
Shedding Light on Targeting Chronic Myeloid Leukemia Stem Cells

 , , , ,
, , , ,
Abstract
1. Introduction
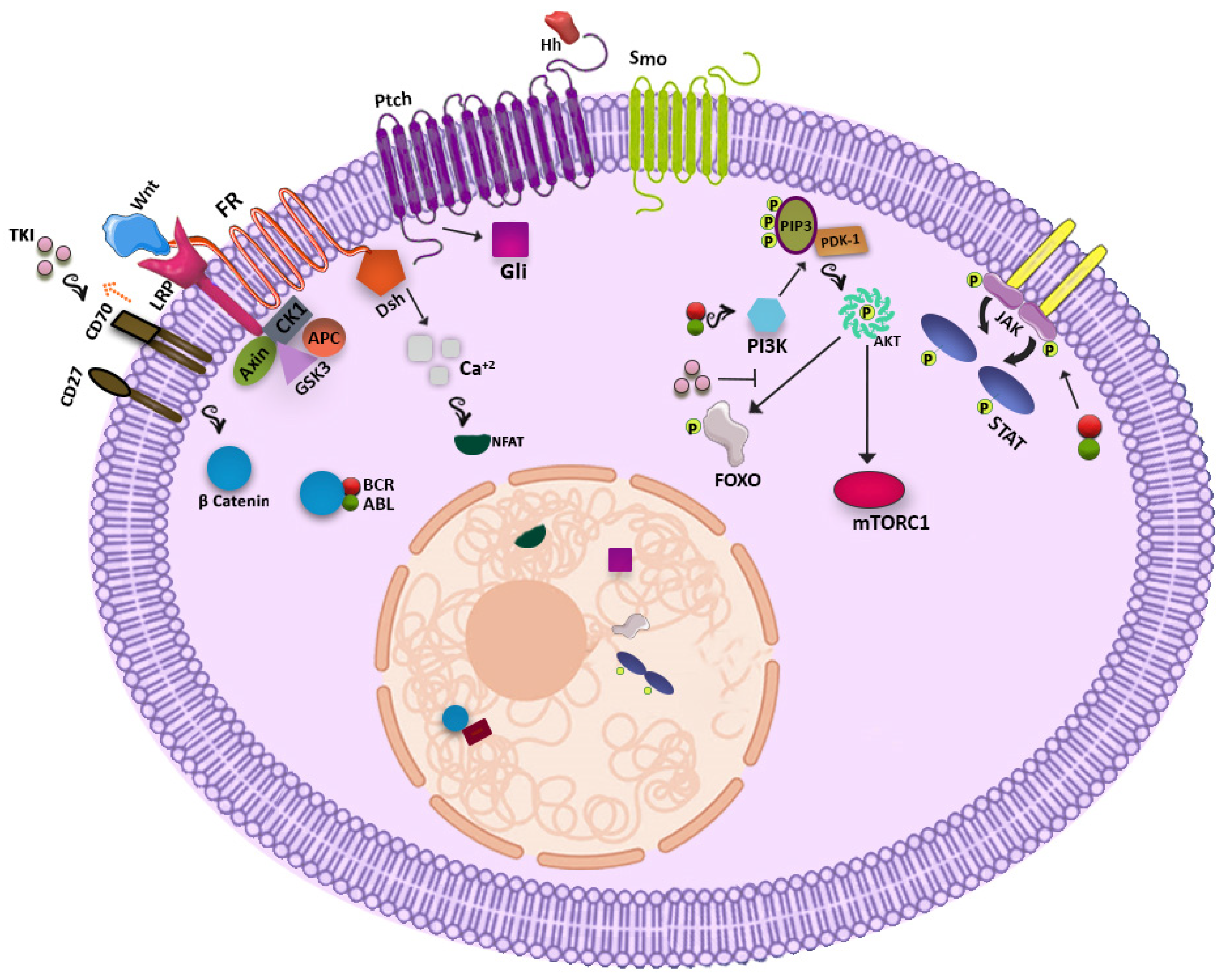
2. Metabolic Pathways Potentially Useful for Targeting CML LSCs
2.1. WNT Signaling Pathway
2.2. Hedgehog Signaling Pathway
2.3. PI3K-AKT Pathway
2.4. JAK-STAT Pathway
2.5. Other Players
2.5.1. Blk
2.5.2. EZH2
2.5.3. Fap-1
2.5.4. HIF-1
2.5.5. PML
2.5.6. PP2A
2.5.7. ALOX5
2.5.8. SIRT1
2.5.9. microRNAs
3. The Environment
Bone Marrow Niche
4. Phenotypic Differences
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saglio, G.; Gale, R.P. Prospects for achieving treatment-free remission in chronic myeloid leukaemia. Br. J. Haematol. 2020, 190, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef]
- Zhang, B.; Li, M.; McDonald, T.; Holyoake, T.L.; Moon, R.T.; Campana, D.; Shultz, L.; Bhatia, R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 2013, 121, 1824–1838. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zang, S.; Liu, Y.; Wang, Y.; Li, W.; Liu, Q.; Ji, M.; Ma, D.; Ji, C. FZD7 regulates BMSCs-mediated protection of CML cells. Oncotarget 2016, 7, 6175–6187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Liu, Q.; Andreeff, M.; Carter, B.Z. Combined inhibition of beta-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065–2074. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, Y.; Douglas, L.; Li, S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 2009, 23, 109–116. [Google Scholar] [CrossRef]
- Hanna, A.; Shevde, L.A. Hedgehog signaling: Modulation of cancer properies and tumor mircroenvironment. Mol. Cancer 2016, 15, 24. [Google Scholar] [CrossRef]
- Su, W.; Meng, F.; Huang, L.; Zheng, M.; Liu, W.; Sun, H. Sonic hedgehog maintains survival and growth of chronic myeloid leukemia progenitor cells through beta-catenin signaling. Exp. Hematol. 2012, 40, 418–427. [Google Scholar] [CrossRef]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef]
- Cea, M.; Cagnetta, A.; Cirmena, G.; Garuti, A.; Rocco, I.; Palermo, C.; Pierri, I.; Reverberi, D.; Nencioni, A.; Ballestrero, A.; et al. Tracking molecular relapse of chronic myeloid leukemia by measuring Hedgehog signaling status. Leuk. Lymphoma 2013, 54, 342–352. [Google Scholar] [CrossRef]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; Grimaldi, C.; Cappellini, A.; Ognibene, A.; McCubrey, J.A. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim. Biophys. Acta 2010, 1803, 991–1002. [Google Scholar] [CrossRef]
- Pellicano, F.; Scott, M.T.; Helgason, G.V.; Hopcroft, L.E.; Allan, E.K.; Aspinall-O'Dea, M.; Copland, M.; Pierce, A.; Huntly, B.J.; Whetton, A.D.; et al. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem. Cells 2014, 32, 2324–2337. [Google Scholar] [CrossRef]
- Airiau, K.; Mahon, F.X.; Josselin, M.; Jeanneteau, M.; Belloc, F. PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor. Cell Death Dis. 2013, 4, e827. [Google Scholar] [CrossRef]
