
Compound Mutation in Cardiac Sarcomere Proteins Is Associated with Increased Risk for Major Arrhythmic Events in Pediatric Onset Hypertrophic Cardiomyopathy

 ,
,
Abstract
:1. Introduction
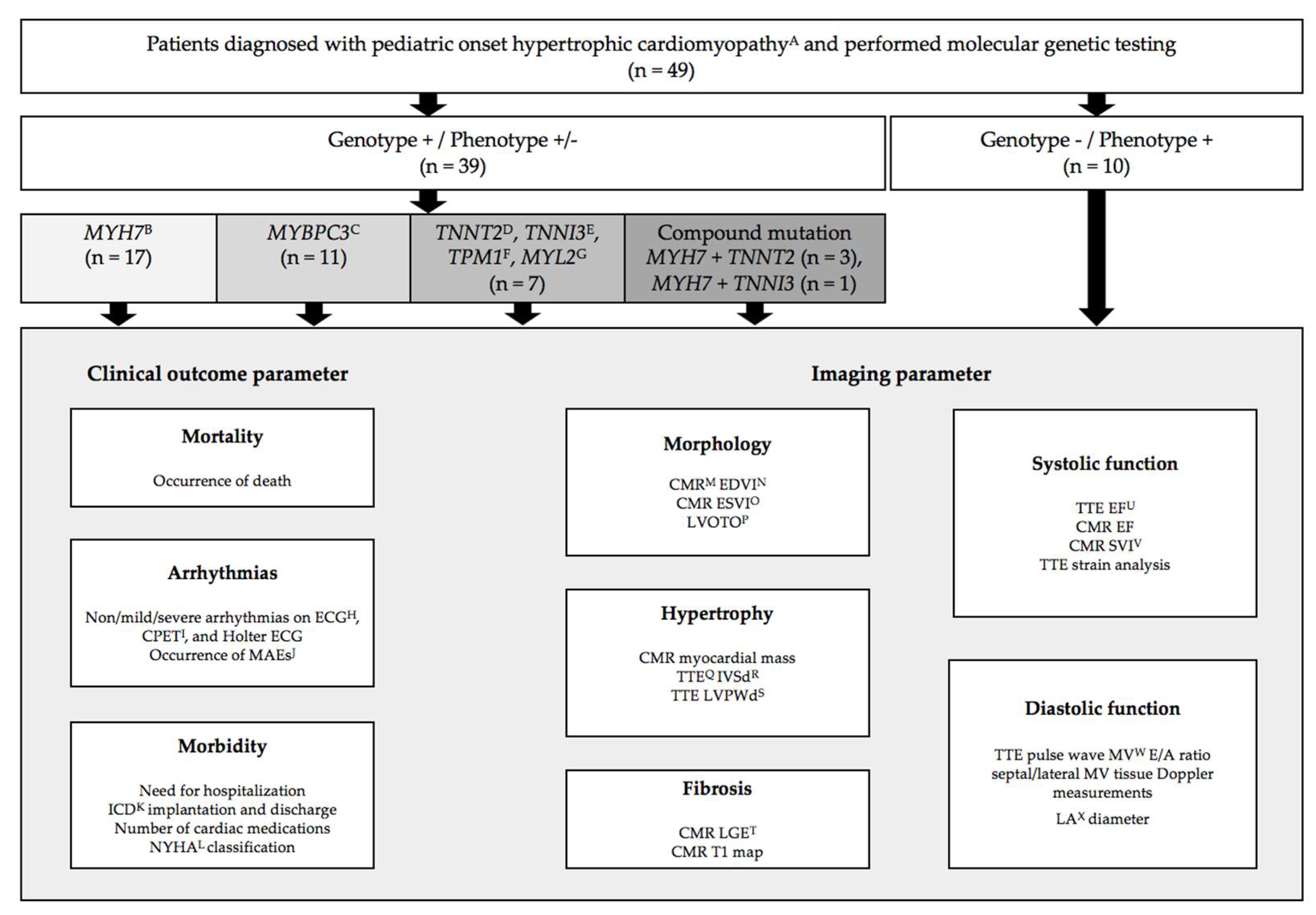
2. Materials and Methods
3. Results
3.1. Patients Characteristics
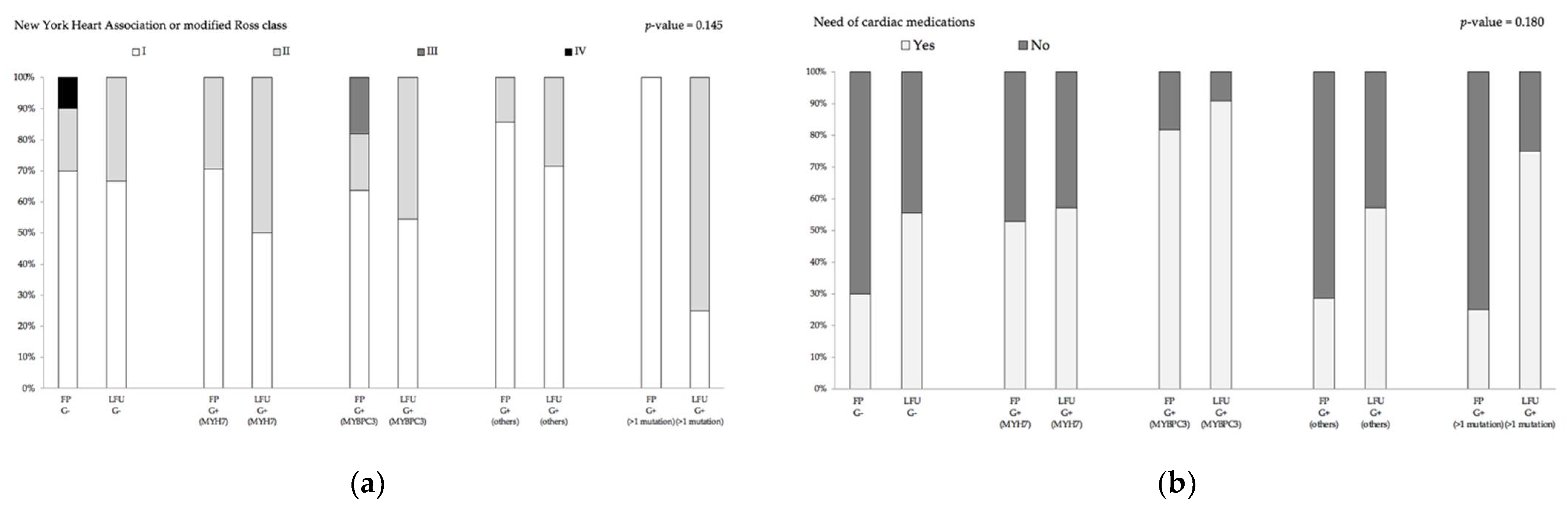
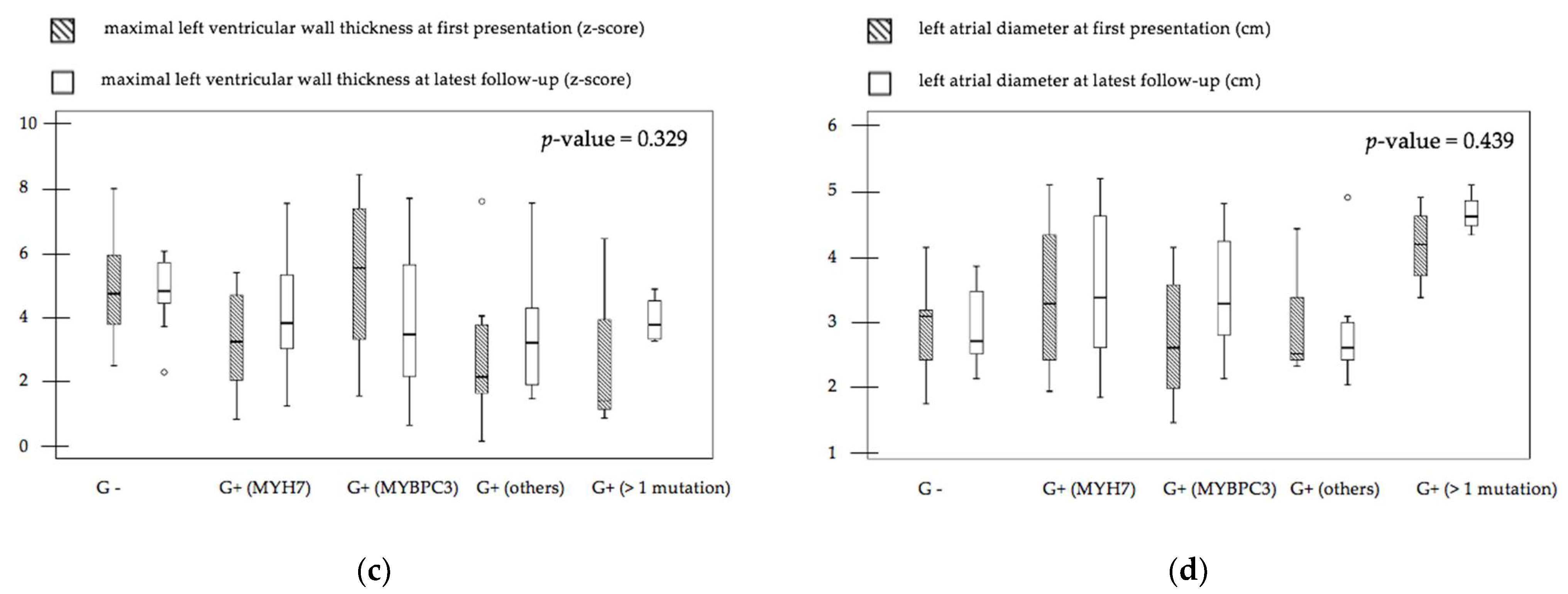
3.2. Genotype–Phenotype Relation of Clinical Outcome and Imaging Parameters
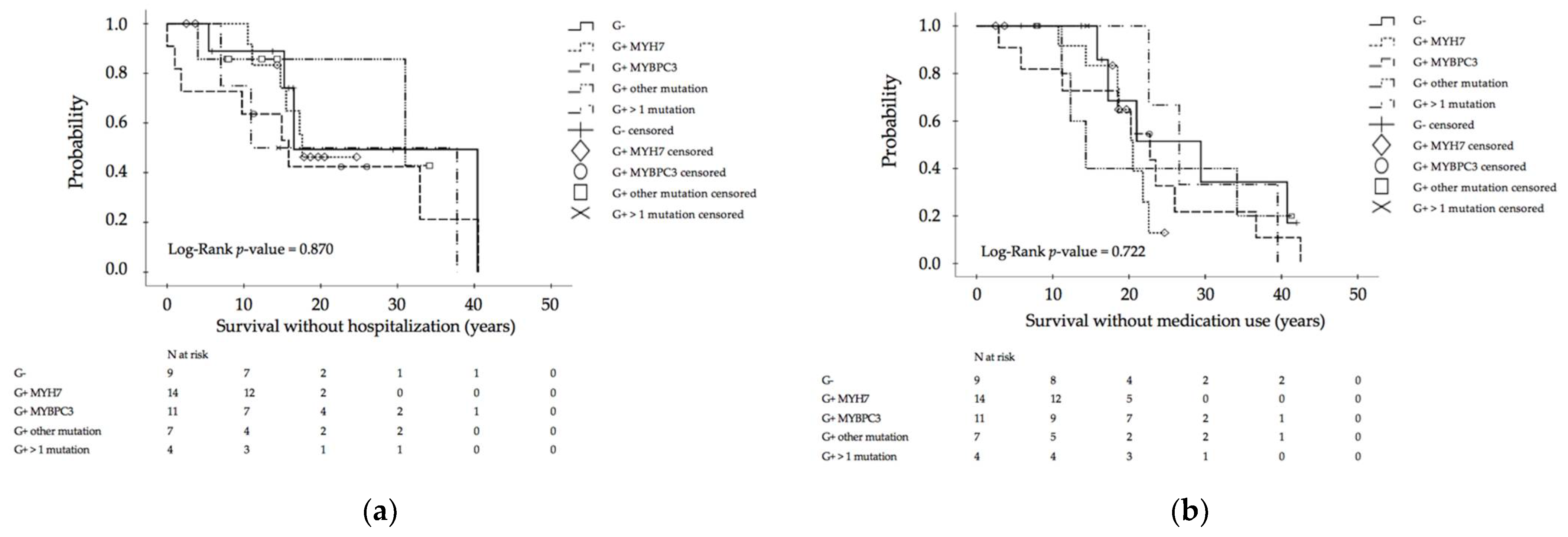
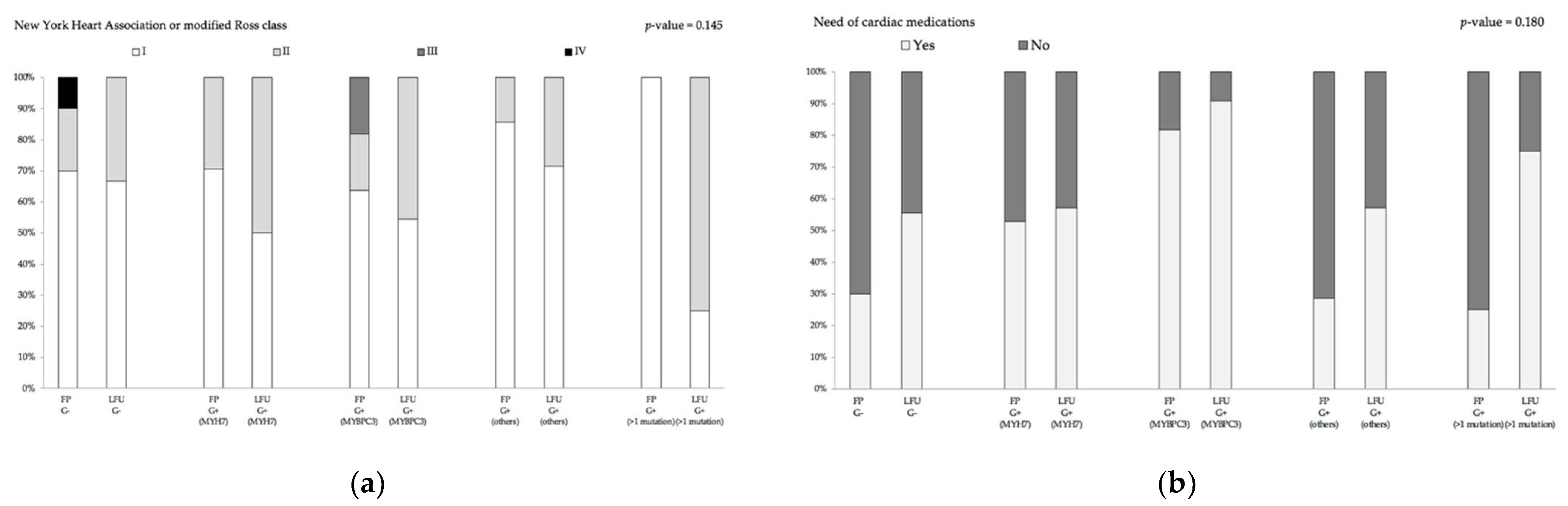
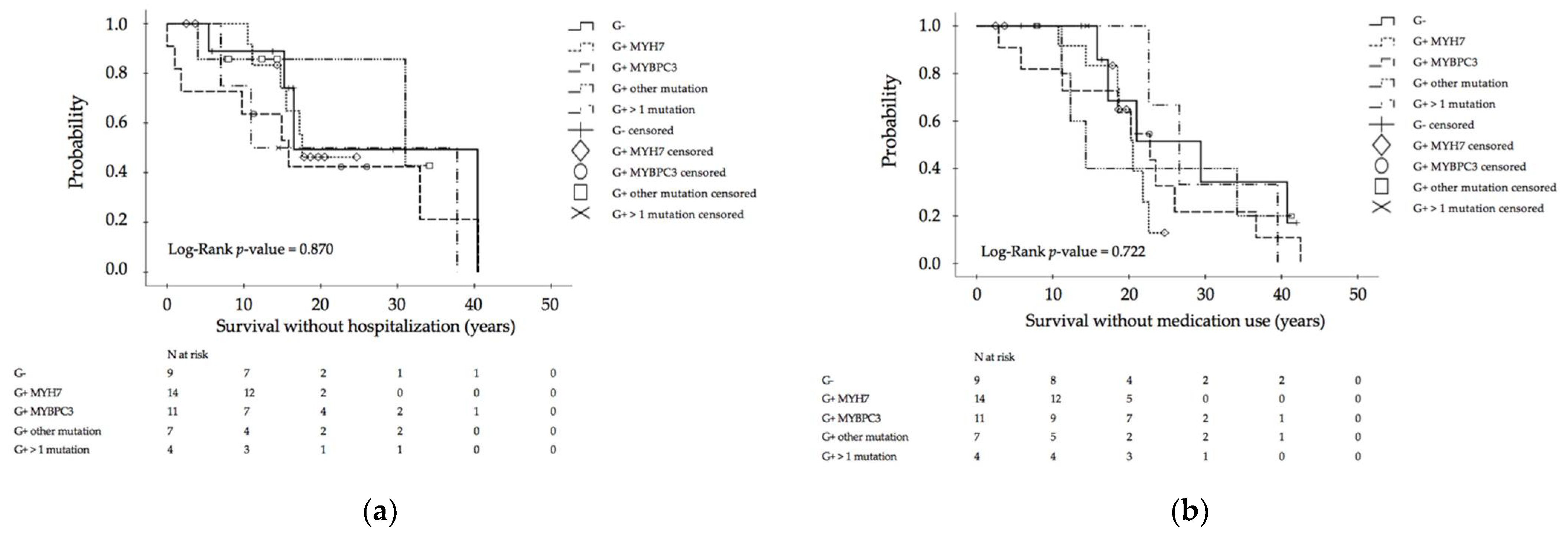
3.3. Genotype–Phenotype Assessment for Disease Progression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maron, B.J. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002, 287, 1308–1320. [Google Scholar] [CrossRef] [Green Version]
- Keren, A.; Syrris, P.; McKenna, W.J. Hypertrophic cardiomyopathy: The genetic determinants of clinical disease expression. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Seidman, C.E.; Seidman, J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, L.; Watkins, H.; MacRae, C.; Lamas, R.; McKenna, W.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994, 77, 701–712. [Google Scholar] [CrossRef]
- Kimura, A.; Harada, H.; Park, J.E.; Nishi, H.; Satoh, M.; Takahashi, M.; Hiroi, S.; Sasaoka, T.; Ohbuchi, N.; Nakamura, T.; et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat. Genet. 1997, 16, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Parvatiyar, M.S.; Pinto, J.R.; Marquardt, M.L.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Potter, J.D.; Ackerman, M.J. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol. 2008, 45, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, C.M. Hypertrophic cardiomyopathy: Genetics and clinical perspectives. Cardiovasc. Diagn. Ther. 2019, 9, S388–S415. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Seidman, J.G.; Seidman, C.E. Phenotypic diversity in hypertrophic cardiomyopathy. Hum. Mol. Genet. 2002, 11, 2499–2506. [Google Scholar] [CrossRef] [PubMed]
