
What Every Internist-Endocrinologist Should Know about Rare Genetic Syndromes in Order to Prevent Needless Diagnostics, Missed Diagnoses and Medical Complications: Five Years of ‘Internal Medicine for Rare Genetic Syndromes’

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Genetic Diagnosis
2.2. Medical Questionnaire
2.3. Biochemical Analysis
2.4. Additional Tests
2.5. Literature Search
2.6. Data Analysis
3. Results
3.1. Case Series: Missed Diagnoses
3.1.1. Case 1: Untreated Diabetes Mellitus in a Patient with PWS
3.1.2. Case 2: Untreated Hypothyroidism, Hypogonadism and Vitamin D Deficiency in a Patient with PWS
3.1.3. Case 3: Untreated Heart Failure in a Patient with PWS
3.2. Case Series: Overtreatment
3.2.1. Case 4: Hypersexuality in a Patient with Cockayne Syndrome
3.2.2. Case 5: Overtreatment with Hydrocortisone in a Patient with PWS
3.3. Case Series: Unnecessary Diagnostic Tests
3.3.1. Case 6: Depression and Behavioral Problems in a Patient with PWS
3.3.2. Case 7: Unnecessary Invasive Testing (Colonoscopy and Magnetic Resonance Cholangio-Pancreatography (MRCP)) in a Patient with Williams-Beuren Syndrome
4. Discussion
4.1. Multidisciplinary Approach
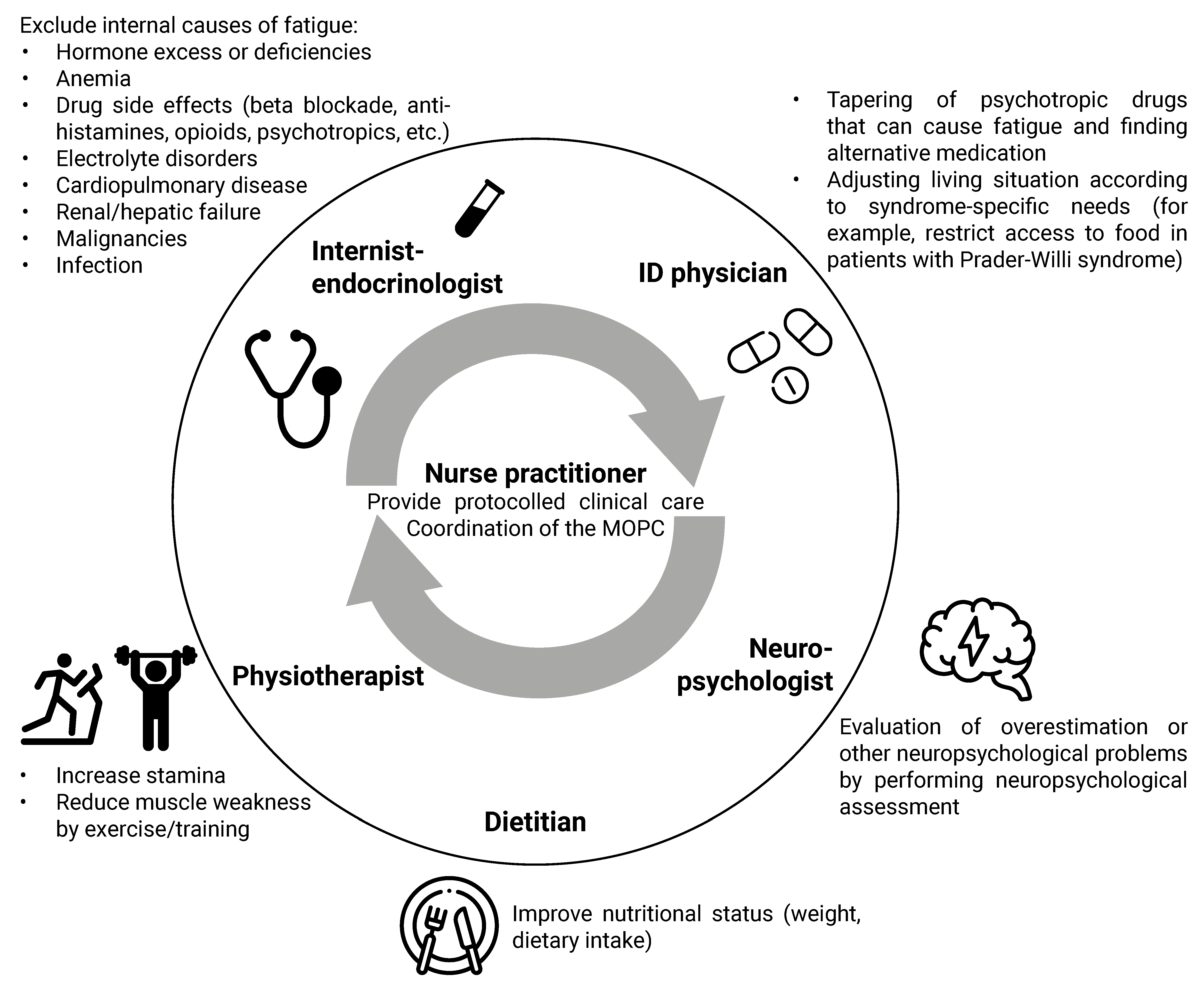
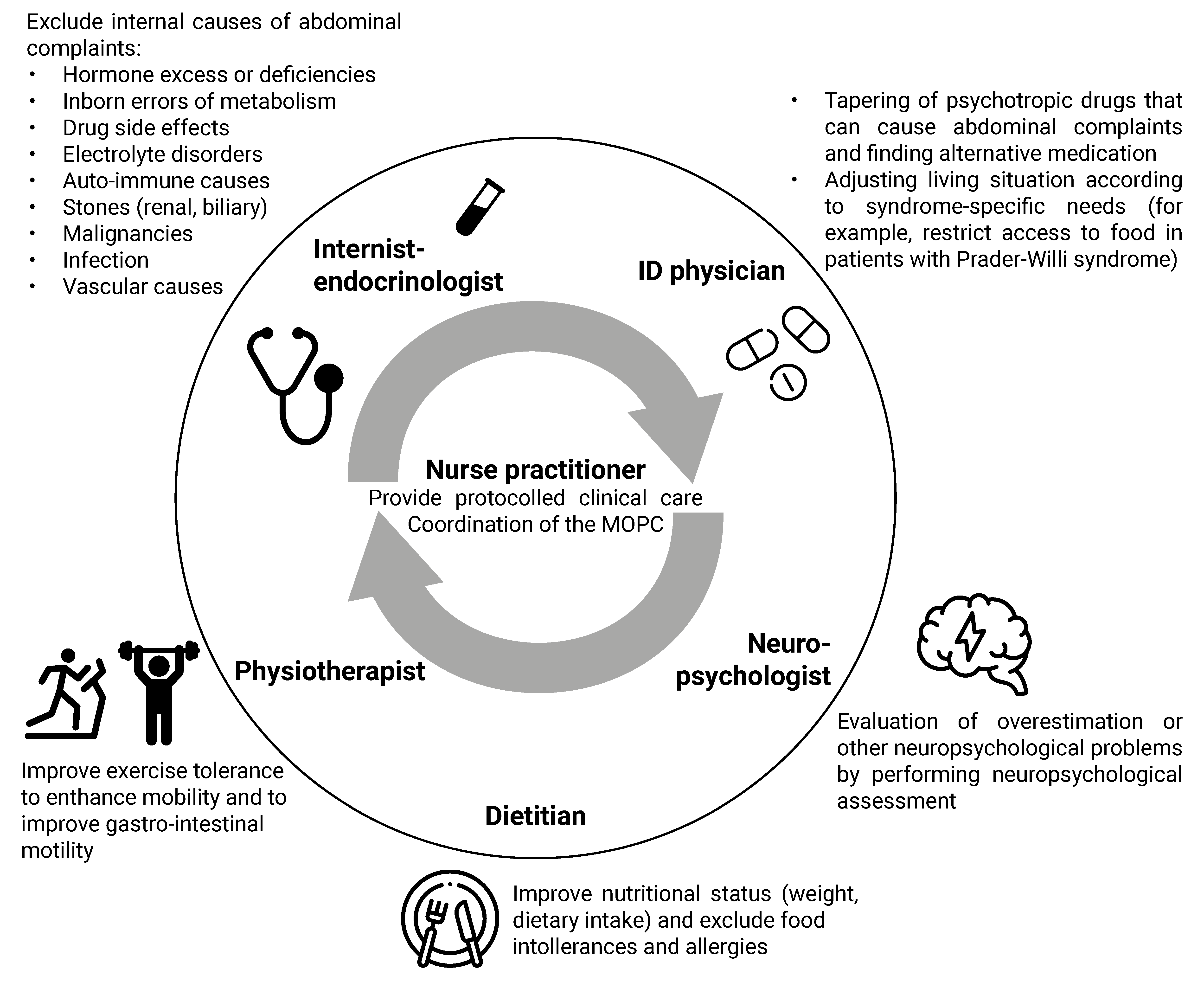
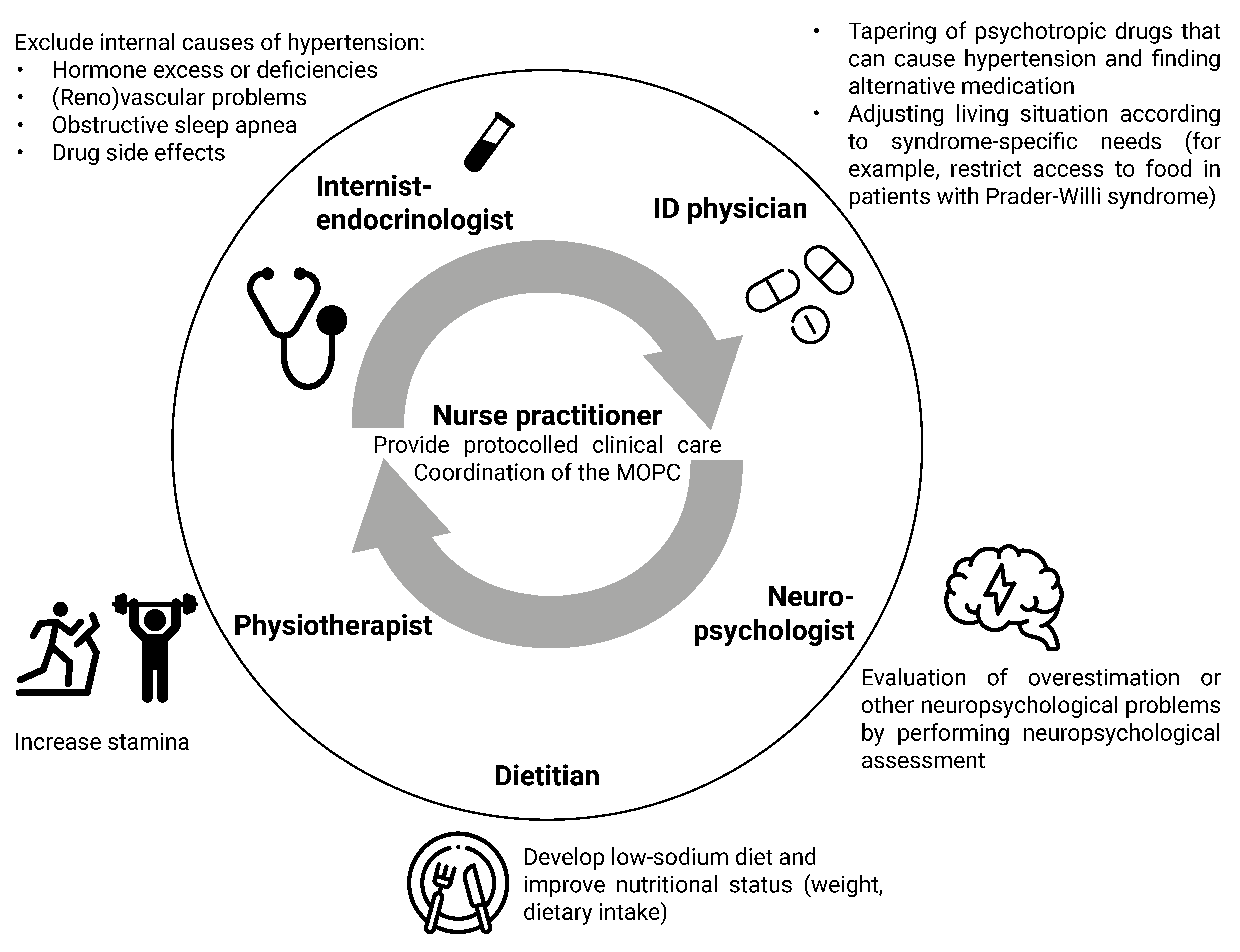
4.2. Clinical Algorithm for the Approach to Physical Symptoms
4.2.1. Syndrome-Specific Complaints
4.2.2. Drug Side Effects
4.2.3. Chronic Stress
4.2.4. Lifestyle
4.2.5. Additional Diagnostic Testing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valk, H.M.J.V.S.L.-D.; Walsh, P.N. Managing health problems in people with intellectual disabilities. BMJ 2008, 337, a2507. [Google Scholar] [CrossRef]
- Liptak, G.S.; O’Donnell, M.; Conaway, M.; Chumlea, W.C.; Wolrey, G.; Henderson, R.C.; Fung, E.; Stallings, V.A.; Samson-Fang, L.; Calvert, R.; et al. Health status of children with moderate to severe cerebral palsy. Dev. Med. Child Neurol. 2001, 43, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Oeseburg, B.; Dijkstra, G.J.; Groothoff, J.W.; Reijneveld, S.A.; Jansen, D.E. Prevalence of chronic health conditions in children with intellectual disability: A systematic literature review. Intellect. Dev. Disabil. 2011, 49, 59–85. [Google Scholar] [CrossRef] [PubMed]
- Sarneel, M.C.; Penning, C.; Roukema, J.; Moll, H.A.; Bindels-de Heus, G.C.B.; Evenhuis, H.M. Inventory of frequency and reasons of visits of children with profound intellectual and multiple disabilities to a university hospital. Ned. Tijdschr. Voor Kindergeneeskd. 2005, 73, 32–33. [Google Scholar]
- Strauss, D.; Brooks, J.; Rosenbloom, L.; Shavelle, R. Life expectancy in cerebral palsy: An update. Dev. Med. Child Neurol. 2008, 50, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Westbom, L.; Bergstrand, L.; Wagner, P.; Nordmark, E. Survival at 19 years of age in a total population of children and young people with cerebral palsy. Dev. Med. Child Neurol. 2011, 53, 808–814. [Google Scholar] [CrossRef]
- Bhaumik, S.; Watson, J.; Barrett, M.; Raju, B.; Burton, T.; Forte, J. Transition for teenagers with intellectual disability: Carer’s perspectives. J. Policy Pract. Intellect. Disabil. 2011, 8, 53–61. [Google Scholar] [CrossRef]
- Bindels-de Heus, K.G.; van Staa, A.; van Vliet, I.; Ewals, F.V.; Hilberink, S.R. Transferring young people with profound intellectual and multiple disabilities from pediatric to adult medical care: Parents’ experiences and recommendations. Intellect. Dev. Disabil. 2013, 51, 176–189. [Google Scholar] [CrossRef]
- Bruin, G. The Transition of Young Persons with Profound Multiple Disabilities from Pediatric to Adult Care; Rotterdam University: Rotterdam, The Netherlands, 2008. [Google Scholar]
- Inspectie voor de Volksgezondheid (IGZ). Profound Intellectual and Multiple Disabilities: What Next? A Research into the Quality of Care for People with PIMD; IGZ (Health Care Inspectorate): The Hague, The Netherlands, 2000. [Google Scholar]
- Neece, C.L.; Kraemer, B.R.; Blacher, J. Transition satisfaction and family well being among parents of young adults with severe intellectual disability. Intellect. Dev. Disabil. 2009, 47, 31–43. [Google Scholar] [CrossRef]
- Schrander-Stumpel, C.T.; Sinnema, M.; van den Hout, L.; Maaskant, M.A.; van Schrojenstein Lantman-de Valk, H.M.; Wagemans, A.; Schrander, J.J.; Curfs, L.M. Healthcare transition in persons with intellectual disabilities: General issues, the Maastricht model, and Prader-Willi syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2007, 145, 241–247. [Google Scholar] [CrossRef]
- Stewart, D. Transition to adult services for young people with disabilities: Current evidence to guide future research. Dev. Med. Child Neurol. 2009, 51 (Suppl. 4), 169–173. [Google Scholar] [CrossRef]
- van Staa, A.L.; Jedeloo, S.; van Meeteren, J.; Latour, J.M. Crossing the transition chasm: Experiences and recommendations for improving transitional care of young adults, parents and providers. Child Care Health Dev. 2011, 37, 821–832. [Google Scholar] [CrossRef]
- Young, N.L.; Barden, W.S.; Mills, W.A.; Burke, T.A.; Law, M.; Boydell, K. Transition to adult-oriented health care: Perspectives of youth and adults with complex physical disabilities. Phys. Occup. Ther. Pediatr. 2009, 29, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Both, P.; Ten Holt, L.; Mous, S.; Patist, J.; Rietman, A.; Dieleman, G.; Ten Hoopen, L.; Vergeer, M.; de Wit, M.C.; Bindels-de Heus, K.; et al. Tuberous sclerosis complex: Concerns and needs of patients and parents from the transitional period to adulthood. Epilepsy Behav. 2018, 83, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Rietman, A.B.; van Helden, H.; Both, P.H.; Taal, W.; Legerstee, J.S.; van Staa, A.; Moll, H.A.; Oostenbrink, R.; van Eeghen, A.M. Worries and needs of adults and parents of adults with neurofibromatosis type 1. Am. J. Med. Genet. Part A 2018, 176, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Rare Diseases. 2021. Available online: https://ec.europa.eu/health/non_communicable_diseases/rare_diseases_en (accessed on 12 November 2021).
- Pellikaan, K.; Rosenberg, A.G.W.; Kattentidt-Mouravieva, A.A.; Kersseboom, R.; Bos-Roubos, A.G.; Veen-Roelofs, J.M.C.; van Wieringen, N.; Hoekstra, F.M.E.; van den Berg, S.A.A.; van der Lely, A.J.; et al. Missed Diagnoses and Health Problems in Adults With Prader-Willi Syndrome: Recommendations for Screening and Treatment. J. Clin. Endocrinol. Metab. 2020, 105, e4671–e4687. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, G.; Bastepe, M.; Monk, D.; de Sanctis, L.; Thiele, S.; Usardi, A.; Ahmed, S.F.; Bufo, R.; Choplin, T.; De Filippo, G.; et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: First international Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 476–500. [Google Scholar] [CrossRef]