- Chai, S.K.; Nichols, G.L.; Rothman, P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J. Immunol. 1997, 159, 4720–4728. [Google Scholar]
- Xie, S.; Wang, Y.; Liu, J.; Sun, T.; Wilson, M.B.; Smithgall, T.E.; Arlinghaus, R.B. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene 2001, 20, 6188–6195. [Google Scholar] [CrossRef]
- Samanta, A.; Perazzona, B.; Chakraborty, S.; Sun, X.; Modi, H.; Bhatia, R.; Priebe, W.; Arlinghaus, R. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia 2011, 25, 463–472. [Google Scholar] [CrossRef]
- Madapura, H.S.; Nagy, N.; Ujvari, D.; Kallas, T.; Krohnke, M.C.L.; Amu, S.; Bjorkholm, M.; Stenke, L.; Mandal, P.K.; McMurray, J.S.; et al. Interferon gamma is a STAT1-dependent direct inducer of BCL6 expression in imatinib-treated chronic myeloid leukemia cells. Oncogene 2017, 36, 4619–4628. [Google Scholar] [CrossRef]
- Patel, S.B.; Nemkov, T.; Stefanoni, D.; Benavides, G.A.; Bassal, M.A.; Crown, B.L.; Matkins, V.R.; Camacho, V.; Kuznetsova, V.; Hoang, A.T.; et al. Metabolic alterations mediated by STAT3 promotes drug persistence in CML. Leukemia 2021. [Google Scholar] [CrossRef]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef]
- Zhang, H.; Peng, C.; Hu, Y.; Li, H.; Sheng, Z.; Chen, Y.; Sullivan, C.; Cerny, J.; Hutchinson, L.; Higgins, A.; et al. The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat. Genet. 2012, 44, 861–871. [Google Scholar] [CrossRef]
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016, 6, 1248–1257. [Google Scholar] [CrossRef]
- Huang, W.; Luan, C.H.; Hjort, E.E.; Bei, L.; Mishra, R.; Sakamoto, K.M.; Platanias, L.C.; Eklund, E.A. The role of Fas-associated phosphatase 1 in leukemia stem cell persistence during tyrosine kinase inhibitor treatment of chronic myeloid leukemia. Leukemia 2016, 30, 1502–1509. [Google Scholar] [CrossRef]
- Cheloni, G.; Tanturli, M.; Tusa, I.; Ho DeSouza, N.; Shan, Y.; Gozzini, A.; Mazurier, F.; Rovida, E.; Li, S.; Dello Sbarba, P. Targeting chronic myeloid leukemia stem cells with the hypoxia-inducible factor inhibitor acriflavine. Blood 2017, 130, 655–665. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef] [PubMed]
- Kakizuka, A.; Miller, W.H., Jr.; Umesono, K.; Warrell, R.P., Jr.; Frankel, S.R.; Murty, V.V.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991, 66, 663–674. [Google Scholar] [CrossRef]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef]
- Hong, C.S.; Ho, W.; Zhang, C.; Yang, C.; Elder, J.B.; Zhuang, Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 2015, 16, 821–833. [Google Scholar] [CrossRef]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Invest. 2013, 123, 4144–4157. [Google Scholar] [CrossRef]