- Ullal, A.J.; Abdelfattah, R.S.; Ashley, E.A.; Froelicher, V.F. Hypertrophic Cardiomyopathy as a Cause of Sudden Cardiac Death in the Young: A Meta-Analysis. Am. J. Med. 2016, 129, 486–496.e482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Authors/Task Force, M.; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2020, 76, e159–e240. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.D. The Ross classification for heart failure in children after 25 years: A review and an age-stratified revision. Pediatr. Cardiol. 2012, 33, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2011, 58, 2703–2738. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef] [PubMed]
- Charron, P.; Arad, M.; Arbustini, E.; Basso, C.; Bilinska, Z.; Elliott, P.; Helio, T.; Keren, A.; McKenna, W.J.; Monserrat, L.; et al. Genetic counselling and testing in cardiomyopathies: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2010, 31, 2715–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Amendola, L.M.; Jarvik, G.P.; Leo, M.C.; McLaughlin, H.M.; Akkari, Y.; Amaral, M.D.; Berg, J.S.; Biswas, S.; Bowling, K.M.; Conlin, L.K.; et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am. J. Hum. Genet. 2016, 99, 247. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landstrom, A.P.; Ackerman, M.J. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation 2010, 122, 2441–2449; discussion 2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fananapazir, L.; Epstein, N.D. Genotype-phenotype correlations in hypertrophic cardiomyopathy. Insights provided by comparisons of kindreds with distinct and identical beta-myosin heavy chain gene mutations. Circulation 1994, 89, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, L.R.; Rahman, M.S.; Elliott, P.M. A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013, 99, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Vasile, V.C.; Ommen, S.R.; Will, M.L.