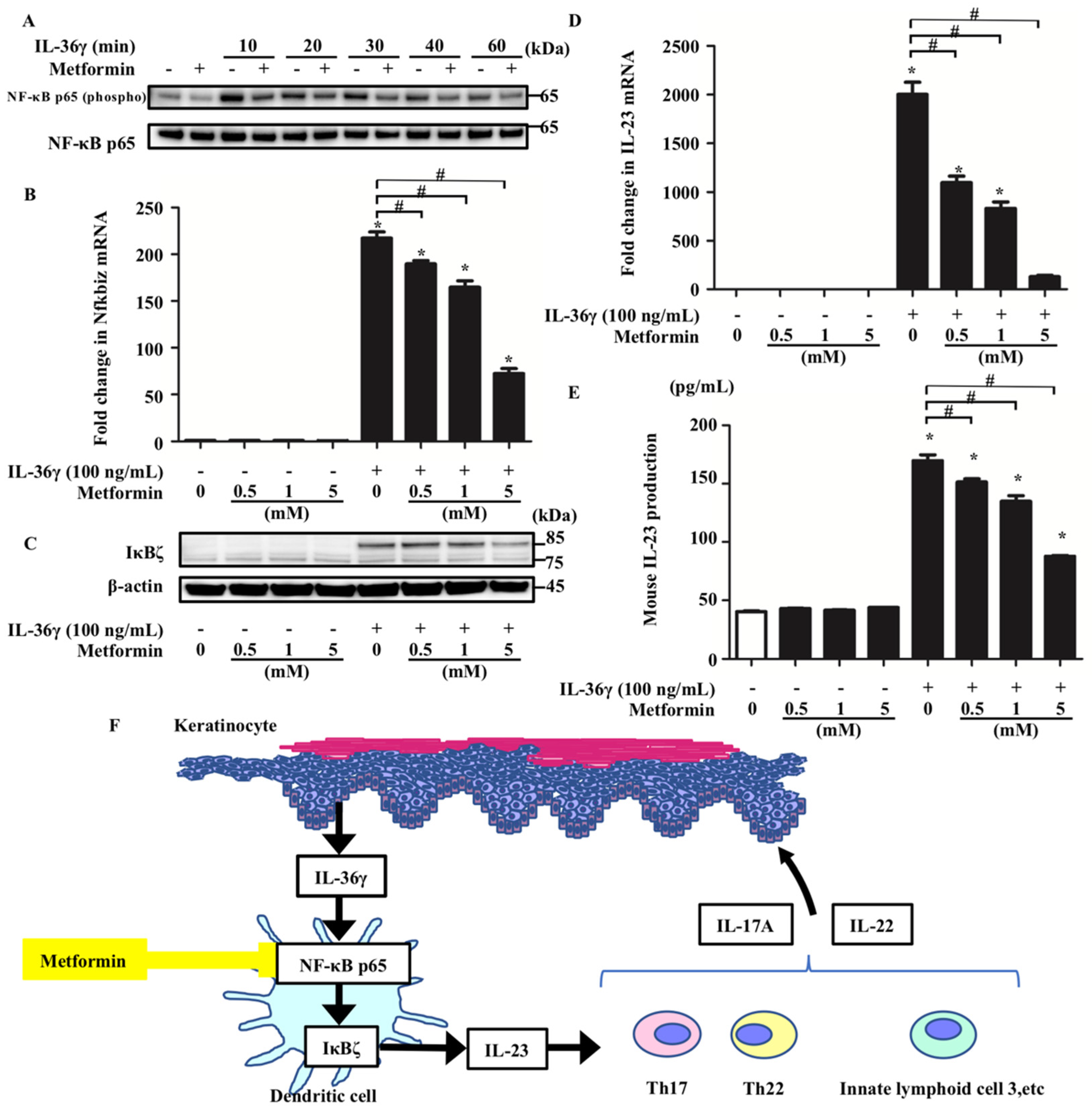
3.4. Metformin Treatment Inhibited the Upregulation of Nfkbiz and IL-23 Induced by IL-36 Stimulation by Impairing NF-κB Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Michalek, I.M.; Loring, B.; John, S.M. A systematic review of worldwide epidemiology of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 205–212. [Google Scholar] [CrossRef]
- Furue, M.; Furue, K.; Tsuji, G.; Nakahara, T. Interleukin-17A and Keratinocytes in Psoriasis. Int. J. Mol. Sci. 2020, 21, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehncke, W.H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Yan, B.; Liu, N.; Li, J.; Li, J.; Zhu, W.; Kuang, Y.; Chen, X.; Peng, C. The role of Langerhans cells in epidermal homeostasis and pathogenesis of psoriasis. J. Cell Mol. Med. 2020, 24, 11646–11655. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef]
- Wohn, C.; Ober-Blöbaum, J.L.; Haak, S.; Pantelyushin, S.; Cheong, C.; Zahner, S.P.; Onderwater, S.; Kant, M.; Weighardt, H.; Holzmann, B.; et al. Langerin(neg) conventional dendritic cells produce IL-23 to drive psoriatic plaque formation in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 10723–10728. [Google Scholar] [CrossRef] [Green Version]
- Riol-Blanco, L.; Ordovas-Montanes, J.; Perro, M.; Naval, E.; Thiriot, A.; Alvarez, D.; Paust, S.; Wood, J.N.; von Andrian, U.H. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 2014, 510, 157–161. [Google Scholar] [CrossRef]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef]
- Di Cesare, A.; Di Meglio, P.; Nestle, F.O. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J. Investig. Dermatol. 2009, 129, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.G.; Wang, T.; Zheng, J.; et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity 2011, 35, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Gordon, K.B.; Duffin, K.C.; Bissonnette, R.; Prinz, J.C.; Wasfi, Y.; Reich, K.; Li, S.; Shen, Y.-K.; Szapary, P.; Randazzo, B.; et al. A Phase 2 Trial of Guselkumab versus Adalimumab for Plaque Psoriasis. N. Engl. J. Med. 2015, 373, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.-P.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S.; et al. Risankizumab versus Ustekinumab for Moderate-to-Severe Plaque Psoriasis. N. Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Warren, R.B.; Iversen, L.; Puig, L.; Pau-Charles, I.; Igarashi, A.; Ohtsuki, M.; Falqués, M.; Harmut, M.; Rozzo, S.; et al. Long-term efficacy and safety of tildrakizumab for moderate-to-severe psoriasis: Pooled analyses of two randomized phase III clinical trials (reSURFACE 1 and reSURFACE 2) through 148 weeks. Br. J. Dermatol. 2020, 182, 605–617. [Google Scholar] [CrossRef] [PubMed]