- Joseph, A.W.; Shoemaker, A.H.; Germain-Lee, E.L. Increased prevalence of carpal tunnel syndrome in albright hereditary osteodystrophy. J. Clin. Endocrinol. Metab. 2011, 96, 2065–2073. [Google Scholar] [CrossRef]
- Schwartz, C.E.; Stevenson, R.E. The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 307–321. [Google Scholar] [CrossRef]
- Remerand, G.; Boespflug-Tanguy, O.; Tonduti, D.; Touraine, R.; Rodriguez, D.; Curie, A.; Perreton, N.; Des Portes, V.; Sarret, C.; Group, R.A.S. Expanding the phenotypic spectrum of Allan-Herndon-Dudley syndrome in patients with SLC16A2 mutations. Dev. Med. Child Neurol. 2019, 61, 1439–1447. [Google Scholar] [CrossRef]
- Ozantürk, A.; Marshall, J.D.; Collin, G.B.; Düzenli, S.; Marshall, R.P.; Candan, Ş.; Tos, T.; Esen, İ.; Taşkesen, M.; Çayır, A.; et al. The phenotypic and molecular genetic spectrum of Alström syndrome in 44 Turkish kindreds and a literature review of Alström syndrome in Turkey. J. Hum. Genet. 2015, 60, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Satta, M.; Castro-Sánchez, S.; Valverde, D. Alström syndrome: Current perspectives. Appl. Clin. Genet. 2015, 8, 171–179. [Google Scholar] [PubMed]
- Larson, A.M.; Shinnick, J.E.; Shaaya, E.A.; Thiele, E.A.; Thibert, R.L. Angelman syndrome in adulthood. Am. J. Med. Genet. Part A 2015, 167, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C. Angelman syndrome: Evolution of the phenotype in adolescents and adults. Dev. Med. Child Neurol. 2001, 43, 476–480. [Google Scholar] [CrossRef]
- Seifi, M.; Walter, M.A. Axenfeld-Rieger syndrome. Clin. Genet. 2018, 93, 1123–1130. [Google Scholar] [CrossRef]
- Jena, A.K.; Kharbanda, O.P. Axenfeld-Rieger syndrome: Report on dental and craniofacial findings. J. Clin. Pediatr. Dent. 2005, 30, 83–88. [Google Scholar] [CrossRef]
- Priya, S.; Nampoothiri, S.; Sen, P.; Sripriya, S. Bardet-Biedl syndrome: Genetics, molecular pathophysiology, and disease management. Indian. J. Ophthalmol. 2016, 64, 620–627. [Google Scholar] [CrossRef]
- Mujahid, S.; Hunt, K.F.; Cheah, Y.S.; Forsythe, E.; Hazlehurst, J.M.; Sparks, K.; Mohammed, S.; Tomlinson, J.W.; Amiel, S.A.; Carroll, P.V.; et al. The Endocrine and Metabolic Characteristics of a Large Bardet-Biedl Syndrome Clinic Population. J. Clin. Endocrinol. Metab. 2018, 103, 1834–1841. [Google Scholar] [CrossRef]
- Guran, T.; Ekinci, G.; Atay, Z.; Turan, S.; Akcay, T.; Bereket, A. Radiologic and hormonal evaluation of pituitary abnormalities in patients with Bardet-Biedl syndrome. Clin. Dysmorphol. 2011, 20, 26–31. [Google Scholar] [CrossRef]
- Shrestha, S.; Chaudhary, N. A rare case of obesity. Can it be Bardet-Biedl Syndrome? Clin. Case Rep. 2019, 7, 1725–1728. [Google Scholar] [CrossRef]
- Beales, P.L.; Elcioglu, N.; Woolf, A.S.; Parker, D.; Flinter, F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446. [Google Scholar] [PubMed]
- Cunniff, C.; Djavid, A.R.; Carrubba, S.; Cohen, B.; Ellis, N.A.; Levy, C.F.; Jeong, S.; Lederman, H.M.; Vogiatzi, M.; Walsh, M.F.; et al. Health supervision for people with Bloom syndrome. Am. J. Med. Genet. Part A 2018, 176, 1872–1881. [Google Scholar] [CrossRef]
- Gécz, J.; Turner, G.; Nelson, J.; Partington, M. The Börjeson-Forssman-Lehman syndrome (BFLS, MIM #301900). Eur. J. Hum. Genet. 2006, 14, 1233–1237. [Google Scholar] [CrossRef]
- Lower, K.M.; Solders, G.; Bondeson, M.-L.; Nelson, J.; Brun, A.; Crawford, J.; Malm, G.; Börjeson, M.; Turner, G.; Partington, M.; et al. 1024C>T (R342X) is a recurrent PHF6 mutation also found in the original Börjeson–Forssman–Lehmann syndrome family. Eur. J. Hum. Genet. 2004, 12, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Küry, S.; van Woerden, G.M.; Besnard, T.; Proietti Onori, M.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef] [PubMed]
- Chia, P.H.; Zhong, F.L.; Niwa, S.; Bonnard, C.; Utami, K.H.; Zeng, R.; Lee, H.; Eskin, A.; Nelson, S.F.; Xie, W.H.; et al. A homozygous loss-of-function CAMK2A mutation causes growth delay, frequent seizures and severe intellectual disability. Elife 2018, 7, e32451. [Google Scholar] [CrossRef] [PubMed]
- Gregory, L.C.; Gevers, E.F.; Baker, J.; Kasia, T.; Chong, K.; Josifova, D.J.; Caimari, M.; Bilan, F.; McCabe, M.J.; Dattani, M.T. Structural pituitary abnormalities associated with CHARGE syndrome. J. Clin. Endocrinol. Metab. 2013, 98, E737–E743. [Google Scholar] [CrossRef]
- Wheeler, P.G.; Quigley, C.A.; Sadeghi-Nejad, A.; Weaver, D.D. Hypogonadism and CHARGE association. Am. J. Med. Genet. 2000, 94, 228–231. [Google Scholar] [CrossRef]
- Forward, K.E.; Cummings, E.A.; Blake, K.D. Risk factors for poor bone health in adolescents and adults with CHARGE syndrome. Am. J. Med. Genet. Part A 2007, 143, 839–845. [Google Scholar] [CrossRef]
- Macdonald, M.; Hudson, A.; Bladon, A.; Ratcliffe, E.; Blake, K. Experiences in feeding and gastrointestinal dysfunction in children with CHARGE syndrome. Am. J. Med. Genet. Part A 2017, 173, 2947–2953. [Google Scholar] [CrossRef]
- Hudson, A.; Macdonald, M.; Friedman, J.N.; Blake, K. CHARGE syndrome gastrointestinal involvement: From mouth to anus. Clin. Genet. 2017, 92, 10–17. [Google Scholar] [CrossRef]
- Blake, K.D.; Prasad, C. CHARGE syndrome. Orphanet J. Rare Dis. 2006, 1, 34. [Google Scholar] [CrossRef]
- Hale, C.L.; Niederriter, A.N.; Green, G.E.; Martin, D.M. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am. J. Med. Genet. Part A 2016, 170, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Trider, C.L.; Corsten, G.; Morrison, D.; Hefner, M.; Davenport, S.; Blake, K. Understanding obstructive sleep apnea in children with CHARGE syndrome. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 947–953. [Google Scholar] [CrossRef]
