
Combined Inhibition of Bcl2 and Bcr-Abl1 Exercises Anti-Leukemia Activity but Does Not Eradicate the Primitive Leukemic Cells

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Isolation and Culture of Ph+ Primary Cells
2.3. Drug Treatment
2.4. Trypan Blue Exclusion Assay
2.5. Long Term Culture-Initiating Cells (LTC-ICs) Assays
2.6. Colony Forming Unit (CFUs) and Secondary Re-Plating (CFUs-r) Assays
2.7. Western Blotting, Immunoblotting and Densitometric Analysis
2.8. Cell Death Assay
2.9. Statistical Analysis
3. Results
3.1. BCL2 and BCR-ABL1 Kinase Dual Inhibition Exercises Anti-Clonogenic and Anti-Proliferation Activity on Committed CP-CML Progenitors
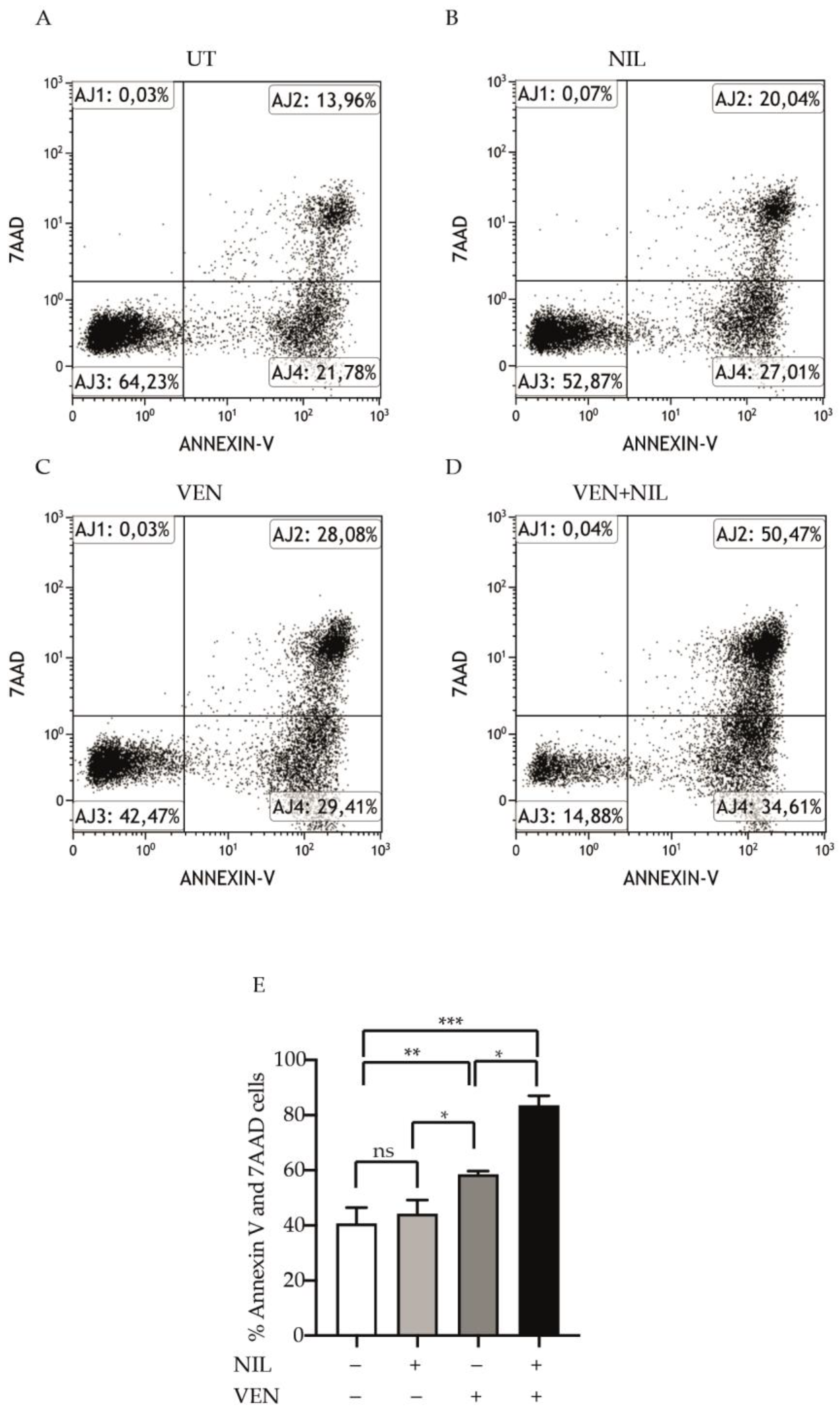
3.2. BCL2 and BCR-ABL1 Kinase Co-Targeting Kills Committed CP-CML Progenitors
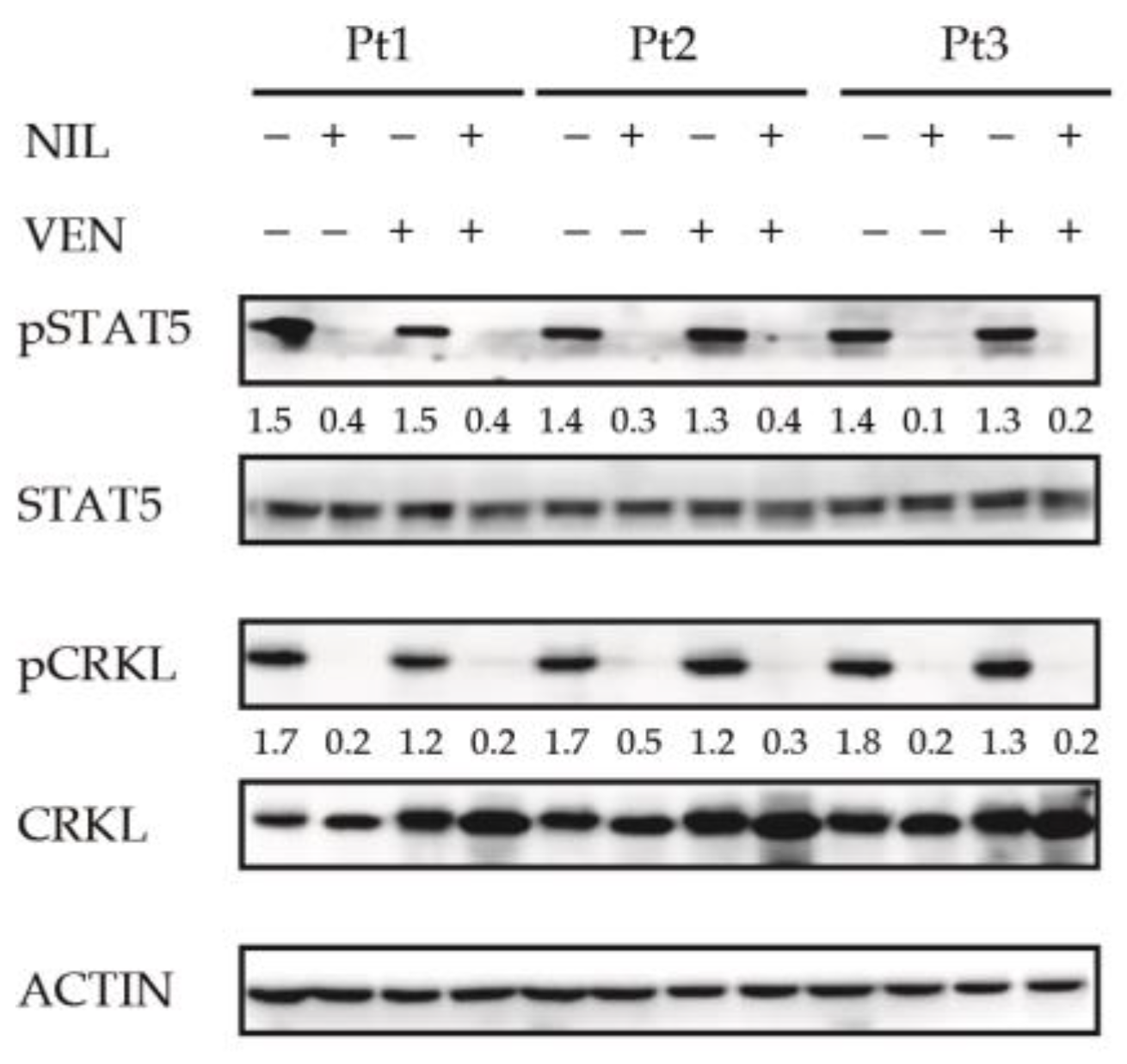
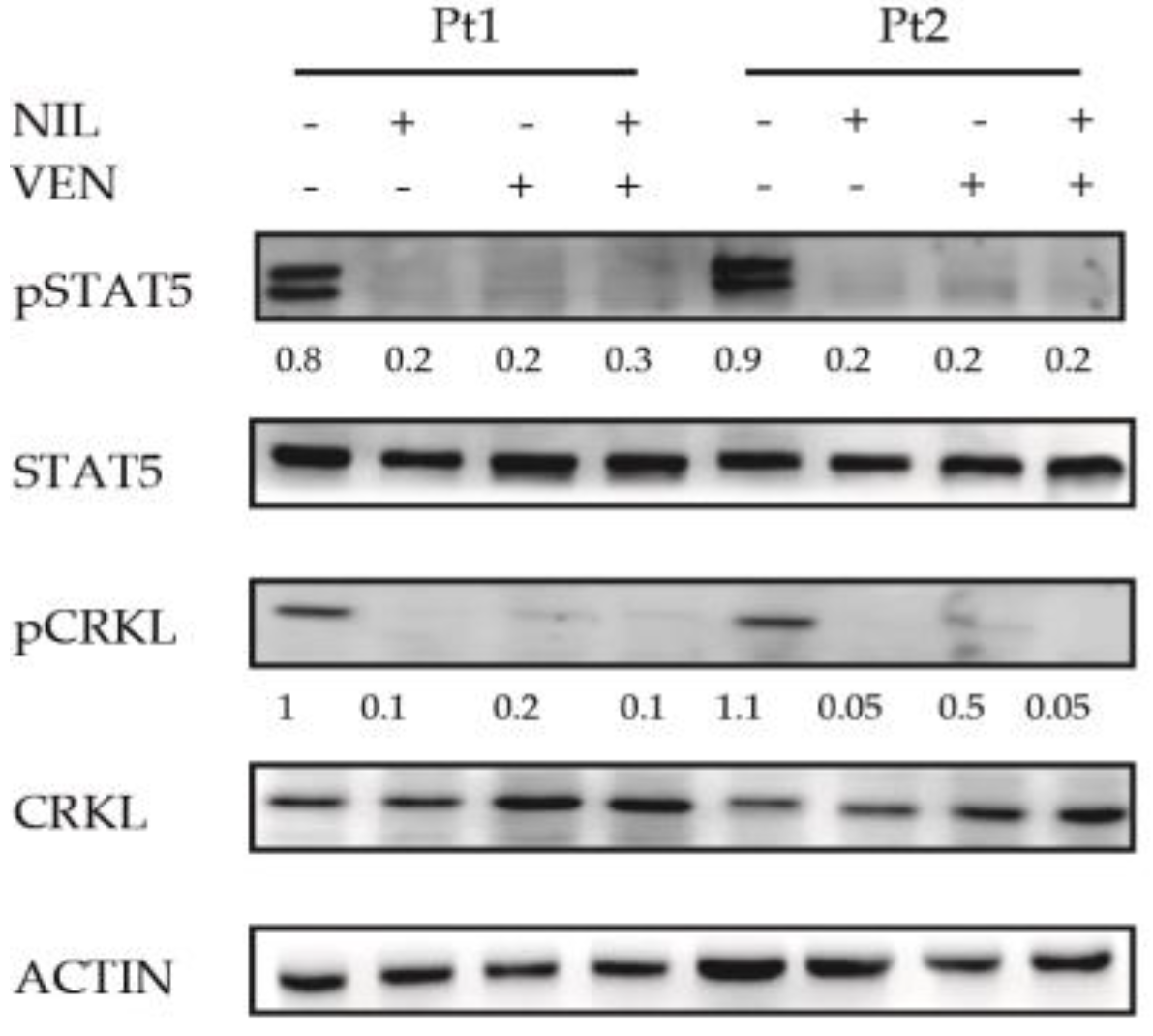
3.3. BCL2 Inhibition Does Not Affect the Activity of BCR-ABL1-Dependent Pro-Survival Mediator STAT5
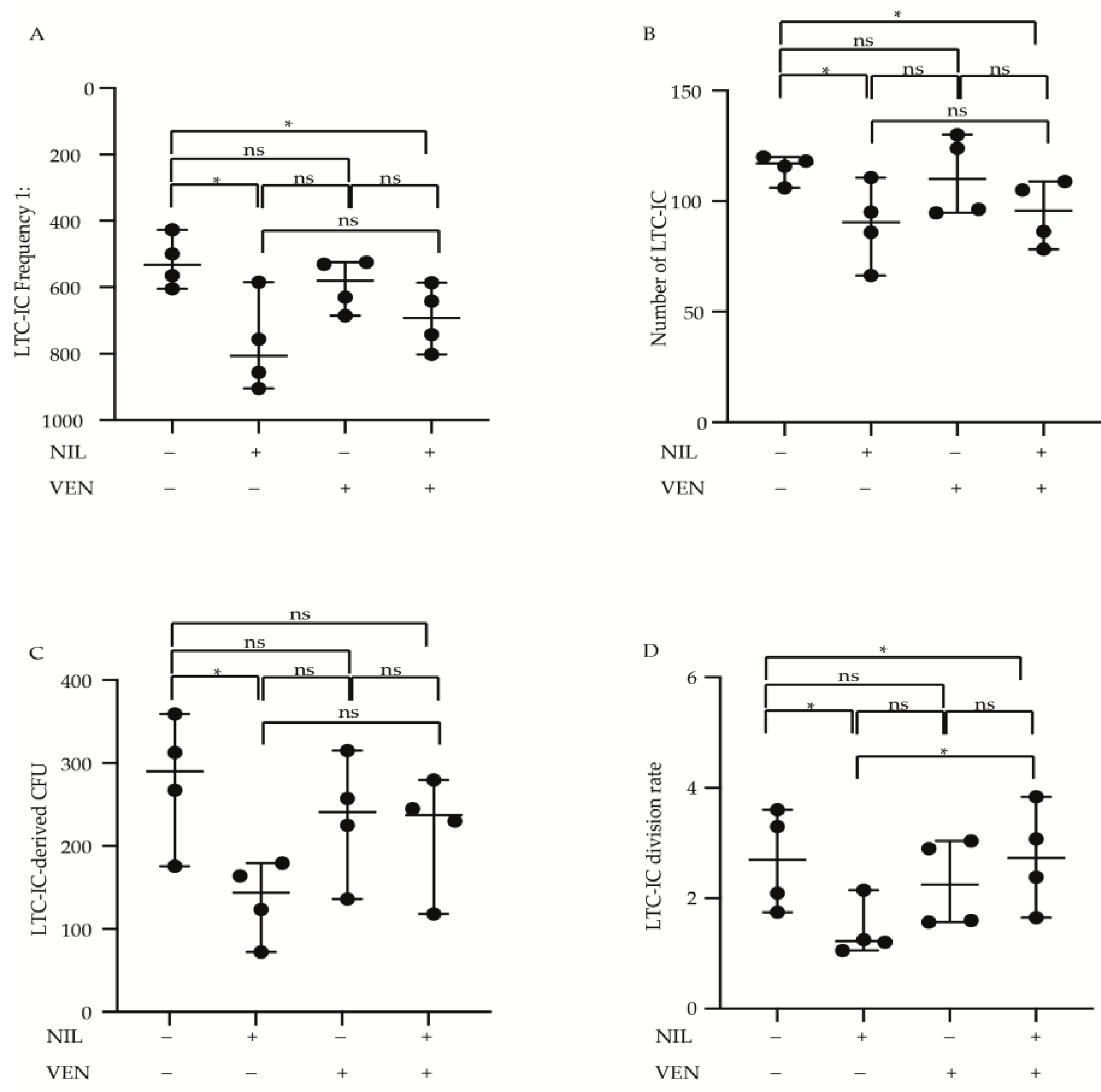
3.4. Primitive CP-CML Cells Are Not Dependent on BCL2 Antiapoptotic Activity for Their Survival
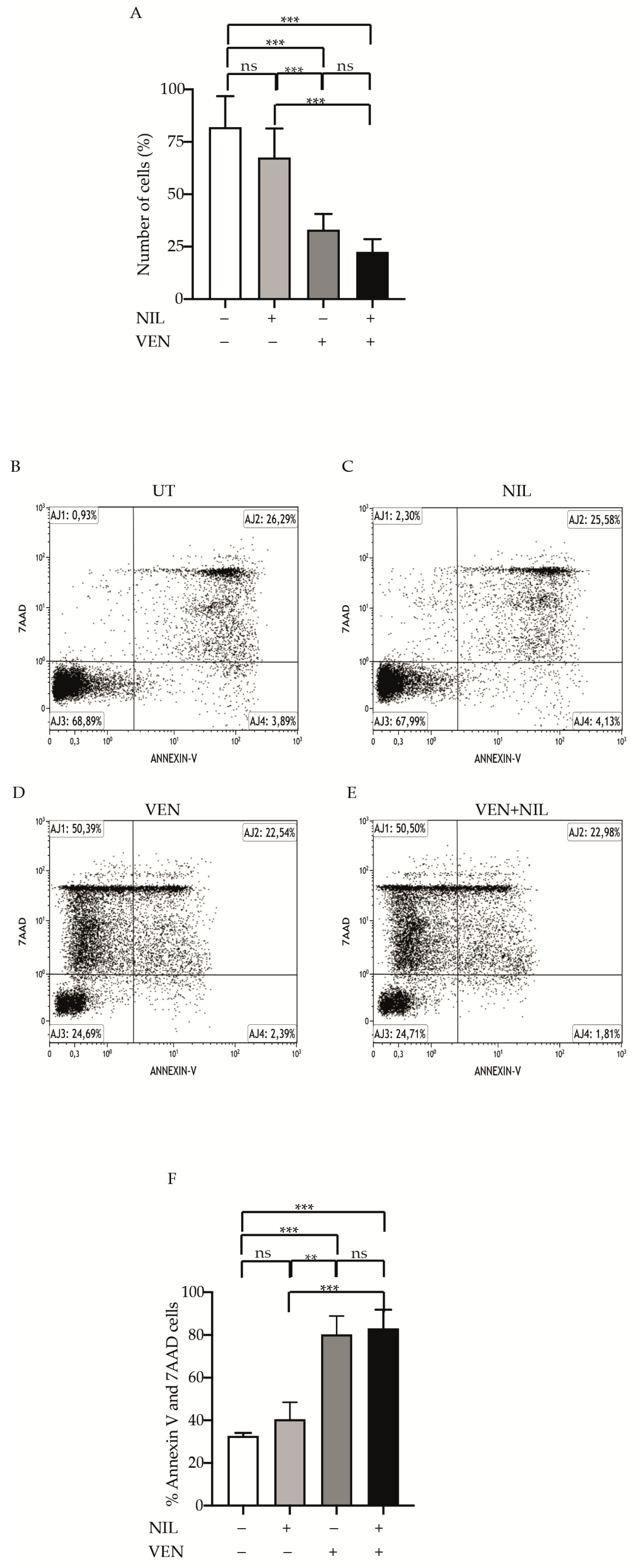
3.5. BCR-ABL1-Positive B-ALL Cells Are Sensitivity to BCL2 Inhibition
3.6. BCL2 Targeting Exercises Antileukemic Activity by Phosphorylation Reduction in Pro-Survival Factor STAT5 in Ph+ Positive B-ALL Cells
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
Ethics Committee or Institutional Review Board Approval
References
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, S.; Vozniak, M.; Rhodes, J.; Forcello, N.; Olszta, D. BCR-ABL1 tyrosine kinase inhibitors for the treatment of chronic myeloid leukemia. J. Oncol. Pharm. Pract. 2018, 24, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Stella, S.; Tirro, E.; Pennisi, M.S.; Vitale, S.R.; Puma, A.; Romano, C.; Di Gregorio, S.; Tomarchio, C.; Di Raimondo, F.; et al. ABL1-Directed Inhibitors for CML: Efficacy, Resistance and Future Perspectives. Anticancer Res. 2020, 40, 2457–2465. [Google Scholar] [CrossRef]
- Massimino, M.; Stella, S.; Tirro, E.; Romano, C.; Pennisi, M.S.; Puma, A.; Manzella, L.; Zanghi, A.; Stagno, F.; Di Raimondo, F.; et al. Non ABL-directed inhibitors as alternative treatment strategies for chronic myeloid leukemia. Mol. Cancer 2018, 17, 56. [Google Scholar] [CrossRef] [PubMed]