- D’Erme, A.M.; Wilsmann-Theis, D.; Wagenpfeil, J.; Hölzel, M.; Ferring-Schmitt, S.; Sternberg, S.; Wittmann, M.; Peters, B.; Bosio, A.; Bieber, T.; et al. IL-36γ (IL-1F9) is a biomarker for psoriasis skin lesions. J. Investig. Dermatol. 2015, 135, 1025–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercurio, L.; Morelli, M.; Scarponi, C.; Eisenmesser, E.Z.; Doti, N.; Pagnanelli, G.; Gubinelli, E.; Mazzanti, C.; Cavani, A.; Ruvo, M.; et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. 2018, 9, 1104. [Google Scholar] [CrossRef]
- Walsh, P.T.; Fallon, P.G. The emergence of the IL-36 cytokine family as novel targets for inflammatory diseases. Ann. N. Y. Acad. Sci. 2018, 1417, 23–34. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Wan, M.T. Psoriasis: A novel risk factor for type 2 diabetes. Lancet Diabetes Endocrinol. 2018, 6, 919–921. [Google Scholar] [CrossRef]
- Brauchli, Y.B.; Jick, S.S.; Curtin, F.; Meier, C.R. Association between use of thiazolidinediones or other oral antidiabetics and psoriasis: A population based case-control study. J. Am. Acad. Dermatol. 2008, 58, 421–429. [Google Scholar] [CrossRef]
- Singh, S.; Bhansali, A. Randomized placebo control study of insulin sensitizers (Metformin and Pioglitazone) in psoriasis patients with metabolic syndrome (Topical Treatment Cohort). BMC Dermatol. 2016, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Bhansali, A. Randomized placebo control study of metformin in psoriasis patients with metabolic syndrome (Systemic Treatment Cohort). Indian J. Endocrinol. Metab. 2017, 21, 581–587. [Google Scholar] [CrossRef]
- Müller, A.; Hennig, A.; Lorscheid, S.; Grondona, P.; Schulze-Osthoff, K.; Kramer, D. IκBζ is a key transcriptional regulator of IL-36-driven psoriasis-related gene expression in keratinocytes. Proc. Natl. Acad. Sci. USA 2018, 115, 10088–10093. [Google Scholar] [CrossRef] [Green Version]
- Ovesen, S.K.; Schulze-Osthoff, K.; Iversen, L.; Johansen, C. IkBζ is a Key Regulator of Tumour Necrosis Factor-a and Interleukin-17A-mediated Induction of Interleukin-36g in Human Keratinocytes. Acta. Derm. Venereol. 2021, 101, adv00386. [Google Scholar] [CrossRef]
- Siegert, I.; Schatz, V.; Prechtel, A.T.; Steinkasserer, A.; Bogdan, C.; Jantsch, J. Electroporation of siRNA into mouse bone marrow-derived macrophages and dendritic cells. Methods Mol. Biol. 2014, 1121, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Willems, M.; Dubois, N.; Musumeci, L.; Bours, V.; Robe, P.A. IκBζ: An emerging player in cancer. Oncotarget 2016, 7, 66310–66322. [Google Scholar] [CrossRef]
- Sultuybek, G.K.; Soydas, T.; Yenmis, G. NF-κB as the mediator of metformin’s effect on ageing and ageing-related diseases. Clin. Exp. Pharmacol. Physiol. 2019, 46, 413–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.-C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+CD3+CD4−CD8− entheseal resident T cells. Nat. Med. 2011, 18, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deshpande, M.; Grisotto, M.; Smaldini, P.; Garcia, R.; He, Z.; Gulko, P.S.; Lira, S.A.; Furtado, G.C. Skin expression of IL-23 drives the development of psoriasis and psoriatic arthritis in mice. Sci. Rep. 2020, 10, 8259. [Google Scholar] [CrossRef] [PubMed]