- Blake, K.D.; Hartshorne, T.S.; Lawand, C.; Dailor, A.N.; Thelin, J.W. Cranial nerve manifestations in CHARGE syndrome. Am. J. Med. Genet. Part A 2008, 146, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Park, D.H.; Shin, J.P.; Kim, I.T. Two cases of CHARGE syndrome with multiple congenital anomalies. Int. Ophthalmol. 2014, 34, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Devriendt, K.; Swillen, A.; Fryns, J.P. Deletion in chromosome region 22q11 in a child with CHARGE association. Clin. Genet. 1998, 53, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Brock, K.E.; Mathiason, M.A.; Rooney, B.L.; Williams, M.S. Quantitative analysis of limb anomalies in CHARGE syndrome: Correlation with diagnosis and characteristic CHARGE anomalies. Am. J. Med. Genet. Part A 2003, 123, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, P.J.; Zachariou, A.; Loveday, C.; Beleza-Meireles, A.; Bertoli, M.; Dean, J.; Douglas, A.G.L.; Ellis, I.; Foster, A.; Graham, J.M.; et al. The CHD8 overgrowth syndrome: A detailed evaluation of an emerging overgrowth phenotype in 27 patients. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 557–564. [Google Scholar] [CrossRef]
- An, Y.; Zhang, L.; Liu, W.; Jiang, Y.; Chen, X.; Lan, X.; Li, G.; Hang, Q.; Wang, J.; Gusella, J.F.; et al. De novo variants in the Helicase-C domain of CHD8 are associated with severe phenotypes including autism, language disability and overgrowth. Hum. Genet. 2020, 139, 499–512. [Google Scholar] [CrossRef]
- Douzgou, S.; Liang, H.W.; Metcalfe, K.; Somarathi, S.; Tischkowitz, M.; Mohamed, W.; Kini, U.; McKee, S.; Yates, L.; Bertoli, M.; et al. The clinical presentation caused by truncating CHD8 variants. Clin. Genet. 2019, 96, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D.; et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008, 359, 1685–1699. [Google Scholar] [CrossRef]
- Bernier, R.; Steinman, K.J.; Reilly, B.; Wallace, A.S.; Sherr, E.H.; Pojman, N.; Mefford, H.C.; Gerdts, J.; Earl, R.; Hanson, E.; et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 2016, 18, 341–349. [Google Scholar] [CrossRef]
- Haldeman-Englert, C.R.; Jewett, T. 1q21.1 Recurrent Microdeletion; University of Washington, Seattle: Seattle, WA, USA, 2011. [Google Scholar]
- Koivisto, M.; Akerblom, H.K.; Remes, M.; de La Chapelle, A. Primary hypothyroidism, growth hormone deficiency and congenital malformations in a child with the karyotype 46,XY,del(1)(q25q32). Acta Paediatr. Scand. 1976, 65, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Wang, Y.; Meng, L.L.; Qin, L.; Ma, D.Y.; Yi, L.; Xu, Z.F. 1q25.2-q31.3 Deletion in a female with mental retardation, clinodactyly, minor facial anomalies but no growth retardation. Mol. Cytogenet. 2013, 6, 30. [Google Scholar] [CrossRef]
- Kostopoulou, E.; Dastamani, A.; Caiulo, S.; Antell, H.; Flanagan, S.E.; Shah, P. Hyperinsulinaemic hypoglycaemia: A new presentation of 16p11.2 deletion syndrome. Clin. Endocrinol. 2019, 90, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Rein, B.; Yan, Z. 16p11.2 Copy Number Variations and Neurodevelopmental Disorders. Trends Neurosci. 2020, 43, 886–901. [Google Scholar] [CrossRef]
- Shiow, L.R.; Paris, K.; Akana, M.C.; Cyster, J.G.; Sorensen, R.U.; Puck, J.M. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a Coronin-1A mutation and a chromosome 16p11.2 deletion. Clin. Immunol. 2009, 131, 24–30. [Google Scholar] [CrossRef]
- Balasubramanian, M.; Smith, K.; Mordekar, S.R.; Parker, M.J. Clinical report: AN INTERSTITIAL deletion of 16p13.11 detected by array CGH in a patient with infantile spasms. Eur. J. Med. Genet. 2011, 54, 314–318. [Google Scholar] [CrossRef]
- Nagamani, S.C.; Erez, A.; Bader, P.; Lalani, S.R.; Scott, D.A.; Scaglia, F.; Plon, S.E.; Tsai, C.H.; Reimschisel, T.; Roeder, E.; et al. Phenotypic manifestations of copy number variation in chromosome 16p13.11. Eur. J. Hum. Genet. 2011, 19, 280–286. [Google Scholar] [CrossRef]
- de Kovel, C.G.; Trucks, H.; Helbig, I.; Mefford, H.C.; Baker, C.; Leu, C.; Kluck, C.; Muhle, H.; von Spiczak, S.; Ostertag, P.; et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 2010, 133, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Landing, B.H.; Sugarman, G.; Dixon, L.G. Eccrine sweat gland anatomy in cockayne syndrome: A possible diagnostic aid. Pediatr. Pathol. 1983, 1, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.T.; Stark, Z.; Sutton, R.E.; Danda, S.; Ekbote, A.V.; Elsayed, S.M.; Gibson, L.; Goodship, J.A.; Jackson, A.P.; Keng, W.T.; et al. The Cockayne Syndrome Natural History (CoSyNH) study: Clinical findings in 102 individuals and recommendations for care. Genet. Med. 2016, 18, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, L.; Laugel, V.; Lazaro, L.; Dollfus, H.; Journel, H.; Edery, P.; Goldenberg, A.; Martin, D.; Heron, D.; Le Merrer, M.; et al. Wide clinical variability among 13 new Cockayne syndrome cases confirmed by biochemical assays. Arch. Dis. Child 2006, 91, 178–182. [Google Scholar] [CrossRef]
- Ovaert, C.; Cano, A.; Chabrol, B. Aortic dilatation in Cockayne syndrome. Am. J. Med. Genet. Part A 2007, 143, 2604–2606. [Google Scholar] [CrossRef] [PubMed]
- Laugel, V. Cockayne Syndrome; University of Washington, Seattle: Seattle, WA, USA, 2000. [Google Scholar]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef]
- Tamhane, S.; Rodriguez-Gutierrez, R.; Iqbal, A.M.; Prokop, L.J.; Bancos, I.; Speiser, P.W.; Murad, M.H. Cardiovascular and Metabolic Outcomes in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2018, 103, 4097–4103. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Noon, S.E.; Krantz, I.D. Cornelia de Lange Syndrome; University of Washington, Seattle: Seattle, WA, USA, 2016. [Google Scholar]
- Mariani, M.; Decimi, V.; Bettini, L.R.; Maitz, S.; Gervasini, C.; Masciadri, M.; Ajmone, P.; Kullman, G.; Dinelli, M.; Panceri, R.; et al. Adolescents and adults affected by Cornelia de Lange syndrome: A report of 73 Italian patients. Am. J. Med. Genet. C Semin. Med. Genet. 2016, 172, 206–213. [Google Scholar] [CrossRef]
- Zambrelli, E.; Fossati, C.; Turner, K.; Taiana, M.; Vignoli, A.; Gervasini, C.; Russo, S.; Furia, F.; Masciadri, M.; Ajmone, P.; et al. Sleep disorders in Cornelia de Lange syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2016, 172, 214–221. [Google Scholar] [CrossRef]
- Ichiyama, T.; Hayashi, T.; Tanaka, H.; Nishikawa, M.; Furukawa, S. Hearing impairment in two boys with Cornelia de Lange syndrome. Brain Dev. 1994, 16, 485–487. [Google Scholar] [CrossRef]
- Decimi, V.; Parma, B.; Panceri, R.; Fossati, C.; Mariani, M.; Russo, S.; Gervasini, C.C.; Cheli, M.; Cereda, A.; Selicorni, A. Use of nutritional devices in Cornelia de Lange syndrome: Data from a large Italian cohort. Am. J. Med. Genet. Part A 2018, 176, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W.; Rauen, R.K.A. Costello Syndrome; University of Washington, Seattle: Seattle, WA, USA, 2019; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1507/ (accessed on 12 October 2020).