- Westerweel, P.E.; Te Boekhorst, P.A.W.; Levin, M.D.; Cornelissen, J.J. New Approaches and Treatment Combinations for the Management of Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 665. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R. Novel approaches to therapy in CML. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lubbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef] [PubMed]
- Lampson, B.L.; Davids, M.S. The Development and Current Use of BCL-2 Inhibitors for the Treatment of Chronic Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2017, 12, 11–19. [Google Scholar] [CrossRef]
- Perini, G.F.; Ribeiro, G.N.; Pinto Neto, J.V.; Campos, L.T.; Hamerschlak, N. BCL-2 as therapeutic target for hematological malignancies. J. Hematol. Oncol. 2018, 11, 65. [Google Scholar] [CrossRef]
- Goff, D.J.; Court Recart, A.; Sadarangani, A.; Chun, H.J.; Barrett, C.L.; Krajewska, M.; Leu, H.; Low-Marchelli, J.; Ma, W.; Shih, A.Y.; et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 2013, 12, 316–328. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Qiu, Y.H.; Post, S.M.; Zhang, Y.; Creighton, C.J.; Cortes, J.; Kornblau, S.M. Reverse phase protein array profiling reveals distinct proteomic signatures associated with chronic myeloid leukemia progression and with chronic phase in the CD34-positive compartment. Cancer 2012, 118, 5283–5292. [Google Scholar] [CrossRef]
- Aichberger, K.J.; Mayerhofer, M.; Krauth, M.T.; Skvara, H.; Florian, S.; Sonneck, K.; Akgul, C.; Derdak, S.; Pickl, W.F.; Wacheck, V.; et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): Evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood 2005, 105, 3303–3311. [Google Scholar] [CrossRef]
- Horita, M.; Andreu, E.J.; Benito, A.; Arbona, C.; Sanz, C.; Benet, I.; Prosper, F.; Fernandez-Luna, J.L. Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-xL. J. Exp. Med. 2000, 191, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.; Barr, P.M.; McAuley, E.M.; Liu, D.; Wiestner, A.; Friedberg, J.W. Fostamatinib inhibits B-cell receptor signaling, cellular activation and tumor proliferation in patients with relapsed and refractory chronic lymphocytic leukemia. Leukemia 2013, 27, 1769–1773. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Okabe, S.; Tauchi, T.; Tanaka, Y.; Ohyashiki, K. Anti-Leukemic Effects of Venetoclax on Philadelphia Chromosome Positive Leukemia Cells. Blood 2016, 128, 5428. [Google Scholar] [CrossRef]