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.K.; Sanders, H.K.; McNamara, J.W.; Jagadeesan, A.; Jahangir, A.; Tajik, A.J.; Sadayappan, S. Hypertrophic cardiomyopathy clinical phenotype is independent of gene mutation and mutation dosage. PLoS ONE 2017, 12, e0187948. [Google Scholar] [CrossRef] [Green Version]
- Mathew, J.; Zahavich, L.; Lafreniere-Roula, M.; Wilson, J.; George, K.; Benson, L.; Bowdin, S.; Mital, S. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin. Genet. 2018, 93, 310–319. [Google Scholar] [CrossRef] [Green Version]
- Ingles, J.; Doolan, A.; Chiu, C.; Seidman, J.; Seidman, C.; Semsarian, C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. J. Med. Genet. 2005, 42, e59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moolman, J.C.; Corfield, V.A.; Posen, B.; Ngumbela, K.; Seidman, C.; Brink, P.A.; Watkins, H. Sudden death due to troponin T mutations. J. Am. Coll. Cardiol. 1997, 29, 549–555. [Google Scholar] [CrossRef]
- Maass, A.H.; Ikeda, K.; Oberdorf-Maass, S.; Maier, S.K.; Leinwand, L.A. Hypertrophy, fibrosis, and sudden cardiac death in response to pathological stimuli in mice with mutations in cardiac troponin T. Circulation 2004, 110, 2102–2109. [Google Scholar] [CrossRef]
- Girolami, F.; Ho, C.Y.; Semsarian, C.; Baldi, M.; Will, M.L.; Baldini, K.; Torricelli, F.; Yeates, L.; Cecchi, F.; Ackerman, M.J.; et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J. Am. Coll. Cardiol. 2010, 55, 1444–1453. [Google Scholar] [CrossRef] [Green Version]