- Cakir, M.; Arici, C.; Tacoy, S.; Karayalcin, U. A case of Costello with parathyroid adenoma and hyperprolactinemia. Am. J. Med. Genet. Part A 2004, 124, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, Y.; Sugai, K.; Fukumizu, M.; Hanaoka, S.; Sasaki, M.; Kaga, M. Three cases of Costello syndrome presenting with intractable epilepsy and profound psychomotor retardation/regression. No To Hattatsu 2001, 33, 430–435. [Google Scholar]
- Cerruti Mainardi, P. Cri du Chat syndrome. Orphanet J. Rare Dis. 2006, 1, 33. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Clark, D.A. Common genetic and epigenetic syndromes. Pediatr. Clin. N. Am. 2015, 62, 411–426. [Google Scholar] [CrossRef]
- Van Buggenhout, G.J.; Pijkels, E.; Holvoet, M.; Schaap, C.; Hamel, B.C.; Fryns, J.P. Cri du chat syndrome: Changing phenotype in older patients. Am. J. Med. Genet. 2000, 90, 203–215. [Google Scholar] [CrossRef]
- Harvard, C.; Malenfant, P.; Koochek, M.; Creighton, S.; Mickelson, E.C.; Holden, J.J.; Lewis, M.E.; Rajcan-Separovic, E. A variant Cri du Chat phenotype and autism spectrum disorder in a subject with de novo cryptic microdeletions involving 5p15.2 and 3p24.3-25 detected using whole genomic array CGH. Clin. Genet. 2005, 67, 341–351. [Google Scholar] [CrossRef]
- Kuechler, A.; Willemsen, M.H.; Albrecht, B.; Bacino, C.A.; Bartholomew, D.W.; van Bokhoven, H.; van den Boogaard, M.J.; Bramswig, N.; Büttner, C.; Cremer, K.; et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: Expanding the mutational and clinical spectrum. Hum. Genet. 2015, 134, 97–109. [Google Scholar] [CrossRef]
- de Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef]
- Verhoeven, W.M.A.; Egger, J.I.M.; Jongbloed, R.E.; van Putten, M.M.; de Bruin-van Zandwijk, M.; Zwemer, A.S.; Pfundt, R.; Willemsen, M.H. A de novo CTNNB1 Novel Splice Variant in an Adult Female with Severe Intellectual Disability. Int. Med. Case Rep. J. 2020, 13, 487–492. [Google Scholar] [CrossRef]
- Menon, V.K.; Sorur, T.M.M.; Al Ghafri, K.A.; Shahin, M.M.H.E. Scoliosis in Dandy-Walker syndrome: A case report and review of literature. J. Spine Surg. 2017, 3, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Society for Maternal-Fetal Medicine (SMFM); Monteagudo, A. Dandy-Walker Malformation. Am. J. Obstet. Gynecol. 2020, 223, B38–B41. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.C.; Donnelly, D.E.; Donnelly, D.K.; Morrison, P.J.; Hunter, S.J. DiGeorge syndrome presenting as late onset hypocalcaemia in adulthood. Ulster Med. J. 2008, 77, 201–202. [Google Scholar] [PubMed]
- Kar, P.S.; Ogoe, B.; Poole, R.; Meeking, D. Di-George syndrome presenting with hypocalcaemia in adulthood: Two case reports and a review. J. Clin. Pathol. 2005, 58, 655–657. [Google Scholar] [CrossRef]
- Cuneo, B.F.; Driscoll, D.A.; Gidding, S.S.; Langman, C.B. Evolution of latent hypoparathyroidism in familial 22q11 deletion syndrome. Am. J. Med. Genet. 1997, 69, 50–55. [Google Scholar] [CrossRef]
- Bassett, A.S.; Chow, E.W.; Husted, J.; Weksberg, R.; Caluseriu, O.; Webb, G.D.; Gatzoulis, M.A. Clinical features of 78 adults with 22q11 Deletion Syndrome. Am. J. Med. Genet. Part A 2005, 138, 307–313. [Google Scholar] [CrossRef]
- Brauner, R.; de Gonneville, A.L.H.; Kindermans, C.; Le Bidois, J.; Prieur, M.; Lyonnet, S.; Souberbielle, J.C. Parathyroid function and growth in 22q11.2 deletion syndrome. J. Pediatr. 2003, 142, 504–508. [Google Scholar] [CrossRef]
- Giardino, G.; Cirillo, E.; Maio, F.; Gallo, V.; Esposito, T.; Naddei, R.; Grasso, F.; Pignata, C. Gastrointestinal involvement in patients affected with 22q11.2 deletion syndrome. Scand. J. Gastroenterol. 2014, 49, 274–279. [Google Scholar] [CrossRef]
- Fagerberg, C.R.; Graakjaer, J.; Heinl, U.D.; Ousager, L.B.; Dreyer, I.; Kirchhoff, M.; Rasmussen, A.A.; Lautrup, C.K.; Birkebaek, N.; Sorensen, K. Heart defects and other features of the 22q11 distal deletion syndrome. Eur. J. Med. Genet. 2013, 56, 98–107. [Google Scholar] [CrossRef]
- Evers, L.J.; van Amelsvoort, T.A.; Candel, M.J.; Boer, H.; Engelen, J.J.; Curfs, L.M. Psychopathology in adults with 22q11 deletion syndrome and moderate and severe intellectual disability. J. Intellect. Disabil. Res. 2014, 58, 915–925. [Google Scholar] [CrossRef]
- Rauch, A.; Zink, S.; Zweier, C.; Thiel, C.T.; Koch, A.; Rauch, R.; Lascorz, J.; Hüffmeier, U.; Weyand, M.; Singer, H.; et al. Systematic assessment of atypical deletions reveals genotype-phenotype correlation in 22q11.2. J. Med. Genet. 2005, 42, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Couser, N.L.; Pande, C.K.; Walsh, J.M.; Tepperberg, J.; Aylsworth, A.S. Camptodactyly and the 22q11.2 deletion syndrome. Am. J. Med. Genet. Part A 2017, 173, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Eaton, C.B.; Thomas, R.H.; Hamandi, K.; Payne, G.C.; Kerr, M.P.; Linden, D.E.J.; Owen, M.J.; Cunningham, A.C.; Bartsch, U.; Struik, S.S.; et al. Epilepsy and seizures in young people with 22q11.2 deletion syndrome: Prevalence and links with other neurodevelopmental disorders. Epilepsia 2019, 60, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Debbané, M.; Bassett, A.S.; Chow, E.W.; Fung, W.L.; van den Bree, M.; Owen, M.; Murphy, K.C.; Niarchou, M.; Kates, W.R.; et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: Results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am. J. Psychiatry 2014, 171, 627–639. [Google Scholar] [CrossRef]
- Fung, W.L.; Butcher, N.J.; Costain, G.; Andrade, D.M.; Boot, E.; Chow, E.W.; Chung, B.; Cytrynbaum, C.; Faghfoury, H.; Fishman, L.; et al. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet. Med. 2015, 17, 599–609. [Google Scholar] [CrossRef]
- Vorstman, J.A.; Breetvelt, E.J.; Thode, K.I.; Chow, E.W.; Bassett, A.S. Expression of autism spectrum and schizophrenia in patients with a 22q11.2 deletion. Schizophr. Res. 2013, 143, 55–59. [Google Scholar] [CrossRef]
- Leader, G.; Murray, M.; O’Súilleabháin, P.S.; Maher, L.; Naughton, K.; Arndt, S.; White, K.; Traina, I.; Mannion, A. Relationship between parent-reported gastrointestinal symptoms, sleep problems, autism spectrum disorder symptoms, and behavior problems in children and adolescents with 22q11.2 deletion syndrome. Res. Dev. Disabil. 2020, 104, 103698. [Google Scholar] [CrossRef]
- Shprintzen, R.J.; Goldberg, R.B.; Lewin, M.L.; Sidoti, E.J.; Berkman, M.D.; Argamaso, R.V.; Young, D. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: Velo-cardio-facial syndrome. Cleft. Palate J. 1978, 15, 56–62. [Google Scholar]
- Witchel, S.F. Disorders of sex development. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 48, 90–102. [Google Scholar] [CrossRef]
- Godfrey, L.M. Mental health outcomes among individuals with 46,XY disorders of sex development: A systematic review. J. Health Psychol. 2021, 26, 40–59. [Google Scholar] [CrossRef]
- Grinspon, R.P.; Bedecarrás, P.; Ballerini, M.G.; Iñiguez, G.; Rocha, A.; Resende, E.A.M.R.; Brito, V.N.; Milani, C.; Figueroa Gacitúa, V.; Chiesa, A.; et al. Early onset of primary hypogonadism revealed by serum anti-Müllerian hormone determination during infancy and childhood in trisomy 21. Int. J. Androl. 2011, 34, e487–e498. [Google Scholar] [CrossRef] [PubMed]
- Whooten, R.; Schmitt, J.; Schwartz, A. Endocrine manifestations of Down syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Capone, G.T.; Chicoine, B.; Bulova, P.; Stephens, M.; Hart, S.; Crissman, B.; Videlefsky, A.; Myers, K.; Roizen, N.; Esbensen, A.; et al. Co-occurring medical conditions in adults with Down syndrome: A systematic review toward the development of health care guidelines. Am. J. Med. Genet. Part A 2018, 176, 116–133. [Google Scholar] [CrossRef] [PubMed]
- Benhaourech, S.; Drighil, A.; Hammiri, A.E. Congenital heart disease and Down syndrome: Various aspects of a confirmed association. Cardiovasc. J. Afr. 2016, 27, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A.; Benfenati, F.; Gasparini, L. Communication breaks-Down: From neurodevelopment defects to cognitive disabilities in Down syndrome. Prog. Neurobiol. 2010, 91, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Grieco, J.; Pulsifer, M.; Seligsohn, K.; Skotko, B.; Schwartz, A. Down syndrome: Cognitive and behavioral functioning across the lifespan. Am. J. Med. Genet. C Semin. Med. Genet. 2015, 169, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Thakker, S.; Persily, J.; Najari, B.B. Kallman syndrome and central non-obstructive azoospermia. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101475. [Google Scholar] [CrossRef]
- Dodé, C.; Hardelin, J.-P. Kallmann syndrome. Eur. J. Hum. Genet. 2009, 17, 139–146. [Google Scholar] [CrossRef]
- Geng, D.; Zhang, H.; Liu, X.; Fei, J.; Jiang, Y.; Liu, R.; Wang, R.; Zhang, G. Identification of KISS1R gene mutations in disorders of non-obstructive azoospermia in the northeast population of China. J. Clin. Lab. Anal. 2020, 34, e23139. [Google Scholar] [CrossRef] [PubMed]
- Moalla, M.; Kacem, F.H.; Al-Mutery, A.F.; Mahfood, M.; Mejdoub-Rekik, N.; Abid, M.; Mnif-Feki, M.; Hadj Kacem, H. Nonstop mutation in the Kisspeptin 1 receptor (KISS1R) gene causes normosmic congenital hypogonadotropic hypogonadism. J. Assist. Reprod. Genet. 2019, 36, 1273–1280. [Google Scholar] [CrossRef]
- Pivnick, E.K.; Velagaleti, G.V.; Wilroy, R.S.; Smith, M.E.; Rose, S.R.; Tipton, R.E.; Tharapel, A.T. Jacobsen syndrome: Report of a patient with severe eye anomalies, growth hormone deficiency, and hypothyroidism associated with deletion 11 (q23q25) and review of 52 cases. J. Med. Genet. 1996, 33, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Mattina, T.; Perrotta, C.S.; Grossfeld, P. Jacobsen syndrome. Orphanet J. Rare Dis. 2009, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, N.; Monpoux, F.; Pedeutour, F.; Fraye, M.; Philip, P.; Ticchioni, M.; Turc-Carel, C.; Mariani, R. Jacobsen’s syndrome, thrombopenia and humoral immunodeficiency. Arch. Pediatr. 1998, 5, 1338–1340. [Google Scholar] [CrossRef]
- von Bubnoff, D.; Kreiss-Nachtsheim, M.; Novak, N.; Engels, E.; Engels, H.; Behrend, C.; Propping, P.; de la Salle, H.; Bieber, T. Primary immunodeficiency in combination with transverse upper limb defect and anal atresia in a 34-year-old patient with Jacobsen syndrome. Am. J. Med. Genet. Part A 2004, 126, 293–298. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Dempsey, J.C.; Bulgheroni, S.; Chen, M.L.; D’Arrigo, S.; Glass, I.A.; Heller, T.; Héon, E.; Hildebrandt, F.; Joshi, N.; et al. Healthcare recommendations for Joubert syndrome. Am. J. Med. Genet. Part A 2020, 182, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Fleming, L.R.; Doherty, D.A.; Parisi, M.A.; Glass, I.A.; Bryant, J.; Fischer, R.; Turkbey, B.; Choyke, P.; Daryanani, K.; Vemulapalli, M.; et al. Prospective Evaluation of Kidney Disease in Joubert Syndrome. Clin. J. Am. Soc. Nephrol. 2017, 12, 1962–1973. [Google Scholar] [CrossRef]
- Hoeve, H.L.J.; Brooks, A.S.; Smit, L.S. JS-X syndrome: A multiple congenital malformation with vocal cord paralysis, ear deformity, hearing loss, shoulder musculature underdevelopment, and X-linked recessive inheritance. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1164–1170. [Google Scholar] [CrossRef]
- Wang, Y.R.; Xu, N.X.; Wang, J.; Wang, X.M. Kabuki syndrome: Review of the clinical features, diagnosis and epigenetic mechanisms. World J. Pediatr. 2019, 15, 528–535. [Google Scholar] [CrossRef]
- Moon, J.E.; Lee, S.J.; Ko, C.W. A de novo KMT2D mutation in a girl with Kabuki syndrome associated with endocrine symptoms: A case report. BMC Med. Genet. 2018, 19, 102. [Google Scholar] [CrossRef]
- Adam, M.P.; Hudgins, L.; Hannibal, M. Kabuki Syndrome; University of Washington, Seattle: Seattle, WA, USA, 2019; Available online: https://www.ncbi.nlm.nih.gov/books/NBK62111/ (accessed on 12 October 2020).