- Maiti, A.; Franquiz, M.J.; Ravandi, F.; Cortes, J.E.; Jabbour, E.J.; Sasaki, K.; Marx, K.; Daver, N.G.; Kadia, T.M.; Konopleva, M.Y.; et al. Venetoclax and BCR-ABL Tyrosine Kinase Inhibitor Combinations: Outcome in Patients with Philadelphia Chromosome-Positive Advanced Myeloid Leukemias. Acta Haematol. 2020, 143, 567–573. [Google Scholar] [CrossRef]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (Venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Zhou, H.; Mak, D.H.; Schober, W.; Leverson, J.D.; Zhang, B.; Bhatia, R.; Huang, X.; et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci. Transl. Med. 2016, 8, 355ra117. [Google Scholar] [CrossRef] [PubMed]
- Stella, S.; Zammit, V.; Vitale, S.R.; Pennisi, M.S.; Massimino, M.; Tirro, E.; Forte, S.; Spitaleri, A.; Antolino, A.; Siracusa, S.; et al. Clinical Implications of Discordant Early Molecular Responses in CML Patients Treated with Imatinib. Int. J. Mol. Sci. 2019, 20, 2226. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Consoli, M.L.; Mesuraca, M.; Stagno, F.; Tirro, E.; Stella, S.; Pennisi, M.S.; Romano, C.; Buffa, P.; Bond, H.M.; et al. IRF5 is a target of BCR-ABL kinase activity and reduces CML cell proliferation. Carcinogenesis 2014, 35, 1132–1143. [Google Scholar] [CrossRef]
- Bradeen, H.A.; Eide, C.A.; O’Hare, T.; Johnson, K.J.; Willis, S.G.; Lee, F.Y.; Druker, B.J.; Deininger, M.W. Comparison of imatinib mesylate, dasatinib (BMS-354825), and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: High efficacy of drug combinations. Blood 2006, 108, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Ponchio, L.; Duma, L.; Oliviero, B.; Gibelli, N.; Pedrazzoli, P.; Robustelli della Cuna, G. Mitomycin C as an alternative to irradiation to inhibit the feeder layer growth in long-term culture assays. Cytotherapy 2000, 2, 281–286. [Google Scholar] [CrossRef]
- Konig, H.; Holtz, M.; Modi, H.; Manley, P.; Holyoake, T.L.; Forman, S.J.; Bhatia, R. Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia 2008, 22, 748–755. [Google Scholar] [CrossRef]
- Liu, M.; Miller, C.L.; Eaves, C.J. Human long-term culture initiating cell assay. Methods Mol. Biol. 2013, 946, 241–256. [Google Scholar] [CrossRef]