- Baudenbacher, F.; Schober, T.; Pinto, J.R.; Sidorov, V.Y.; Hilliard, F.; Solaro, R.J.; Potter, J.D.; Knollmann, B.C. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Investig. 2008, 118, 3893–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafreniere-Roula, M.; Bolkier, Y.; Zahavich, L.; Mathew, J.; George, K.; Wilson, J.; Stephenson, E.A.; Benson, L.N.; Manlhiot, C.; Mital, S. Family screening for hypertrophic cardiomyopathy: Is it time to change practice guidelines? Eur. Heart J. 2019, 40, 3672–3681. [Google Scholar] [CrossRef] [PubMed]
- Miron, A.; Lafreniere-Roula, M.; Steve Fan, C.P.; Armstrong, K.R.; Dragulescu, A.; Papaz, T.; Manlhiot, C.; Kaufman, B.; Butts, R.J.; Gardin, L.; et al. A Validated Model for Sudden Cardiac Death Risk Prediction in Pediatric Hypertrophic Cardiomyopathy. Circulation 2020, 142, 217–229. [Google Scholar] [CrossRef]
- Varnava, A.M.; Elliott, P.M.; Baboonian, C.; Davison, F.; Davies, M.J.; McKenna, W.J. Hypertrophic cardiomyopathy: Histopathological features of sudden death in cardiac troponin T disease. Circulation 2001, 104, 1380–1384. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.C.; Michels, V.V.; Pellikka, P.A.; Ballew, J.D.; Karst, M.L.; Herron, K.J.; Nelson, S.M.; Rodeheffer, R.J.; Olson, T.M. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin. Genet. 2008, 74, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokado, H.; Shimizu, M.; Yoshio, H.; Ino, H.; Okeie, K.; Emoto, Y.; Matsuyama, T.; Yamaguchi, M.; Yasuda, T.; Fujino, N.; et al. Clinical features of hypertrophic cardiomyopathy caused by a Lys183 deletion mutation in the cardiac troponin I gene. Circulation 2000, 102, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Sedaghat-Hamedani, F.; Kayvanpour, E.; Tugrul, O.F.; Lai, A.; Amr, A.; Haas, J.; Proctor, T.; Ehlermann, P.; Jensen, K.; Katus, H.A.; et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: A meta-analysis on 7675 individuals. Clin. Res. Cardiol. 2018, 107, 30–41. [Google Scholar] [CrossRef]