- Philip, N.; Meinecke, P.; David, A.; Dean, J.; Ayme, S.; Clark, R.; Gross-Kieselstein, E.; Hosenfeld, D.; Moncla, A.; Muller, D. Kabuki make-up (Niikawa-Kuroki) syndrome: A study of 16 non-Japanese cases. Clin. Dysmorphol. 1992, 1, 63–77. [Google Scholar] [CrossRef]
- Atalay, Y.O.; Kaya, C.; Ustun, Y.B.; Sahinoglu, A.H. Anesthesia management in a patient with kabuki syndrome. Med. Arch. 2014, 68, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Goudie, D.; Blair, E.; Chandler, K.; Joss, S.; McKay, V.; Green, A.; Armstrong, R.; Lees, M.; Kamien, B.; et al. KAT6A Syndrome: Genotype-phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. 2019, 21, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Urreizti, R.; Lopez-Martin, E.; Martinez-Monseny, A.; Pujadas, M.; Castilla-Vallmanya, L.; Pérez-Jurado, L.A.; Serrano, M.; Natera-de Benito, D.; Martínez-Delgado, B.; Posada-de-la-Paz, M.; et al. Five new cases of syndromic intellectual disability due to KAT6A mutations: Widening the molecular and clinical spectrum. Orphanet J. Rare Dis. 2020, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Salzano, A.; D’Assante, R.; Heaney, L.M.; Monaco, F.; Rengo, G.; Valente, P.; Pasquali, D.; Bossone, E.; Gianfrilli, D.; Lenzi, A.; et al. Klinefelter syndrome, insulin resistance, metabolic syndrome, and diabetes: Review of literature and clinical perspectives. Endocrine 2018, 61, 194–203. [Google Scholar] [CrossRef]
- Han, S.J.; Kim, K.S.; Kim, W.; Kim, J.H.; Lee, Y.H.; Nam, J.S.; Seo, J.A.; Kim, B.K.; Lee, J.; Chung, J.O.; et al. Obesity and Hyperglycemia in Korean Men with Klinefelter Syndrome: The Korean Endocrine Society Registry. Endocrinol. Metab. 2016, 31, 598–603. [Google Scholar] [CrossRef]
- Ferlin, A.; Schipilliti, M.; Foresta, C. Bone density and risk of osteoporosis in Klinefelter syndrome. Acta Paediatr. 2011, 100, 878–884. [Google Scholar] [CrossRef]
- Akcan, N.; Poyrazoğlu, Ş.; Baş, F.; Bundak, R.; Darendeliler, F. Klinefelter Syndrome in Childhood: Variability in Clinical and Molecular Findings. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 100–107. [Google Scholar] [CrossRef]
- Salzano, A.; Arcopinto, M.; Marra, A.M.; Bobbio, E.; Esposito, D.; Accardo, G.; Giallauria, F.; Bossone, E.; Vigorito, C.; Lenzi, A.; et al. Klinefelter syndrome, cardiovascular system, and thromboembolic disease: Review of literature and clinical perspectives. Eur. J. Endocrinol. 2016, 175, R27–R40. [Google Scholar] [CrossRef]
- Beuers, U.; Richter, W.O.; Ritter, M.M.; Wiebecke, B.; Schwandt, P. Klinefelter’s syndrome and liver adenoma. J. Clin. Gastroenterol. 1991, 13, 214–216. [Google Scholar] [CrossRef]
- Fentiman, I.S. The endocrinology of male breast cancer. Endocr.-Relat. Cancer 2018, 25, R365–R373. [Google Scholar] [CrossRef]
- Gregory, L.C.; Shah, P.; Sanner, J.R.F.; Arancibia, M.; Hurst, J.; Jones, W.D.; Spoudeas, H.; Le Quesne Stabej, P.; Williams, H.J.; Ocaka, L.A.; et al. Mutations in MAGEL2 and L1CAM Are Associated With Congenital Hypopituitarism and Arthrogryposis. J. Clin. Endocrinol. Metab. 2019, 104, 5737–5750. [Google Scholar] [CrossRef]
- Isik, E.; Onay, H.; Atik, T.; Akgun, B.; Cogulu, O.; Ozkinay, F. Clinical and genetic features of L1 syndrome patients: Definition of two novel mutations. Clin. Neurol. Neurosurg. 2018, 172, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Stumpel, C.; Vos, Y.J. L1 Syndrome; University of Washington, Seattle: Seattle, WA, USA, 2004; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1484/ (accessed on 12 October 2020).
- Burglen, L.; Héron, D.; Moerman, A.; Dieux-Coeslier, A.; Bourguignon, J.P.; Bachy, A.; Carel, J.C.; Cormier-Daire, V.; Manouvrier, S.; Verloes, A. Myhre syndrome: New reports, review, and differential diagnosis. J. Med. Genet. 2003, 40, 546. [Google Scholar] [CrossRef]
- García-Cruz, D.; Figuera, L.E.; Feria-Velazco, A.; Sánchez-Corona, J.; García-Cruz, M.O.; Ramírez-Duenãs, R.M.; Hernandez-Córdova, A.; Ruiz, M.X.; Bitar-Alatorre, W.E.; Ramírez-Dueñas, M.L. The Myhre syndrome: Report of two cases. Clin. Genet. 1993, 44, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, C.; Michot, C.; Cormier-Daire, V. Myhre syndrome. Clin. Genet. 2014, 85, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.S.; Levy, H.P.; Sloan, J.; Dariotis, J.; Biesecker, B.B. Depression among adults with neurofibromatosis type 1: Prevalence and impact on quality of life. Clin. Genet. 2015, 88, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Sani, I.; Albanese, A. Endocrine Long-Term Follow-Up of Children with Neurofibromatosis Type 1 and Optic Pathway Glioma. Horm. Res. Paediatr. 2017, 87, 179–188. [Google Scholar] [CrossRef]
- Favere, A.M.; Tsukumo, D.M.; Matos, P.S.; Santos, S.L.; Lalli, C.A. Association between atypical parathyroid adenoma and neurofibromatosis. Arch. Endocrinol. Metab. 2015, 59, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Fossali, E.; Signorini, E.; Intermite, R.C.; Casalini, E.; Lovaria, A.; Maninetti, M.M.; Rossi, L.N. Renovascular disease and hypertension in children with neurofibromatosis. Pediatr. Nephrol. 2000, 14, 806–810. [Google Scholar] [CrossRef]
- Heervä, E.; Koffert, A.; Jokinen, E.; Kuorilehto, T.; Peltonen, S.; Aro, H.T.; Peltonen, J. A controlled register-based study of 460 neurofibromatosis 1 patients: Increased fracture risk in children and adults over 41 years of age. J. Bone Miner. Res. 2012, 27, 2333–2337. [Google Scholar] [CrossRef]
- Kinori, M.; Hodgson, N.; Zeid, J.L. Ophthalmic manifestations in neurofibromatosis type 1. Surv. Ophthalmol. 2018, 63, 518–533. [Google Scholar] [CrossRef]
- Lidzba, K.; Granström, S.; Lindenau, J.; Mautner, V.F. The adverse influence of attention-deficit disorder with or without hyperactivity on cognition in neurofibromatosis type 1. Dev. Med. Child Neurol. 2012, 54, 892–897. [Google Scholar] [CrossRef]
- Leschziner, G.D.; Golding, J.F.; Ferner, R.E. Sleep disturbance as part of the neurofibromatosis type 1 phenotype in adults. Am. J. Med. Genet. Part A 2013, 161, 1319–1322. [Google Scholar] [CrossRef]
- Terry, A.R.; Jordan, J.T.; Schwamm, L.; Plotkin, S.R. Increased Risk of Cerebrovascular Disease Among Patients With Neurofibromatosis Type 1: Population-Based Approach. Stroke 2016, 47, 60–65. [Google Scholar] [CrossRef]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef]
- Tartaglia, M.; Gelb, B.D.; Zenker, M. Noonan syndrome and clinically related disorders. Best Pract Res. Clin. Endocrinol. Metab. 2011, 25, 161–179. [Google Scholar] [CrossRef]
- Johnston, J.J.; van der Smagt, J.J.; Rosenfeld, J.A.; Pagnamenta, A.T.; Alswaid, A.; Baker, E.H.; Blair, E.; Borck, G.; Brinkmann, J.; Craigen, W.; et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet. Med. 2018, 20, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Hufnagel, R.B.; Züchner, S. PNPLA6-Related Disorders; University of Washington, Seattle: Seattle, WA, USA, 2014; Available online: https://www.ncbi.nlm.nih.gov/books/NBK247161/ (accessed on 12 October 2020).
- Yehia, L.; Eng, C. PTEN Hamartoma Tumor Syndrome; University of Washington, Seattle: Seattle, WA, USA, 2001; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1488/ (accessed on 12 October 2020).
- Pal, A.; Barber, T.M.; Van de Bunt, M.; Rudge, S.A.; Zhang, Q.; Lachlan, K.L.; Cooper, N.S.; Linden, H.; Levy, J.C.; Wakelam, M.J.O.; et al. PTEN Mutations as a Cause of Constitutive Insulin Sensitivity and Obesity. N. Engl. J. Med. 2012, 367, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Eng, C. 65 YEARS OF THE DOUBLE HELIX: One gene, many endocrine and metabolic syndromes: PTEN-opathies and precision medicine. Endocr.-Relat. Cancer 2018, 25, T121–T140. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Diene, G.; Mimoun, E.; Feigerlova, E.; Caula, S.; Molinas, C.; Grandjean, H.; Tauber, M.; PWS, F.R.C.f. Endocrine disorders in children with Prader-Willi syndrome--data from 142 children of the French database. Horm. Res. Paediatr. 2010, 74, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.G.W.; Pellikaan, K.; Poitou, C.; Goldstone, A.P.; Høybye, C.; Markovic, T.; Grugni, G.; Crinò, A.; Caixàs, A.; Coupaye, M.; et al. Central Adrenal Insufficiency Is Rare in Adults With Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 2020, 105, e2563–e2571. [Google Scholar] [CrossRef] [PubMed]
- l’Allemand, D.; Eiholzer, U.; Schlumpf, M.; Steinert, H.; Riesen, W. Cardiovascular risk factors improve during 3 years of growth hormone therapy in Prader-Willi syndrome. Eur. J. Pediatr. 2000, 159, 835–842. [Google Scholar] [CrossRef]
- Purtell, L.; Viardot, A.; Campbell, L.V. Vitamin D levels in primary growth hormone deficiency disorder Prader-Willi syndrome. Endocrine 2016, 53, 619–620. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, L.; Joensson, I.M.; Froekjaer, J.B.; Krogh, K.; Farholt, S. A descriptive study of colorectal function in adults with Prader-Willi Syndrome: High prevalence of constipation. BMC Gastroenterol. 2014, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Steinhausen, H.C.; Eiholzer, U.; Hauffa, B.P.; Malin, Z. Behavioural and emotional disturbances in people with Prader-Willi Syndrome. J. Intellect. Disabil. Res. 2004, 48, 47–52. [Google Scholar] [CrossRef]
- Bantim, Y.C.V.; Kussaba, S.T.; de Carvalho, G.P.; Garcia-Junior, I.R.; Roman-Torres, C.V.G. Oral health in patients with Prader-Willi syndrome: Current perspectives. Clin. Cosmet. Investig. Dent. 2019, 11, 163–170. [Google Scholar] [CrossRef]
- Kawano, H.; Ikeda, T.; Shimazaki, K.; Arakawa, S.; Matsumoto, Y.; Hayano, M.; Maemura, K. Successful treatment of heart failure in an adult patient with Prader-Willi syndrome. Intern. Med. 2013, 52, 771–776. [Google Scholar] [CrossRef][Green Version]
- Sinnema, M.; Boer, H.; Collin, P.; Maaskant, M.A.; van Roozendaal, K.E.; Schrander-Stumpel, C.T.; Curfs, L.M. Psychiatric illness in a cohort of adults with Prader-Willi syndrome. Res. Dev. Disabil. 2011, 32, 1729–1735. [Google Scholar] [CrossRef]
- Percy, A.K.; Lane, J.B. Rett syndrome: Clinical and molecular update. Curr. Opin. Pediatr. 2004, 16, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Christodoulou, J. MECP2 Disorders; University of Washington, Seattle: Seattle, WA, USA, 2019; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1497/ (accessed on 12 October 2020).