- Sutherland, H.J.; Lansdorp, P.M.; Henkelman, D.H.; Eaves, A.C.; Eaves, C.J. Functional characterization of individual human hematopoietic stem cells cultured at limiting dilution on supportive marrow stromal layers. Proc. Natl. Acad. Sci. USA 1990, 87, 3584–3588. [Google Scholar] [CrossRef] [PubMed]
- Pettengell, R.; Luft, T.; Henschler, R.; Hows, J.M.; Dexter, T.M.; Ryder, D.; Testa, N.G. Direct comparison by limiting dilution analysis of long-term culture-initiating cells in human bone marrow, umbilical cord blood, and blood stem cells. Blood 1994, 84, 3653–3659. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Tirro, E.; Stella, S.; Manzella, L.; Pennisi, M.S.; Romano, C.; Vitale, S.R.; Puma, A.; Tomarchio, C.; Di Gregorio, S.; et al. Impact of the Breakpoint Region on the Leukemogenic Potential and the TKI Responsiveness of Atypical BCR-ABL1 Transcripts. Front. Pharmacol. 2021, 12, 669469. [Google Scholar] [CrossRef]
- Heaney, N.B.; Pellicano, F.; Zhang, B.; Crawford, L.; Chu, S.; Kazmi, S.M.; Allan, E.K.; Jorgensen, H.G.; Irvine, A.E.; Bhatia, R.; et al. Bortezomib induces apoptosis in primitive chronic myeloid leukemia cells including LTC-IC and NOD/SCID repopulating cells. Blood 2010, 115, 2241–2250. [Google Scholar] [CrossRef]
- Koller, M.R.; Manchel, I.; Smith, A.K. Quantitative long-term culture-initiating cell assays require accessory cell depletion that can be achieved by CD34-enrichment or 5-fluorouracil exposure. Blood 1998, 91, 4056–4064. [Google Scholar] [CrossRef]
- Chen, S.; Gao, R.; Kobayashi, M.; Yu, H.; Yao, C.; Kapur, R.; Yoder, M.C.; Liu, Y. Pharmacological inhibition of AKT activity in human CD34(+) cells enhances their ability to engraft immunodeficient mice. Exp. Hematol. 2017, 45, 74–84. [Google Scholar] [CrossRef]
- von Palffy, S.; Landberg, N.; Sanden, C.; Zacharaki, D.; Shah, M.; Nakamichi, N.; Hansen, N.; Askmyr, M.; Lilljebjorn, H.; Rissler, M.; et al. A high-content cytokine screen identifies myostatin propeptide as a positive regulator of primitive chronic myeloid leukemia cells. Haematologica 2020, 105, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Buffa, P.; Romano, C.; Pandini, A.; Massimino, M.; Tirro, E.; Di Raimondo, F.; Manzella, L.; Fraternali, F.; Vigneri, P.G. BCR-ABL residues interacting with ponatinib are