- Vigne, S.; Palmer, G.; Lamacchia, C.; Martin, P.; Talabot-Ayer, D.; Rodriguez, E.; Ronchi, F.; Sallusto, F.; Dinh, H.; Sims, J.E.; et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood 2011, 118, 5813–5823. [Google Scholar] [CrossRef] [PubMed]
- Kovach, M.A.; Singer, B.; Martinez-Colon, G.; Newstead, M.W.; Zeng, X.; Mancuso, P.; Moore, T.A.; Kunkel, S.L.; Peters-Golden, M.; Moore, B.B.; et al. IL-36γ is a crucial proximal component of protective type-1-mediated lung mucosal immunity in Gram-positive and -negative bacterial pneumonia. Mucosal Immunol. 2017, 10, 1320–1334. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Yeung, H.; Takeshita, J.; Mehta, N.N.; Kimmel, S.E.; Ogdie, A.; Margolis, D.J.; Shin, D.B.; Attor, R.; Troxel, A.B.; Gelfand, J.M. Psoriasis severity and the prevalence of major medical comorbidity: A population-based study. JAMA Dermatol. 2013, 149, 1173–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, M.T.; Shin, D.B.; Hubbard, R.A.; Noe, M.H.; Mehta, N.N.; Gelfand, J.M. Psoriasis and the risk of diabetes: A prospective population-based cohort study. J. Am. Acad. Dermatol. 2018, 78, 315–322. [Google Scholar] [CrossRef]
- Nyambuya, T.M.; Dludla, P.V.; Mxinwa, V.; Mokgalaboni, K.; Ngcobo, S.R.; Nkambule, B.B. The impact of metformin and aspirin on T-cell mediated inflammation: A systematic review of in vitro and in vivo findings. Life Sci. 2020, 255, 117854. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kwak, H.J.; Cha, J.Y.; Jeong, Y.S.; Rhee, S.D.; Kim, K.R.; Cheon, H.G. Metformin suppresses lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages via activating transcription factor-3 (ATF-3) induction. J. Biol. Chem. 2014, 289, 23246–23255. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuda-Taniguchi, T.; Takemura, M.; Nakahara, T.; Hashimoto-Hachiya, A.; Takai-Yumine, A.; Furue, M.; Tsuji, G. The Antidiabetic Agent Metformin Inhibits IL-23 Production in Murine Bone-Marrow-Derived Dendritic Cells. J. Clin. Med. 2021, 10, 5610. https://doi.org/10.3390/jcm10235610
Matsuda-Taniguchi T, Takemura M, Nakahara T, Hashimoto-Hachiya A, Takai-Yumine A, Furue M, Tsuji G. The Antidiabetic Agent Metformin Inhibits IL-23 Production in Murine Bone-Marrow-Derived Dendritic Cells. Journal of Clinical Medicine. 2021; 10(23):5610. https://doi.org/10.3390/jcm10235610
Chicago/Turabian StyleMatsuda-Taniguchi, Tomoyo, Masaki Takemura, Takeshi Nakahara, Akiko Hashimoto-Hachiya, Ayako Takai-Yumine, Masutaka Furue, and Gaku Tsuji. 2021. "The Antidiabetic Agent Metformin Inhibits IL-23 Production in Murine Bone-Marrow-Derived Dendritic Cells" Journal of Clinical Medicine 10, no. 23: 5610. https://doi.org/10.3390/jcm10235610
APA StyleMatsuda-Taniguchi, T., Takemura, M., Nakahara, T., Hashimoto-Hachiya, A., Takai-Yumine, A., Furue, M., & Tsuji, G. (2021). The Antidiabetic Agent Metformin Inhibits IL-23 Production in Murine Bone-Marrow-Derived Dendritic Cells. Journal of Clinical Medicine, 10(23), 5610. https://doi.org/10.3390/jcm10235610