- Ho, C.Y.; Lopez, B.; Coelho-Filho, O.R.; Lakdawala, N.K.; Cirino, A.L.; Jarolim, P.; Kwong, R.; Gonzalez, A.; Colan, S.D.; Seidman, J.G.; et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N. Engl. J. Med. 2010, 363, 552–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, C.M.; Berul, C.I. Molecular mechanisms of inherited arrhythmias. Curr. Genom. 2008, 9, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Saeed, M.; Link, M.S.; Mahapatra, S.; Mouded, M.; Tzeng, D.; Jung, V.; Contreras, R.; Swygman, C.; Homoud, M.; Estes, N.A., 3rd; et al. Analysis of intracardiac electrograms showing monomorphic ventricular tachycardia in patients with implantable cardioverter-defibrillators. Am. J. Cardiol. 2000, 85, 580–587. [Google Scholar] [CrossRef]
- Schober, T.; Huke, S.; Venkataraman, R.; Gryshchenko, O.; Kryshtal, D.; Hwang, H.S.; Baudenbacher, F.J.; Knollmann, B.C. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ. Res. 2012, 111, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients Characteristics and Clinical Outcome Parameters | Genotype- Positive (MYH7) | Genotype- Positive (MYBPC3) | Genotype- Positive (Others) | Genotype- Positive (>1 Mutation) | Genotype-Negative/Phenotype-Positive | p-Value |
|---|---|---|---|---|---|---|
| Patients, n 1 (% of total) | 17 (34.7) | 11 (22.4) | 7 (14.3) | 4 (8.2) | 10 (20.4) | |
| Male, n (%) | 9 (52.9) | 10 (90.9) | 1 (14.3) | 3 (75.0) | 6 (60.0) | 0.025 10 |
| Age at first diagnosis (years) | 13.0 (0.0–19.0) | 9.0 (0.0–18.0) | 6.8 (0.0–15.0) | 3.5 (0.0–7.0) | 8.3 (0.0–15.6) | 0.674 11 |
| Age at last follow-up (years) | 18.7 (2.6–51.7) | 22.7 (3.0–42.5) | 12.4 (7.9–41.4) | 24.6 (14.6–39.6) | 18.0 (5.9–42.0) | 0.521 11 |
| Follow-up time (years) | 5.8 (0.2–22.6) | 8.8 (2.3–31.7) | 10.3 (1.2–27.3) | 21.1 (13.5–33.5) | 7.0 (0.2–38.0) | 0.188 11 |