critical to preserve the tumorigenic potential of the oncoprotein. FASEB J. 2014, 28, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, P.; Stagno, F.; Stella, S.; Cupri, A.; Forte, S.; Massimino, M.; Antolino, A.; Siragusa, S.; Mannina, D.; Impera, S.S.; et al. High BCR-ABL/GUS(IS) Levels at Diagnosis of Chronic Phase CML Are Associated with Unfavorable Responses to Standard-Dose Imatinib. Clin. Cancer Res. 2017, 23, 7189–7198. [Google Scholar] [CrossRef]
- Chandrasekhar, C.; Kumar, P.S.; Sarma, P. Novel mutations in the kinase domain of BCR-ABL gene causing imatinib resistance in chronic myeloid leukemia patients. Sci. Rep. 2019, 9, 2412. [Google Scholar] [CrossRef]
- Castagnetti, F.; Gugliotta, G.; Breccia, M.; Iurlo, A.; Levato, L.; Albano, F.; Vigneri, P.; Abruzzese, E.; Rossi, G.; Rupoli, S.; et al. The BCR-ABL1 transcript type influences response and outcome in Philadelphia chromosome-positive chronic myeloid leukemia patients treated frontline with imatinib. Am. J. Hematol. 2017, 92, 797–805. [Google Scholar] [CrossRef]
- Loscocco, F.; Visani, G.; Galimberti, S.; Curti, A.; Isidori, A. BCR-ABL Independent Mechanisms of Resistance in Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 939. [Google Scholar] [CrossRef] [PubMed]
- Jordanides, N.E.; Jorgensen, H.G.; Holyoake, T.L.; Mountford, J.C. Functional ABCG2 is overexpressed on primary CML CD34+ cells and is inhibited by imatinib mesylate. Blood 2006, 108, 1370–1373. [Google Scholar] [CrossRef]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Venables, A.; Zrim, S.; Zannettino, A.; Lynch, K.; Manley, P.W.; Hughes, T. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: Higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood 2007, 110, 4064–4072. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Nhiayi, M.K.; Tse, E.; Cheng, J.; Massimino, M.; Durden, D.L.; Vigneri, P.; Wang, J.Y. Knockout Serum Replacement Promotes Cell Survival by Preventing BIM from Inducing Mitochondrial Cytochrome C Release. PLoS ONE 2015, 10, e0140585. [Google Scholar] [CrossRef]