- Lai, D.; Chen, M.; Su, J.; Liu, X.; Rothe, K.; Hu, K.; Forrest, D.L.; Eaves, C.J.; Morin, G.B.; Jiang, X. PP2A inhibition sensitizes cancer stem cells to ABL tyrosine kinase inhibitors in BCR-ABL(+) human leukemia. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Massoumi, R.; Sjolander, A. The role of leukotriene receptor signaling in inflammation and cancer. Sci. World J. 2007, 7, 1413–1421. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Zhang, H.; Peng, C.; Li, S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat. Genet. 2009, 41, 783–792. [Google Scholar] [CrossRef]
- Dolinska, M.; Piccini, A.; Wong, W.M.; Gelali, E.; Johansson, A.S.; Klang, J.; Xiao, P.; Yektaei-Karin, E.; Stromberg, U.O.; Mustjoki, S.; et al. Leukotriene signaling via ALOX5 and cysteinyl leukotriene receptor 1 is dispensable for in vitro growth of CD34(+)CD38(-) stem and progenitor cells in chronic myeloid leukemia. Biochem. Biophys. Res. Commun. 2017, 490, 378–384. [Google Scholar] [CrossRef]
- Rahman, S.; Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell Commun. Signal. 2011, 9, 11. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, Z.; Li, L.; Zhang, H.; Modi, H.; Horne, D.; Bhatia, R.; Chen, W. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 2012, 119, 1904–1914. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, H.; Roth, M.; Stark, J.M.; Bhatia, R.; Chen, W.Y. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene 2013, 32, 589–598. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef]
- Houshmand, M.; Nakhlestani Hagh, M.; Soleimani, M.; Hamidieh, A.A.; Abroun, S.; Nikougoftar Zarif, M. MicroRNA Microarray Profiling during Megakaryocyte Differentiation of Cord Blood CD133+ Hematopoietic Stem Cells. Cell J. 2018, 20, 195–203. [Google Scholar] [CrossRef]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A brief review on the mechanisms of miRNA regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Zhang, B.; Nguyen, L.X.T.; Li, L.; Zhao, D.; Kumar, B.; Wu, H.; Lin, A.; Pellicano, F.; Hopcroft, L.; Su, Y.L.; et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 2018, 24, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem. Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Wang, W.Z.; Pu, Q.H.; Lin, X.H.; Liu, M.Y.; Wu, L.R.; Wu, Q.Q.; Chen, Y.H.; Liao, F.F.; Zhu, J.Y.; Jin, X.B. Silencing of miR-21 sensitizes CML CD34+ stem/progenitor cells to imatinib-induced apoptosis by blocking PI3K/AKT pathway. Leuk. Res. 2015, 39, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, L.; Zhao, M.; Zhu, S.; Kang, R.; Vernon, P.; Tang, D.; Cao, L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012, 26, 1752–1760. [Google Scholar] [CrossRef]
- Houshmand, M.; Blanco, T.M.; Circosta, P.; Yazdi, N.; Kazemi, A.; Saglio, G.; Zarif, M.N. Bone marrow microenvironment: The guardian of leukemia stem cells. World J. Stem. Cells 2019, 11, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Tabe, Y.; Konoplev, S.; Xu, Y.; Leysath, C.E.; Lu, H.; Kimura, S.; Ohsaka, A.; Rios, M.B.; Calvert, L.; et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol. Cancer Ther. 2008, 7, 48–58. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, Y.; Smith, C.; Gasparetto, M.; Turhan, A.; Eaves, A.; Eaves, C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia 2007, 21, 926–935. [Google Scholar] [CrossRef]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rulicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef]