| Family History | ||||||
| Negative, n (%) | 3/17 (17.6) | 4/11 (22.4) | 1/7 (14.3) | 0/4 (0.0) | 7/10 (70.0) | |
| HCM 2, n (%) | 12/17 (70.6) | 5/11 (45.5) | 2/7 (28.6) | 1/4 (25.0) | 3/10 (30.0) | |
| SCD 3, n (%) | 2/17 (11.8) | 2/11 (18.2) | 4/7 (57.1) | 3/4 (75.0) | 0/10 (0.0) | 0.003 10 |
| Mortality | ||||||
| Death, n (%) | 0/17 (0.0) | 0/11 (0.0) | 0/7 (0.0) | 1/4 (25.0) | 0/10 (0.0) | 0.022 10 |
| Arrhythmia 4 | ||||||
| None, n (%) | 7/14 (50.0) | 3/11 (27.3) | 4/7 (57.1) | 1/4 (25.0) | 6/9 (66.7) | |
| Mild 5, n (%) | 5/14 (35.7) | 4/11 (36.4) | 1/7 (14.3) | 0/4 (0.0) | 3/9 (33.3) | |
| Severe 6, n (%) | 2/14 (14.3) | 4/11 (36.4) | 2/7 (28.6) | 3/4 (75.0) | 0/9 (0.0) | |
| MAEs 7, n (%) | 1/17 (5.9) | 3/11 (27.3) | 1/7 (14.3) | 3/4 (75.0) | 0/10 (0.0) | 0.006 10 |
| Morbidity | ||||||
| Hospitalization, n (%) | 6/17 (35.3) | 8/11 (72.7) | 2/7 (28.6) | 3/4 (75.0) | 4/10 (40.0) | 0.180 10 |
| Age (years), median (range) | 6.5 (1.1–17.3) | 1.0 (0.0–27.9) | 10.5 (4.0–17.0) | 10.9 (0.0–31.8) | 9.0 (0.0–34.5) | 0.722 11 |
| ICD 8, n (%) | 2/17 (11.8) | 4/11 (36.4) | 2/7 (28.6) | 3/4 (75.0) | 2/10 (20.0) | 0.112 10 |
| Age (years), median (range) | 15.2 (14.8–15.5) | 23.9 (9.9–40.5) | 17.7 (4.3–31.0) | 14.3 (10.9–37.8) | 28.5 (16.5–40.4) | 0.700 11 |
| Primary prevention, n (%) | 2/2 (100.0) | 4/4 (100.0) | 2/2 (100.0) | 1/3 (33.3) | 2/2 (100.0) | 0.096 10 |
| Secondary prevention, n (%) | 0/2 (0.0) | 0/4 (0.0) | 0/2 (0.0) | 2/3 (66.6) | 0/2 (0.0) | 0.096 10 |
| Appropriate discharge, n (%) | 1/2 (50.0) | 2/4 (50.0) | 1/2 (50.0) | 2/3 (66.7) | 0/2 (0.0) | 0.686 10 |
| Number of cardiac medication, n (%) | 1 (0–2) | 1 (0–2) | 1 (0–2) | 1 (0–2) | 1 (0–2) | 0.624 10 |
| NYHA 9/Ross class | ||||||
| I, n (%) | 7/14 (50.0) | 6/11 (54.5) | 5/7 (71.4) | 1/4 (25.0) | 6/9 (66.7) | 0.582 10 |