- Morotti, A.; Panuzzo, C.; Fava, C.; Saglio, G. Kinase-inhibitor-insensitive cancer stem cells in chronic myeloid leukemia. Expert. Opin. Biol. Ther. 2014, 14, 287–299. [Google Scholar] [CrossRef]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Tirro, E.; Stella, S.; Frasca, F.; Vella, V.; Sciacca, L.; Pennisi, M.S.; Vitale, S.R.; Puma, A.; Romano, C.; et al. Effect of Combined Epigenetic Treatments and Ectopic NIS Expression on Undifferentiated Thyroid Cancer Cells. Anticancer Res. 2018, 38, 6653–6662. [Google Scholar] [CrossRef]
- Baudino, T.A. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef]
- Peters, G.J. From ‘Targeted Therapy’ to Targeted Therapy. Anticancer Res. 2019, 39, 3341–3345. [Google Scholar] [CrossRef] [PubMed]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019, 11, 1758835919830826. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Kizilbash, S.H.; Daniels, D.J.; Sarkaria, J.N. Editorial: Targeted Therapies for Glioblastoma: A Critical Appraisal. Front. Oncol. 2019, 9, 1216. [Google Scholar] [CrossRef] [PubMed]
- Masoud, V.; Pages, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef]
- Manzella, L.; Massimino, M.; Stella, S.; Tirro, E.; Pennisi, M.S.; Martorana, F.; Motta, G.; Vitale, S.R.; Puma, A.; Romano, C.; et al. Activation of the IGF Axis in Thyroid Cancer: Implications for Tumorigenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 3258. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.J.F.; Davids, M.S. Breaking through BCL-2 inhibition in CLL. Blood 2020, 135, 709–711. [Google Scholar] [CrossRef]
- McBride, A.; Houtmann, S.; Wilde, L.; Vigil, C.; Eischen, C.M.; Kasner, M.; Palmisiano, N. The Role of Inhibition of Apoptosis in Acute Leukemias and Myelodysplastic Syndrome. Front. Oncol. 2019, 9, 192. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Ko, T.K.; Chuah, C.; Huang, J.; Ng, K.P.; Ong, S.T. The BCL-2 Inhibitor ABT-199 Enhances Imatinib-Induced Cell Death In Chronic Phase CML Progenitors. Blood 2013, 122, 3978. [Google Scholar] [CrossRef]
- Inoue, C.; Sobue, S.; Aoyama, Y.; Mizutani, N.; Kawamoto, Y.; Nishizawa, Y.; Ichihara, M.; Abe, A.; Hayakawa, F.; Suzuki, M.; et al. BCL2 inhibitor ABT-199 and JNK inhibitor SP600125 exhibit synergistic cytotoxicity against imatinib-resistant Ph+ ALL cells. Biochem. Biophys. Rep. 2018, 15, 69–75. [Google Scholar] [CrossRef]