- Weisberg, E.; Azab, A.K.; Manley, P.W.; Kung, A.L.; Christie, A.L.; Bronson, R.; Ghobrial, I.M.; Griffin, J.D. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 2012, 26, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Saieva, L.; Raimondo, S.; Santoro, A.; De Leo, G.; Alessandro, R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J. Cell Mol. Med. 2016, 20, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Grockowiak, E.; Laperrousaz, B.; Jeanpierre, S.; Voeltzel, T.; Guyot, B.; Gobert, S.; Nicolini, F.E.; Maguer-Satta, V. Immature CML cells implement a BMP autocrine loop to escape TKI treatment. Blood 2017, 130, 2860–2871. [Google Scholar] [CrossRef] [PubMed]
- Traer, E.; Javidi-Sharifi, N.; Agarwal, A.; Dunlap, J.; English, I.; Martinez, J.; Tyner, J.W.; Wong, M.; Druker, B.J. Ponatinib overcomes FGF2-mediated resistance in CML patients without kinase domain mutations. Blood 2014, 123, 1516–1524. [Google Scholar] [CrossRef]
- Bourgeais, J.; Ishac, N.; Medrzycki, M.; Brachet-Botineau, M.; Desbourdes, L.; Gouilleux-Gruart, V.; Pecnard, E.; Rouleux-Bonnin, F.; Gyan, E.; Domenech, J.; et al. Oncogenic STAT5 signaling promotes oxidative stress in chronic myeloid leukemia cells by repressing antioxidant defenses. Oncotarget 2017, 8, 41876–41889. [Google Scholar] [CrossRef]
- Aggoune, D.; Tosca, L.; Sorel, N.; Bonnet, M.L.; Dkhissi, F.; Tachdjian, G.; Bennaceur-Griscelli, A.; Chomel, J.C.; Turhan, A.G. Modeling the influence of stromal microenvironment in the selection of ENU-induced BCR-ABL1 mutants by tyrosine kinase inhibitors. Oncoscience 2014, 1, 57–68. [Google Scholar] [CrossRef][Green Version]
- Aggoune, D.; Sorel, N.; Bonnet, M.L.; Goujon, J.M.; Tarte, K.; Herault, O.; Domenech, J.; Rea, D.; Legros, L.; Johnson-Ansa, H.; et al. Bone marrow mesenchymal stromal cell (MSC) gene profiling in chronic myeloid leukemia (CML) patients at diagnosis and in deep molecular response induced by tyrosine kinase inhibitors (TKIs). Leuk. Res. 2017, 60, 94–102. [Google Scholar] [CrossRef]
- Civini, S.; Jin, P.; Ren, J.; Sabatino, M.; Castiello, L.; Jin, J.; Wang, H.; Zhao, Y.; Marincola, F.; Stroncek, D. Leukemia cells induce changes in human bone marrow stromal cells. J. Transl. Med. 2013, 11, 298. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem. Cell 2016, 19, 23–37. [Google Scholar] [CrossRef]