| II, n (%) | 7/14 (50.0) | 5/11 (45.5) | 2/7 (28.6) | 3/4 (75.0) | 3/9 (33.3) | |
| III, n (%) | 0/14 (0.0) | 0/13 (0.0) | 0/7 (0.0) | 0/4 (0.0) | 0/9 (0.0) | |
| IV, n (%) | 0/14 (0.0) | 0/13 (0.0) | 0/7 (0.0) | 0/4 (0.0) | 0/9 (0.0) |
| Imaging Parameter | Genotype- Positive (MYH7) n = 17 | Genotype- Positive (MYBPC3) n = 11 | Genotype- Positive (Others) n = 7 | Genotype- Positive (>1 Mutation) n = 4 | Genotype-Negative/Phenotype-Positive n = 10 | p-Value |
|---|---|---|---|---|---|---|
| Morphology | ||||||
| CMR 1 EDVI 2 (mL/m2), median (range) | 62.0 (50.0–89.0) | 57.0 (36.0–91.0) | 60.0 (53.0–65.0) | 63.5 (63.0–64.0) | 65.0 (48.0–76.0) | 0.949 18 |
| CMR ESVI 3 (mL/m2), median (range) | 19.5 (8.0–27.0) | 17.0 (7.0–35.0) | 19.0 (11.0–22.0) | 16.0 (16.0–16.0) | 17.0 (8.0–27.0) | 0.978 18 |
| LVOTO 4, n 5 (%) | 7/14 (50.0) | 3/11 (27.3) | 0/7(0.0) | 0/4(0.0) | 2/9 (22.2) | 0.092 19 |
| Hypertrophy | ||||||
| CMR myocardial mass (g/m2), median (range) | 100.5 (39.0–168.0) | 83.0 (39.0–213.0) | 58.0 (48.0–95.0) | 80.0 (43.0–117.0) | 90.0 (56.0–126.0) | 0.602 18 |
| TTE 6 IVSd 7 z-score, median (range) | 3.7 (0.6–7.6) | 3.4 (0.2–7.8) | 2.2 (0.1–7.6) | 3.7 (3.1–4.9) | 4.8 (2.2–6.1) | 0.536 18 |
| TTE LVPWd 8 z-score, median (range) | 2.5 (0.7–3.8) | 1.7 (-1.0–5.1) | 2.1 (1.2–5.3) | 2.9 (1.7–3.3) | 2.6 (1.1–4.5) | 0.891 18 |
| Fibrosis | ||||||
| CMR LGE 9, n (%) | 8/10 (80.0) | 8/9 (88.9) | 3/3 (100.0) | 2/2 (100.0) | 4/7 (57.1) | 0.396 19 |
| CMR LGE localization | 0.204 19 | |||||
| Negative, n (%) | 3/10 (30.0) | 3/9 (33.3) | 1/3 (33.3) | 0/2 (0.0) | 3/7 (42.9) | |
| Uncertain detection, n (%) | 3/10 (30.0) | 0/9 (0.0) | 1/3 (33.3) | 1/2 (50.0) | 1/7 (14.3) | |
| Septum, n (%) | 4/10 (40.0) | 4/9 (44.4) | 1/3 (33.3) | 0/2 (0.0) | 2/7 (28.6) | |