- Ko, T.K.; Chuah, C.T.; Huang, J.W.; Ng, K.P.; Ong, S.T. The BCL2 inhibitor ABT-199 significantly enhances imatinib-induced cell death in chronic myeloid leukemia progenitors. Oncotarget 2014, 5, 9033–9038. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Tirro, E.; Stella, S.; Pennisi, M.S.; Vitale, S.R.; Puma, A.; Romano, C.; Di Gregorio, S.; Romeo, M.A.; Di Raimondo, F.; et al. Targeting BCL-2 as a Therapeutic Strategy for Primary (p210)BCR-ABL1-positive B-ALL Cells. In Vivo 2020, 34, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garcia, I.; Grutz, G. Tumorigenic activity of the BCR-ABL oncogenes is mediated by BCL2. Proc. Natl. Acad. Sci. USA 1995, 92, 5287–5291. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; McGahon, A.J.; Nishioka, W.K.; Afar, D.E.; Witte, O.N.; Green, D.R. Bcl-2-independent Bcr-Abl-mediated resistance to apoptosis: Protection is correlated with up regulation of Bcl-xL. Oncogene 1998, 16, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Malagola, M.; Papayannidis, C.; Baccarani, M. Tyrosine kinase inhibitors in Ph+ acute lymphoblastic leukaemia: Facts and perspectives. Ann. Hematol. 2016, 95, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Jin, S.; Li, X.; Wang, D. The involvement of Bcl-2 family proteins in AKT-regulated cell survival in cisplatin resistant epithelial ovarian cancer. Oncotarget 2017, 8, 1354–1368. [Google Scholar] [CrossRef]
- Asnaghi, L.; Calastretti, A.; Bevilacqua, A.; D’Agnano, I.; Gatti, G.; Canti, G.; Delia, D.; Capaccioli, S.; Nicolin, A. Bcl-2 phosphorylation and apoptosis activated by damaged microtubules require mTOR and are regulated by Akt. Oncogene 2004, 23, 5781–5791. [Google Scholar] [CrossRef] [PubMed]
- Mortenson, M.M.; Galante, J.G.; Gilad, O.; Schlieman, M.G.; Virudachalam, S.; Kung, H.J.; Bold, R.J. BCL-2 functions as an activator of the AKT signaling pathway in pancreatic cancer. J. Cell Biochem. 2007, 102, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Miskimen, K.L.; Wang, Z.; Xie, X.Y.; Brenzovich, J.; Ryan, J.J.; Tse, W.; Moriggl, R.; Bunting, K.D. STAT5 requires the N-domain for suppression of miR15/16, induction of bcl-2, and survival signaling in myeloproliferative disease. Blood 2010, 115, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Waibel, M.; Solomon, V.S.; Knight, D.A.; Ralli, R.A.; Kim, S.K.; Banks, K.M.; Vidacs, E.; Virely, C.; Sia, K.C.; Bracken, L.S.; et al. Combined targeting of JAK2 and Bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Rep. 2013, 5, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Simpson, H.M.; Furusawa, A.; Sadashivaiah, K.; Civin, C.I.; Banerjee, A. STAT5 inhibition induces TRAIL/DR4 dependent apoptosis in peripheral T-cell lymphoma. Oncotarget 2018, 9, 16792–16806. [Google Scholar] [CrossRef][Green Version]