- Eisterer, W.; Jiang, X.; Christ, O.; Glimm, H.; Lee, K.H.; Pang, E.; Lambie, K.; Shaw, G.; Holyoake, T.L.; Petzer, A.L.; et al. Different subsets of primary chronic myeloid leukemia stem cells engraft immunodeficient mice and produce a model of the human disease. Leukemia 2005, 19, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Cerny-Reiterer, S.; Gleixner, K.V.; Blatt, K.; Herndlhofer, S.; Rabitsch, W.; Jager, E.; Mitterbauer-Hohendanner, G.; Streubel, B.; Selzer, E.; et al. CD34(+)/CD38(-) stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica 2012, 97, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Landberg, N.; von Palffy, S.; Askmyr, M.; Lilljebjorn, H.; Sanden, C.; Rissler, M.; Mustjoki, S.; Hjorth-Hansen, H.; Richter, J.; Agerstam, H.; et al. CD36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica 2018, 103, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Sadovnik, I.; Hoelbl-Kovacic, A.; Herrmann, H.; Eisenwort, G.; Cerny-Reiterer, S.; Warsch, W.; Hoermann, G.; Greiner, G.; Blatt, K.; Peter, B.; et al. Identification of CD25 as STAT5-Dependent Growth Regulator of Leukemic Stem Cells in Ph+ CML. Clin. Cancer Res. 2016, 22, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Landberg, N.; Hansen, N.; Askmyr, M.; Agerstam, H.; Lassen, C.; Rissler, M.; Hjorth-Hansen, H.; Mustjoki, S.; Jaras, M.; Richter, J.; et al. IL1RAP expression as a measure of leukemic stem cell burden at diagnosis of chronic myeloid leukemia predicts therapy outcome. Leukemia 2016, 30, 253–257. [Google Scholar] [CrossRef]
- Zhao, K.; Yin, L.L.; Zhao, D.M.; Pan, B.; Chen, W.; Cao, J.; Cheng, H.; Li, Z.Y.; Li, D.P.; Sang, W.; et al. IL1RAP as a surface marker for leukemia stem cells is related to clinical phase of chronic myeloid leukemia patients. Int. J. Clin. Exp. Med. 2014, 7, 4787–4798. [Google Scholar]
- Matteucci, E.; Giampietro, O. Dipeptidyl peptidase-4 (CD26): Knowing the function before inhibiting the enzyme. Curr. Med. Chem. 2009, 16, 2943–2951. [Google Scholar] [CrossRef]
- Houshmand, M.; Garello, F.; Stefania, R.; Gaidano, V.; Cignetti, A.; Spinelli, M.; Fava, C.; Nikougoftar Zarif, M.; Galimberti, S.; Pungolino, E.; et al. Targeting Chronic Myeloid Leukemia Stem/Progenitor Cells Using Venetoclax-Loaded Immunoliposome. Cancers 2021, 13, 1311. [Google Scholar] [CrossRef]
- Bocchia, M.; Sicuranza, A.; Abruzzese, E.; Iurlo, A.; Sirianni, S.; Gozzini, A.; Galimberti, S.; Aprile, L.; Martino, B.; Pregno, P.; et al. Residual Peripheral Blood CD26(+) Leukemic Stem Cells in Chronic Myeloid Leukemia Patients During TKI Therapy and During Treatment-Free Remission. Front. Oncol 2018, 8, 194. [Google Scholar] [CrossRef]
- Culen, M.; Borsky, M.; Nemethova, V.; Razga, F.; Smejkal, J.; Jurcek, T.; Dvorakova, D.; Zackova, D.; Weinbergerova, B.; Semerad, L.; et al. Quantitative assessment of the CD26+ leukemic stem cell compartment in chronic myeloid leukemia: Patient-subgroups, prognostic impact, and technical aspects. Oncotarget 2016, 7, 33016–33024. [Google Scholar] [CrossRef]