- Gold, W.A.; Krishnarajy, R.; Ellaway, C.; Christodoulou, J. Rett Syndrome: A Genetic Update and Clinical Review Focusing on Comorbidities. ACS Chem. Neurosci. 2018, 9, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Jaryal, A.; Gulati, S.; Chakrabarty, B.; Singh, A.; Deepak, K.K.; Pandey, R.M.; Gupta, N.; Sapra, S.; Kabra, M.; et al. Cardiovascular Autonomic Dysfunction in Children and Adolescents With Rett Syndrome. Pediatr. Neurol. 2017, 70, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Killian, J.T.; Lane, J.B.; Lee, H.S.; Skinner, S.A.; Kaufmann, W.E.; Glaze, D.G.; Neul, J.L.; Percy, A.K. Scoliosis in Rett Syndrome: Progression, Comorbidities, and Predictors. Pediatr. Neurol. 2017, 70, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Carotenuto, A.; Trivisano, M.; Cappelletti, S.; Digilio, C.; Capolino, R.; Di Capua, M.; Fusco, L.; Vigevano, F. Ring 21 chromosome presenting with epilepsy and intellectual disability: Clinical report and review of the literature. Am. J. Med Genet. Part A 2011, 155, 911–914. [Google Scholar] [CrossRef]
- Norman, M.; Wainstein, B.; Anazodo, A.; Turner, A.; Ma, C.; Payne, K.; Tangye, S.G.; Gray, P. Combined Immunodeficiency with Ring Chromosome 21. J. Clin. Immunol. 2018, 38, 251–256. [Google Scholar] [CrossRef]
- Gallagher, E.R.; Ratisoontorn, C.; Cunningham, M.L. Saethre-Chotzen Syndrome; University of Washington, Seattle: Seattle, WA, USA, 2019; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1189/ (accessed on 12 October 2020).
- Pelc, A.; Mikulewicz, M. Saethre-Chotzen syndrome: Case report and literature review. Dent. Med. Probl. 2018, 55, 217–225. [Google Scholar] [CrossRef]
- Kilcoyne, S.; Luscombe, C.; Scully, P.; Jayamohan, J.; Magdum, S.; Wall, S.; Johnson, D.; Wilkie, A.O.M. Language Development, Hearing Loss, and Intracranial Hypertension in Children With TWIST1-Confirmed Saethre-Chotzen Syndrome. J. Craniofac. Surg. 2019, 30, 1506–1511. [Google Scholar] [CrossRef]
- Campeau, P.M.; Lu, J.T.; Dawson, B.C.; Fokkema, I.F.; Robertson, S.P.; Gibbs, R.A.; Lee, B.H. The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum. Mutat. 2012, 33, 1520–1525. [Google Scholar] [CrossRef]
- Day, R.; Beckett, B.; Donnai, D.; Fryer, A.; Heidenblad, M.; Howard, P.; Kerr, B.; Mansour, S.; Maye, U.; McKee, S.; et al. A clinical and genetic study of the Say/Barber/Biesecker/Young-Simpson type of Ohdo syndrome. Clin. Genet. 2008, 74, 434–444. [Google Scholar] [CrossRef]
- White, S.M.; Adès, L.C.; Amor, D.; Liebelt, J.; Bankier, A.; Baker, E.; Wilson, M.; Savarirayan, R. Two further cases of Ohdo syndrome delineate the phenotypic variability of the condition. Clin. Dysmorphol. 2003, 12, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Terhal, P.A.; Cohen, L.; Bruccoleri, M.; Irving, M.; Martinez, A.F.; Rosenfeld, J.A.; Machol, K.; Yang, Y.; Liu, P.; et al. De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am. J. Hum. Genet. 2016, 99, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Lazar, H.P.; Kurolap, A.; Martinez, A.F.; Paperna, T.; Cohen, L.; Smeland, M.F.; Whalen, S.; Heide, S.; Keren, B.; et al. The CHD4-related syndrome: A comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis. Genet. Med. 2020, 22, 389–397. [Google Scholar] [CrossRef]
- Anderson, J.; Viskochil, D.; O’Gorman, M.; Gonzales, C. Gastrointestinal complications of Russell-Silver syndrome: A pilot study. Am. J. Med. Genet. 2002, 113, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Marsaud, C.; Rossignol, S.; Tounian, P.; Netchine, I.; Dubern, B. Prevalence and management of gastrointestinal manifestations in Silver-Russell syndrome. Arch. Dis. Child 2015, 100, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Al Kaissi, A.; Ganger, R.; Mindler, G.; Karner, C.; Klaushofer, K.; Grill, F. Correction of the axial and appendicular deformities in a patient with Silver-Russel syndrome. Afr. J. Paediatr. Surg. 2015, 12, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.J.; Deshpande, C.; Rankin, J.; Wilson, L.C.; Balasubramanian, M.; Hall, C.M.; Wagner, B.E.; Pollitt, R.; Dalton, A.; Bishop, N.J. Type 1 collagenopathy presenting with a Russell-Silver phenotype. Am. J. Med. Genet. Part A 2011, 155, 1414–1418. [Google Scholar] [CrossRef]
- Prasad, N.R.; Reddy, P.A.; Karthik, T.S.; Chakravarthy, M.; Ahmed, F. A rare case of Silver-Russell syndrome associated with growth hormone deficiency and urogenital abnormalities. Indian J. Endocrinol. Metab. 2012, 16, S307–S309. [Google Scholar] [CrossRef]
- Donoghue, S.E.; Pitt, J.J.; Boneh, A.; White, S.M. Smith-Lemli-Opitz syndrome: Clinical and biochemical correlates. J. Pediatr. Endocrinol. Metab. 2018, 31, 451–459. [Google Scholar] [CrossRef]
- Andersson, H.C.; Frentz, J.; Martínez, J.E.; Tuck-Muller, C.M.; Bellizaire, J. Adrenal insufficiency in Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. 1999, 82, 382–384. [Google Scholar] [CrossRef]
- Nowaczyk, M.J.; Wassif, C.A. Smith-Lemli-Opitz Syndrome; University of Washington, Seattle: Seattle, WA, USA, 2020; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1143/ (accessed on 12 October 2020).
- Tint, G.S.; Irons, M.; Elias, E.R.; Batta, A.K.; Frieden, R.; Chen, T.S.; Salen, G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 1994, 330, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Tierney, E.; Nwokoro, N.A.; Porter, F.D.; Freund, L.S.; Ghuman, J.K.; Kelley, R.I. Behavior phenotype in the RSH/Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. 2001, 98, 191–200. [Google Scholar] [CrossRef]
- Shayota, B.J.; Elsea, S.H. Behavior and sleep disturbance in Smith-Magenis syndrome. Curr. Opin. Psychiatry 2019, 32, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Sotos, J.F.; Dodge, P.R.; Muirhead, D.; Crawford, J.D.; Talbot, N.B. Cerebral gigantism in childhood. a syndrome of excessively rapid growth and acromegalic features and a nonprogressive neurologic disorder. N. Engl. J. Med. 1964, 271, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Baujat, G.; Cormier-Daire, V. Sotos syndrome. Orphanet J. Rare Dis. 2007, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.; Zachariou, A.; Loveday, C.; Ashraf, T.; Blair, E.; Clayton-Smith, J.; Dorkins, H.; Fryer, A.; Gener, B.; Goudie, D.; et al. The phenotype of Sotos syndrome in adulthood: A review of 44 individuals. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 502–508. [Google Scholar] [CrossRef]
- Goldstein, D.J.; Ward, R.E.; Moore, E.; Fremion, A.S.; Wappner, R.S. Overgrowth, congenital hypotonia, nystagmus, strabismus, and mental retardation: Variant of dominantly inherited Sotos sequence? Am. J. Med. Genet. 1988, 29, 783–792. [Google Scholar] [CrossRef]
- Balci, T.B.; Strong, A.; Kalish, J.M.; Zackai, E.; Maris, J.M.; Reilly, A.; Surrey, L.F.; Wertheim, G.B.; Marcadier, J.L.; Graham, G.E.; et al. Tatton-Brown-Rahman syndrome: Six individuals with novel features. Am. J. Med. Genet. A 2020, 182, 673–680. [Google Scholar] [CrossRef]
- Tatton-Brown, K.; Zachariou, A.; Loveday, C.; Renwick, A.; Mahamdallie, S.; Aksglaede, L.; Baralle, D.; Barge-Schaapveld, D.; Blyth, M.; Bouma, M.; et al. The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. 2018, 3, 46. [Google Scholar] [CrossRef]
- Heinen, C.A.; Losekoot, M.; Sun, Y.; Watson, P.J.; Fairall, L.; Joustra, S.D.; Zwaveling-Soonawala, N.; Oostdijk, W.; van den Akker, E.L.; Alders, M.; et al. Mutations in TBL1X Are Associated With Central Hypothyroidism. J. Clin. Endocrinol. Metab. 2016, 101, 4564–4573. [Google Scholar] [CrossRef]
- García, M.; Barreda-Bonis, A.C.; Jiménez, P.; Rabanal, I.; Ortiz, A.; Vallespín, E.; Del Pozo, Á.; Martínez-San Millán, J.; González-Casado, I.; Moreno, J.C. Central Hypothyroidism and Novel Clinical Phenotypes in Hemizygous Truncation of TBL1X. J. Endocr. Soc. 2019, 3, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.H.; Ma, D.; Wang, K.; Hedges, D.J.; Jaworski, J.M.; Gilbert, J.R.; Cuccaro, M.L.; Wright, H.H.; Abramson, R.K.; Konidari, I.; et al. An X chromosome-wide association study in autism families identifies TBL1X as a novel autism spectrum disorder candidate gene in males. Mol. Autism. 2011, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Kara, C.; Üstyol, A.; Yılmaz, A.; Altundağ, E.; Oğur, G. Premature ovarian failure due to tetrasomy X in an adolescent girl. Eur. J. Pediatr. 2014, 173, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Uppal, S.; Jee, Y.H.; Lightbourne, M.; Han, J.C.; Stratakis, C.A. Combined pituitary hormone deficiency in a girl with 48, XXXX and Rathke’s cleft cyst. Hormones 2017, 16, 92–98. [Google Scholar] [CrossRef]
- Alvaro-Gracia, J.M.; Humbria, A.; García-Vicuña, R.; Ariza, A.; García-Vadillo, A.; Laffón, A. Systemic lupus erythematosus and tetrasomy-X. J. Rheumatol. 1989, 16, 1486–1488. [Google Scholar]
- Slae, M.; Heshin-Bekenstein, M.; Simckes, A.; Heimer, G.; Engelhard, D.; Eisenstein, E.M. Female polysomy-X and systemic lupus erythematosus. Semin. Arthritis Rheum. 2014, 43, 508–512. [Google Scholar] [CrossRef]
- Linden, M.G.; Bender, B.G.; Robinson, A. Sex chromosome tetrasomy and pentasomy. Pediatrics 1995, 96, 672–682. [Google Scholar]
- Jayaraman, D.; Carvalho, K.S.; Hasbani, D.M. A case report of hypersomnia in tetrasomy X improved with medical therapy. Clin. Case Rep. 2018, 6, 893–895. [Google Scholar] [CrossRef]
- Tartaglia, N.R.; Howell, S.; Sutherland, A.; Wilson, R.; Wilson, L. A review of trisomy X (47,XXX). Orphanet J. Rare Dis. 2010, 5, 8. [Google Scholar] [CrossRef]
- Linden, M.G.; Bender, B.G.; Harmon, R.J.; Mrazek, D.A.; Robinson, A. 47,XXX: What is the prognosis? Pediatrics 1988, 82, 619–630. [Google Scholar] [CrossRef]
- McCray, B.A.; Schindler, A.; Hoover-Fong, J.E.; Sumner, C.J. Autosomal Dominant TRPV4 Disorders; University of Washington, Seattle: Seattle, WA, USA, 2014; Available online: https://www.ncbi.nlm.nih.gov/books/NBK201366/ (accessed on 12 October 2020).