- Zhang, J.; Gao, X.; Schmit, F.; Adelmant, G.; Eck, M.J.; Marto, J.A.; Zhao, J.J.; Roberts, T.M. CRKL Mediates p110beta-Dependent PI3K Signaling in PTEN-Deficient Cancer Cells. Cell Rep. 2017, 20, 549–557. [Google Scholar] [CrossRef]
- Ota, J.; Kimura, F.; Sato, K.; Wakimoto, N.; Nakamura, Y.; Nagata, N.; Suzu, S.; Yamada, M.; Shimamura, S.; Motoyoshi, K. Association of CrkL with STAT5 in hematopoietic cells stimulated by granulocyte-macrophage colony-stimulating factor or erythropoietin. Biochem. Biophys. Res. Commun. 1998, 252, 779–786. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massimino, M.; Vigneri, P.; Stella, S.; Tirrò, E.; Pennisi, M.S.; Parrinello, L.N.; Vetro, C.; Manzella, L.; Stagno, F.; Di Raimondo, F. Combined Inhibition of Bcl2 and Bcr-Abl1 Exercises Anti-Leukemia Activity but Does Not Eradicate the Primitive Leukemic Cells. J. Clin. Med. 2021, 10, 5606. https://doi.org/10.3390/jcm10235606
Massimino M, Vigneri P, Stella S, Tirrò E, Pennisi MS, Parrinello LN, Vetro C, Manzella L, Stagno F, Di Raimondo F. Combined Inhibition of Bcl2 and Bcr-Abl1 Exercises Anti-Leukemia Activity but Does Not Eradicate the Primitive Leukemic Cells. Journal of Clinical Medicine. 2021; 10(23):5606. https://doi.org/10.3390/jcm10235606
Chicago/Turabian StyleMassimino, Michele, Paolo Vigneri, Stefania Stella, Elena Tirrò, Maria Stella Pennisi, Laura Nunziatina Parrinello, Calogero Vetro, Livia Manzella, Fabio Stagno, and Francesco Di Raimondo. 2021. "Combined Inhibition of Bcl2 and Bcr-Abl1 Exercises Anti-Leukemia Activity but Does Not Eradicate the Primitive Leukemic Cells" Journal of Clinical Medicine 10, no. 23: 5606. https://doi.org/10.3390/jcm10235606
APA StyleMassimino, M., Vigneri, P., Stella, S., Tirrò, E., Pennisi, M. S., Parrinello, L. N., Vetro, C., Manzella, L., Stagno, F., & Di Raimondo, F. (2021). Combined Inhibition of Bcl2 and Bcr-Abl1 Exercises Anti-Leukemia Activity but Does Not Eradicate the Primitive Leukemic Cells. Journal of Clinical Medicine, 10(23), 5606. https://doi.org/10.3390/jcm10235606