- Kinstrie, R.; Horne, G.A.; Morrison, H.; Irvine, D.; Munje, C.; Castaneda, E.G.; Moka, H.A.; Dunn, K.; Cassels, J.E.; Parry, N.; et al. CD93 is expressed on chronic myeloid leukemia stem cells and identifies a quiescent population which persists after tyrosine kinase inhibitor therapy. Leukemia 2020, 34, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Riether, C.; Radpour, R.; Kallen, N.M.; Burgin, D.T.; Bachmann, C.; Schurch, C.M.; Luthi, U.; Arambasic, M.; Hoppe, S.; Albers, C.E.; et al. Metoclopramide treatment blocks CD93-signaling-mediated self-renewal of chronic myeloid leukemia stem cells. Cell Rep. 2021, 34, 108663. [Google Scholar] [CrossRef]
- Sadovnik, I.; Herrmann, H.; Eisenwort, G.; Blatt, K.; Hoermann, G.; Mueller, N.; Sperr, W.R.; Valent, P. Expression of CD25 on leukemic stem cells in BCR-ABL1(+) CML: Potential diagnostic value and functional implications. Exp. Hematol. 2017, 51, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Sadovnik, I.; Eisenwort, G.; Rulicke, T.; Blatt, K.; Herndlhofer, S.; Willmann, M.; Stefanzl, G.; Baumgartner, S.; Greiner, G.; et al. Delineation of target expression profiles in CD34+/CD38- and CD34+/CD38+ stem and progenitor cells in AML and CML. Blood Adv. 2020, 4, 5118–5132. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Eng, W.S.; Hu, W.; Khalaj, M.; Garrett-Bakelman, F.E.; Tavakkoli, M.; Levine, R.L.; Carroll, M.; Klimek, V.M.; Melnick, A.M.; et al. CD99 is a therapeutic target on disease stem cells in myeloid malignancies. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem. Cell 2010, 7, 708–717. [Google Scholar] [CrossRef]


{kind=link}
| CML Clinical Trial with New Therapeutic Agents (Non-TKIs) | |||||
|---|---|---|---|---|---|
| Generic Name | Brand Name | Clinical Trial Identifier | Target | Start Date | Status |
| Sirolimus | Rapamune | NCT00101088 | mTOR inhibitors | 10-Jan-05 | Terminated |
| Sorafenib | Nexavar | NCT00661180 | Multikinase inhibitors, VEGF/VEGFR inhibitors | 18-Apr-08 | Completed |
| Sunitinib | Sutent | NCT00387426 | Multikinase inhibitors, VEGF/VEGFR inhibitors | 13-Oct-06 | Completed |
| Ruxolitinib | Jakafi | NCT02253277 | JAK/STAT inhibitors | 1-Oct-14 | Completed |
| Axitinib | Inlyta | NCT02782403 | Multikinase inhibitors, VEGF/VEGFR inhibitors | 25-May-16 | Terminated |
| Ibrutinib | Imbruvica | NCT03267186 | BTK inhibitors | 30-Aug-17 | Ongoing |
| Midostaurin | Rydapt | NCT02115295 | Multikinase inhibitors | 16-Apr-14 | Ongoing |
| PRI-724 | - | NCT01606579 | Wnt/β-catenin inhibitors | 25-May-12 | Completed |
| BP1001 | - | NCT02923986 | Grb2 | 5-Oct-16 | Withdrawn |
| Tipifarnib | Zarnestra | NCT00040105 | Farnesyl transferase | 21-Jun-02 | Completed |
| Lonafarnib | SCH66336 | NCT00047502 | Farnesyl transferase | 9-Oct-02 | Completed |
| Rapamycin | Sirolimus | NCT00780104 | mTOR | 27-Oct-08 | Completed |
| RAD001 | Everolimus | NCT01188889 | mTOR | 26-Aug-10 | Withdrawn |
| Panobinostat | LBH589 | NCT00451035 | Histone deacetylase | 22-Mar-07 | Terminated |
| Azacytidine | Vidaza | NCT03895671 | Hypomethylating agents | 29-Mar-19 | Ongoing |
| MK-0457 | Tozasertib | NCT00405054 | Aurora kinase pathway inhibitors | 29-Nov-06 | Terminated |