- Henske, E.P.; Jóźwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2016, 2, 16035. [Google Scholar] [CrossRef]
- Jabir, S.; Al-Hyassat, S. Histological diagnosis of cardiac lipoma in an adult with tuberous sclerosis. BMJ Case Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70. [Google Scholar] [CrossRef]
- De Groote, K.; Demulier, L.; De Backer, J.; De Wolf, D.; De Schepper, J.; T’sjoen, G.; De Backer, T. Arterial hypertension in Turner syndrome: A review of the literature and a practical approach for diagnosis and treatment. J. Hypertens. 2015, 33, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Twite, M.D.; Stenquist, S.; Ing, R.J. Williams syndrome. Paediatr. Anaesth. 2019, 29, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Nordstrøm, M.; Paus, B.; Andersen, L.F.; Kolset, S.O. Dietary aspects related to health and obesity in Williams syndrome, Down syndrome, and Prader-Willi syndrome. Food Nutr. Res. 2015, 59, 25487. [Google Scholar] [CrossRef]
- Güven, A. Seven cases with Williams-Beuren syndrome: Endocrine evaluation and long-term follow-up. J. Pediatr. Endocrinol. Metab. 2017, 30, 159–165. [Google Scholar] [CrossRef]
- Nicita, F.; Garone, G.; Spalice, A.; Savasta, S.; Striano, P.; Pantaleoni, C.; Spartà, M.V.; Kluger, G.; Capovilla, G.; Pruna, D.; et al. Epilepsy is a possible feature in Williams-Beuren syndrome patients harboring typical deletions of the 7q11.23 critical region. Am. J. Med. Genet. Part A 2016, 170, 148–155. [Google Scholar] [CrossRef]
- Huang, Y.C.; Lee, C.T.; Wu, M.Z.; Liu, S.Y.; Tung, Y.C.; Ho, H.N.; Tsai, W.Y. The spectrum of 45,X/46,XY mosaicism in Taiwanese children: The experience of a single center. J. Formos. Med. Assoc. 2019, 118, 450–456. [Google Scholar] [CrossRef]
- Telvi, L.; Lebbar, A.; Del Pino, O.; Barbet, J.P.; Chaussain, J.L. 45,X/46,XY mosaicism: Report of 27 cases. Pediatrics 1999, 104, 304–308. [Google Scholar] [CrossRef]
- Tartaglia, N.; Ayari, N.; Howell, S.; D’Epagnier, C.; Zeitler, P. 48,XXYY, 48,XXXY and 49,XXXXY syndromes: Not just variants of Klinefelter syndrome. Acta Paediatr. 2011, 100, 851–860. [Google Scholar] [CrossRef]
- Blumling, A.A.; Martyn, K.; Talboy, A.; Close, S. Rare sex chromosome variation 48,XXYY: An integrative review. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 386–403. [Google Scholar] [CrossRef]
- Tartaglia, N.; Davis, S.; Hench, A.; Nimishakavi, S.; Beauregard, R.; Reynolds, A.; Fenton, L.; Albrecht, L.; Ross, J.; Visootsak, J.; et al. A new look at XXYY syndrome: Medical and psychological features. Am. J. Med. Genet. A 2008, 146, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, N.R.; Wilson, R.; Miller, J.S.; Rafalko, J.; Cordeiro, L.; Davis, S.; Hessl, D.; Ross, J. Autism Spectrum Disorder in Males with Sex Chromosome Aneuploidy: XXY/Klinefelter Syndrome, XYY, and XXYY. J. Dev. Behav. Pediatr. 2017, 38, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Krauser, D.G.; Lloyd-Jones, D.M.; Chae, C.U.; Cameron, R.; Anwaruddin, S.; Baggish, A.L.; Chen, A.; Tung, R.; Januzzi, J.L., Jr. Effect of body mass index on natriuretic peptide levels in patients with acute congestive heart failure: A ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) substudy. Am. Heart J. 2005, 149, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Nederland, Z. Farmacotherapeutisch Kompas. Available online: https://farmacotherapeutischkompas.nl (accessed on 12 October 2020).
- Mervis, C.B.; Velleman, S.L. Children with Williams Syndrome: Language, Cognitive, and Behavioral Characteristics and their Implications for Intervention. Perspect. Lang. Learn. Educ. 2011, 18, 98–107. [Google Scholar] [CrossRef]
- van’t Leven, M.; Zielhuis, G.A.; van der Meer, J.W.; Verbeek, A.L.; Bleijenberg, G. Fatigue and chronic fatigue syndrome-like complaints in the general population. Eur. J. Public Health 2009, 20, 251–257. [Google Scholar] [CrossRef]
- Drossman, D.A.; Li, Z.; Andruzzi, E.; Temple, R.D.; Talley, N.J.; Thompson, W.G.; Whitehead, W.E.; Janssens, J.; Funch-Jensen, P.; Corazziari, E.; et al. U.S. householder survey of functional gastrointestinal disorders. Prevalence, sociodemography, and health impact. Dig. Dis. Sci. 1993, 38, 1569–1580. [Google Scholar] [CrossRef]
- Centraal Bureau Voor de Statistiek. Gezondheid en Zorggebruik; Persoonskenmerken. 2021. Available online: https://opendata.cbs.nl/statline/#/CBS/nl/dataset/83005NED/table?dl=4486F (accessed on 11 March 2021).
- Lionti, T.; Reid, S.M.; White, S.M.; Rowell, M.M. A population-based profile of 160 Australians with Prader-Willi syndrome: Trends in diagnosis, birth prevalence and birth characteristics. Am. J. Med. Genet. Part A 2015, 167, 371–378. [Google Scholar] [CrossRef]
- Bar, C.; Diene, G.; Molinas, C.; Bieth, E.; Casper, C.; Tauber, M. Early diagnosis and care is achieved but should be improved in infants with Prader-Willi syndrome. Orphanet J. Rare Dis. 2017, 12, 118. [Google Scholar] [CrossRef]
- López-Bastida, J.; Linertová, R.; Oliva-Moreno, J.; Posada-de-la-Paz, M.; Serrano-Aguilar, P.; Kanavos, P.; Taruscio, D.; Schieppati, A.; Iskrov, G.; Baji, P.; et al. Social/economic costs and health-related quality of life in patients with Prader-Willi syndrome in Europe. Eur. J. Health Econ. 2016, 17 (Suppl. 1), 99–108. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bastida, J.; Oliva-Moreno, J.; Linertova, R.; Serrano-Aguilar, P. Social/economic costs and health-related quality of life in patients with rare diseases in Europe. Eur. J. Health Econ. 2016, 17 (Suppl. 1), 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chevreul, K.; Berg Brigham, K.; Clement, M.C.; Poitou, C.; Tauber, M.; Members of the BURQOL-RD Research Network listed in the Online Appendix. Economic burden and health-related quality of life associated with Prader-Willi syndrome in France. J. Intellect. Disabil. Res. 2016, 60, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Wijeysundera, H.C.; Machado, M.; Wang, X.; Van Der Velde, G.; Sikich, N.; Witteman, W.; Tu, J.V.; Lee, D.S.; Goodman, S.G.; Petrella, R.; et al. Cost-effectiveness of specialized multidisciplinary heart failure clinics in Ontario, Canada. Value Health 2010, 13, 915–921. [Google Scholar] [CrossRef]
- Komenda, P.; Levin, A. Analysis of cardiovascular disease and kidney outcomes in multidisciplinary chronic kidney disease clinics: Complex disease requires complex care models. Curr. Opin. Nephrol. Hypertens. 2006, 15, 61–66. [Google Scholar] [CrossRef]
- Wallace, R.A. Clinical audit of gastrointestinal conditions occurring among adults with Down syndrome attending a specialist clinic. J. Intellect. Dev. Disabil. 2007, 32, 45–50. [Google Scholar] [CrossRef]
- Real de Asua, D.; Quero, M.; Moldenhauer, F.; Suarez, C. Clinical profile and main comorbidities of Spanish adults with Down syndrome. Eur. J. Intern. Med. 2015, 26, 385–391. [Google Scholar] [CrossRef]
- Farquhar, M.; Jacobson, M.; Braun, C.; Wolfman, W.; Kelly, C.; Allen, L.M.; Lega, I.C. Medical and gynecological comorbidities in adult women with Turner syndrome: Our multidisciplinary clinic experience. Climacteric 2019, 23, 32–37. [Google Scholar] [CrossRef]
- Freriks, K.; Timmermans, J.; Beerendonk, C.C.; Verhaak, C.M.; Netea-Maier, R.T.; Otten, B.J.; Braat, D.D.; Smeets, D.F.; Kunst, D.H.; Hermus, A.R.; et al. Standardized multidisciplinary evaluation yields significant previously undiagnosed morbidity in adult women with Turner syndrome. J. Clin. Endocrinol. Metab. 2011, 96, E1517–E1526. [Google Scholar] [CrossRef]
- Kahlert, E.; Blaschke, M.; Brockmann, K.; Freiberg, C.; Janssen, O.E.; Stahnke, N.; Strik, D.; Merkel, M.; Mann, A.; Liesenkötter, K.P.; et al. Deficient knowledge in adult Turner syndrome care as an incentive to found Turner centers in Germany. Endocr. Connect. 2019, 8, 1483–1492. [Google Scholar] [CrossRef]
- Vincent, A.J.; Nguyen, H.H.; Ranasinha, S.; Vollenhoven, B. Increased detection of co-morbidities with evaluation at a dedicated adult Turner syndrome clinic. Climacteric 2017, 20, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Penninx, B.; Lange, S.M.M. Metabolic syndrome in psychiatric patients: Overview, mechanisms, and implications. Dialogues. Clin. Neurosci. 2018, 20, 63–73. [Google Scholar] [PubMed]
- Steenbergen, H.A.; Van der Schans, C.P.; Van Wijck, R.; De Jong, J.; Waninge, A. Lifestyle Approaches for People With Intellectual Disabilities: A Systematic Multiple Case Analysis. J. Am. Med. Dir. Assoc. 2017, 18, 980–987.e3. [Google Scholar] [CrossRef] [PubMed]
- Willems, M.; Waninge, A.; Hilgenkamp, T.I.M.; van Empelen, P.; Krijnen, W.P.; van der Schans, C.P.; Melville, C.A. Effects of lifestyle change interventions for people with intellectual disabilities: Systematic review and meta-analysis of randomized controlled trials. J. Appl. Res. Intellect. Disabil. 2018, 31, 949–961. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Syndrome | N | Syndrome | N | Syndrome | N |
|---|---|---|---|---|---|
| Albright hereditary osteodystrophy | <5 | Dandy-Walker syndrome | <5 | Ring chromosome 21 | <5 |
| Allan-Herndon-Dudley syndrome | <5 | DiGeorge syndrome (22q11.2 deletion) | 8 | Saethre-Chotzen syndrome | <5 |
| Alström syndrome | <5 | Disorders of Sex Development 1 | 18 | Say-Barber-Biesecker-Young-Simpson syndrome (KAT6B mutation) | <5 |
| Angelman syndrome | <5 | Down syndrome (trisomy 21) | <5 | Sifrim-Hitz-Weiss syndrome | <5 |
| Axenfeld-Rieger syndrome | <5 | Hypogonadotropic hypogonadism with anosmia (Kallmann syndrome) | 6 | Silver-Russell syndrome | 5 |
| Bardet-Biedl syndrome | 5 | Hypogonadotropic hypogonadism without anosmia (Kiss1R mutation) | <5 | Smith-Lemli-Opitz syndrome | <5 |
| Bloom syndrome | <5 | Jacobsen syndrome | <5 | Smith-Magenis syndrome | <5 |
| Börjeson-Forssman-Lehmann syndrome | <5 | Joubert syndrome | <5 | Sotos-like syndrome | <5 |
| CAMK2A variants | <5 | JS-X syndrome | <5 | Tatton-Brown-Rahman syndrome | <5 |
| CHARGE syndrome | 10 | Kabuki syndrome | <5 | TBL1X mutation | <5 |
| CHD8 syndrome | <5 | KAT6A syndrome | <5 | Tetra-X syndrome | <5 |
| Chromosome 1q21 deletion syndrome | <5 | Klinefelter syndrome | 41 | Triple-X syndrome | <5 |
| Chromosome 1q25-32 deletion | <5 | L1CAM mutation | <5 | TRPV4 mutation | <5 |
| Chromosome 16p11.2 deletion syndrome | <5 | Myhre syndrome | <5 | Tuberous sclerosis complex | 49 |
| Chromosome 16p3.11 deletion syndrome | <5 | Neurofibromatosis type 1 | 120 | Turner syndrome | 184 |
| Cockayne syndrome | <5 | Noonan syndrome | 9 | Williams-Beuren syndrome | 10 |
| Congenital adrenal hyperplasia | 11 | PNPLA6 gene mutation | <5 | 45,X/46,XY mixed gonadal dysgenesis | <5 |
| Cornelia de Lange syndrome | <5 | PTEN hamartoma tumor syndrome | <5 | 48,XXXY syndrome | <5 |
| Costello (like) syndrome | <5 | Prader-Willi like syndrome | 9 | 48,XXYY syndrome | <5 |
| Cri-du-chat syndrome | <5 | Prader-Willi syndrome | 135 | Unknown syndrome | 41 |
| CTNNB1 syndrome | <5 | Rett syndrome | <5 | Total | 720 |
| Endocrine Manifestations | Internal Medicine—Other | Other Disciplines | |
|---|---|---|---|
| Albright hereditary osteodystrophy [20,21] |       |   |       |
| Allan-Herndon- Dudley syndrome [22,23] | | |       |
| Alström syndrome [24,25] |  |     |      |
| Angelman syndrome [26,27] |  |  |   |
| Axenfeld-Rieger syndrome [28,29] | | | |
| Bardet-Biedl syndrome [30,31,32,33,34] |    |  | |
| Bloom syndrome [35] | |   | |
| Börjeson-Forssman-Lehmann syndrome [36,37] |  |  | |
| CAMK2A variants [38,39] |  | | |
| CHARGE syndrome [40,41,42,43,44,45,46,47,48,49,50,51] | | |    |
| CHD8 syndrome [52,53,54] | | |  |
| Chromosome 1q21 deletion syndrome [55,56,57] | | |    |
| Chromosome 1q25-32 deletion [58,59] | | | |
| Chromosome 16p11.2 deletion syndrome [60,61,62] |  | | |
| Chromosome 16p13.11 deletion syndrome [63,64,65] | | | |
| Cockayne syndrome [66,67,68,69,70] | |  |  |
| Congenital adrenal hyperplasia [71,72] |   | | |
| Cornelia de Lange syndrome [73,74,75,76,77] | | |   |
| Costello (like) syndrome [78,79,80] | | | |
| Cri-du-chat syndrome [81,82,83,84] | | | |
| CTNNB1 syndrome (NEDSDV syndrome) [85,86,87] | | | |
| Dandy-Walker syndrome [88,89] | | | |
| DiGeorge syndrome (22q11.2 deletion) [90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105] |  |    |  |
| Disorders of Sex Development 1 [106,107] | |  | |
| Down syndrome (trisomy 21) [108,109,110,111,112,113] | | |  |
| Hypogonadotropic hypogonadism with anosmia (Kallmann syndrome) [114,115] | | |  |
| Hypogonadotropic hypogonadism without anosmia (Kiss 1R mutation) [116,117] | | ||
| Jacobsen syndrome (11q terminal deletion syndrome) [118,119,120,121] | | | |
| Joubert syndrome [122,123] | | | |
| JS-X syndrome [124] |  | ||
| Kabuki syndrome [125,126,127,128,129] | | | |
| KAT6A syndrome [130,131] | | | |
| Klinefelter syndrome [132,133,134,135,136,137,138] | | | |
| L1CAM mutation [139,140,141] | | | |
| Myhre syndrome [142,143,144] | | | |
| Neurofibromatosis type 1 [145,146,147,148,149,150,151,152,153,154] |  |  |  |
| Noonan syndrome [155,156,157] | | | |
| PNPLA6 gene mutation [158] | | | |
| PTEN hamartoma tumor syndrome [159,160,161] | | | |
| Prader-Willi (like) Syndrome [19,162,163,164,165,166,167,168,169,170,171] | | | |
| Rett syndrome [172,173,174,175,176] | |  | |
| Ring chromosome 21 [177,178] | | | |
| Saethre-Chotzen syndrome [179,180,181] | | | |
| Say-Barber-Biesecker-Young-Simpson syndrome (KAT6B mutation) [182,183,184] | | | |
| Sifrim-Hitz-Weiss Syndrome [185,186] | | | |
| Silver-Russell syndrome [82,187,188,189,190,191] | | |  |
| Smith-Lemli-Opitz syndrome [192,193,194,195,196] | | | |
| Smith-Magenis Syndrome [82,197] | | | |
| Sotos-like syndrome [198,199,200,201] | | | |
| Tatton-Brown- Rahman syndrome [202,203] | | | |
| TBL1X mutation [204,205,206] | | | |
| Tetra-X syndrome (48,XXXX) [207,208,209,210,211,212] | | | |
| Triple-X syndrome (47,XXX) [213,214] | | | |
| TRPV4 mutation [215] | | |  |
| Tuberous sclerosis complex [216,217] | | | |
| Turner syndrome [218,219] | |  | |
| Williams-Beuren syndrome [220,221,222,223] | | | |
| 45,X/46,XY mixed gonadal dysgenesis [224,225] | | | |
| 48,XXXY syndrome [226] | | | |
| 48,XXYY syndrome [226,227,228,229] | | | |
obesity diabetes mellitus hypoglycemia metabolic syndrome hypothyroidism hyperthyroidism (pseudo)hypoparathyroidism hyperparathyroidism hypercalcitoninemia hyperaldosteronism impaired cortisol synthesis disturbed gonadal axis hyperprolactinemia hypopituitarism/pituitary anomalies gynecomastia osteopenia/osteoporosis short stature/growth hormone deficiency tall stature/overgrowth hepatic disease/anomalies renal disease/anomalies gallbladder disease pulmonary problems obstructive sleep apnea cardio(vascular) disease/anomalies hypertension hypotension dyslipidemia hematologic anomalies gastrointestinal problems/anomalies splenomegalie increased risk of malignancies immune problems/anomalies celiac disease thymic hypoplasia electrolyte disorders vitamin D deficiency carpal tunnel syndrome limb anomalies skeletal anomalies joint problems urogenital tract anomalies hypotonia/low muscle mass/muscle atrophy hypertonia neuromuscular problems brain anomalies ataxia hypomyelination/cranial nerve anomalies/(poly)neuropathy hemiparesis tremor/parkinsonism Alzheimer disease increased risk of stroke intellectual disability/developmental disorders psychological problems/challenging behavior epilepsy/seizures poor balance scoliosis decreased pain sensitivity temperature intolerance sleeping problems decreased sweat production increased sweat production ectopic ossification visual problems/anomalies oral problems/anomalies laryngeal obstruction hearing problems/anomalies craniofacial anomalies skin anomalies hair problems hyposmia/anosmia macrocephaly microcephaly feeding difficulties. A more detailed overview of the clinical manifestations of these complex rare genetic disorders is given in the Supplementary Data, Table S1. In reality, some manifestations might have a similar prevalence as in the general population, because of publication bias. Although the literature was thoroughly searched, the overview might be incomplete. 1 There are many different types of Disorders of Sex Development. Therefore, we advise checking the specific type of disorder in the literature for the specific clinical manifestations. Icons were derived from flaticon.com (accessed on 30 September 2021) (Freepik, DinosoftLabs, Pixel perfect, Flat Icons, Smashicons, Good Ware, smalllikeart, Vitaly Gorbachev, monkik, Kiranshastry, Eucalyp, surang, lcongeek26, Chanut) and from the Noun Project (icondfield, VectorsPoint, tezar tantular) on 30 September 2021.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosenberg, A.G.W.; Pater, M.R.A.; Pellikaan, K.; Davidse, K.; Kattentidt-Mouravieva, A.A.; Kersseboom, R.; Bos-Roubos, A.G.; van Eeghen, A.; Veen, J.M.C.; van der Meulen, J.J.; et al. What Every Internist-Endocrinologist Should Know about Rare Genetic Syndromes in Order to Prevent Needless Diagnostics, Missed Diagnoses and Medical Complications: Five Years of ‘Internal Medicine for Rare Genetic Syndromes’. J. Clin. Med. 2021, 10, 5457. https://doi.org/10.3390/jcm10225457
Rosenberg AGW, Pater MRA, Pellikaan K, Davidse K, Kattentidt-Mouravieva AA, Kersseboom R, Bos-Roubos AG, van Eeghen A, Veen JMC, van der Meulen JJ, et al. What Every Internist-Endocrinologist Should Know about Rare Genetic Syndromes in Order to Prevent Needless Diagnostics, Missed Diagnoses and Medical Complications: Five Years of ‘Internal Medicine for Rare Genetic Syndromes’. Journal of Clinical Medicine. 2021; 10(22):5457. https://doi.org/10.3390/jcm10225457
Chicago/Turabian StyleRosenberg, Anna G. W., Minke R. A. Pater, Karlijn Pellikaan, Kirsten Davidse, Anja A. Kattentidt-Mouravieva, Rogier Kersseboom, Anja G. Bos-Roubos, Agnies van Eeghen, José M. C. Veen, Jiske J. van der Meulen, and et al. 2021. "What Every Internist-Endocrinologist Should Know about Rare Genetic Syndromes in Order to Prevent Needless Diagnostics, Missed Diagnoses and Medical Complications: Five Years of ‘Internal Medicine for Rare Genetic Syndromes’" Journal of Clinical Medicine 10, no. 22: 5457. https://doi.org/10.3390/jcm10225457
APA StyleRosenberg, A. G. W., Pater, M. R. A., Pellikaan, K., Davidse, K., Kattentidt-Mouravieva, A. A., Kersseboom, R., Bos-Roubos, A. G., van Eeghen, A., Veen, J. M. C., van der Meulen, J. J., van Aalst-van Wieringen, N., Hoekstra, F. M. E., van der Lely, A. J., & de Graaff, L. C. G. (2021). What Every Internist-Endocrinologist Should Know about Rare Genetic Syndromes in Order to Prevent Needless Diagnostics, Missed Diagnoses and Medical Complications: Five Years of ‘Internal Medicine for Rare Genetic Syndromes’. Journal of Clinical Medicine, 10(22), 5457. https://doi.org/10.3390/jcm10225457