| Venetoclax | Venclexta | NCT02689440 | BCL-2 inhibitors | 24-Feb-16 | Ongoing |
| Temsirolimus | Torisel | NCT00101088 | mTOR | 10-Jan-05 | Terminated |
| Abemaciclib | Verzenio | NCT03878524 | CDK 4/6 inhibitors | 18-Mar-19 | Ongoing |
| Alemtuzumab | Lemtrada/campath | NCT00626626 | CD52 monoclonal antibodies | 29-Feb-08 | Terminated |
| Bevacizumab | Avastin | NCT00023920 | VEGF/VEGFR inhibitors | 27-Jan-03 | Terminated |
| Blinatumomab | Blincyto | NCT02790515 | Miscellaneous antineoplastic | 6-Jun-16 | Ongoing |
| Ipilimumab | Yervoy | NCT00732186 | Anti-CTLA-4 monoclonal antibodies | 11-Aug-08 | Withdrawn |
| Nivolumab | Opdivo | NCT02011945 | Anti-PD-1 monoclonal antibodies | 16-Dec-13 | Completed |
| Rituximab | Rituxan | NCT03455517 | Antirheumatics, CD20 monoclonal antibodies | 6-Mar-18 | Terminated |
| Marker Name | CD Name | CML Cells | Normal Cells | References | ||
|---|---|---|---|---|---|---|
| LSCs | Progenitor Cells | Stem Cells | Progenitor Cells | |||
| CD34+/CD38− | CD34+/CD38+ | CD34+/CD38− | CD34+/CD38+ | |||
| IL-2Rα | CD25 | + | +/− | − | +/− | [75] |
| DPPIV | CD26 | + | +/− | − | − | [53] |
| Siglec-3 | CD33 | + | + | +/− | +/− | [76] |
| SCARB3 | CD36 | + | + | +/− | + | [65] |
| Pgp-1 | CD44 | + | + | + | + | [76] |
| IAP | CD47 | + | + | + | + | [77] |
| Campath-1 | CD52 | + | − | +/− | +/− | [76] |
| MXRA4 (C1qR1) | CD93 | + | +/− | +/− | +/− | [73,76] |
| MIC2 | CD99 | − | unknown | + | + | [78] |
| SCFR (KIT) | CD117 | + | + | + | + | [76] |
| IL-3Rα | CD123 | + | + | + | + | [76] |
| AC133 | CD133 | + | +/− | + | + | [76] |
| BST1 | CD157 | + | + | + | + | [76] |
| CLL-1 | CD371 | − | + | − | + | [76] |
| TIM-3 | - | − | unknown | +/− | +/− | [79] |
| IL-1RAP | - | + | + | − | + | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Houshmand, M.; Kazemi, A.; Anjam Najmedini, A.; Ali, M.S.; Gaidano, V.; Cignetti, A.; Fava, C.; Cilloni, D.; Saglio, G.; Circosta, P. Shedding Light on Targeting Chronic Myeloid Leukemia Stem Cells. J. Clin. Med. 2021, 10, 5805. https://doi.org/10.3390/jcm10245805
Houshmand M, Kazemi A, Anjam Najmedini A, Ali MS, Gaidano V, Cignetti A, Fava C, Cilloni D, Saglio G, Circosta P. Shedding Light on Targeting Chronic Myeloid Leukemia Stem Cells. Journal of Clinical Medicine. 2021; 10(24):5805. https://doi.org/10.3390/jcm10245805
Chicago/Turabian StyleHoushmand, Mohammad, Alireza Kazemi, Ali Anjam Najmedini, Muhammad Shahzad Ali, Valentina Gaidano, Alessandro Cignetti, Carmen Fava, Daniela Cilloni, Giuseppe Saglio, and Paola Circosta. 2021. "Shedding Light on Targeting Chronic Myeloid Leukemia Stem Cells" Journal of Clinical Medicine 10, no. 24: 5805. https://doi.org/10.3390/jcm10245805
APA StyleHoushmand, M., Kazemi, A., Anjam Najmedini, A., Ali, M. S., Gaidano, V., Cignetti, A., Fava, C., Cilloni, D., Saglio, G., & Circosta, P. (2021). Shedding Light on Targeting Chronic Myeloid Leukemia Stem Cells. Journal of Clinical Medicine, 10(24), 5805. https://doi.org/10.3390/jcm10245805