| Entire myocardium, n (%) | 0/10 (0.0) | 0/9 (0.0) | 0/3 (0.0) | 1/2 (50.0) | 0/7 (0.0) | |
| Papillary muscle + RVOT 10, n (%) | 0/10 (0.0) | 1/9 (11.1) | 0/3 (0.0) | 0/2 (0.0) | 0/7 (0.0) | |
| LV 11 front wall + septum, n (%) | 0/10 (0.0) | 0/9 (0.0) | 0/3 (0.0) | 0/2 (0.0) | 1/7 (14.3) | |
| Diffuse distribution, n (%) | 0/10 (0.0) | 1/9 (11.1) | 0/3 (0.0) | 0/2 (0.0) | 0/7 (0.0) | |
| CMR LGE mean, median (range) | 7.3 (0.6–14.0) | 20.2 (0.4–23.8) | 7.8 (7.8–7.8) | 4.8 (4.8–4.8) | 1.2 (1.2–1.2) | 0.278 18 |
| CMR ECV 12 total mean, median (range) | 26.8 (25.9–30.6) | 28.6 (24.9–34.4) | 28.5 (28.5–28.5) | 23.1 (23.1–23.1) | 24.8 (24.8–24.8) | 0.289 18 |
| CMR ECV septal mean, median (range) | 31.1 (27.8–34.0) | 28.7 (26.9–34.9) | 31.3 (31.3–31.3) | 22.1 (22.1–22.1) | 26.3 (26.3–26.3) | 0.926 18 |
| Systolic function | ||||||
| TTE EF 13 (%), median (range) | 72.0 (44.0–88.0) | 73.0 (55.0–95.0) | 76.0 (61.0–88.0) | 59.0 (41.0–78.0) | 83.0 (59.0–89.0) | 0.156 18 |
| CMR EF (%), median (range) | 72.5 (62.0.–83.0) | 73.0 (61.0–80.0) | 69.0 (66.0–80.0) | 75.0 (75.0–75.0) | 73.0 (65.0–88.0) | 0.987 18 |
| CMR SVI 14 (mL/m2), median (range) | 42.5 (35.0–67.0) | 45.0 (29.0–59.0) | 43.0 (41.0–44.0) | 47.0 (47.0–47.0) | 46.5 (37.0–59.0) | 0.918 18 |
| GLS 15 average, median (range) | −16.3 (−22.5–−8.8) | −14.7 (−27.8–−7.4) | −10.1 (−24.1–−7.6) | −8.2 (−8.2–−8.2) | −16.25 −22.2–−11.9) | 0.535 18 |
| GLS dispersion, median (range) | −14.5 (−36.0–−4.0) | −11.0 (−24.0–−6.0) | −8.0 (−13.0–−5.0) | −4.0 (−18.0–−2.0) | −11.0 (−19.0–−5.0) | 0.299 18 |
| GLS minimum, median (range) | −24.0 (−38.0–−8.0) | −15.0 (−24.0− −6.0) | −18.0 (-33.0–−10.0) | −13.0 (−17.0–−9.0) | −20.0 (−33.0–−9.0 | 0.34518 |
| GLS maximum, median (range) | −8.5 (−20.0–7.0) | −7.0 (−17.0–3.0) | −6.0 (−22.0–−4.0) | −7.0 (−13.0–5.0) | −9.0 (−15.0–10.0) | 0.576 18 |
| GLS septal basal, median (range) | −9.5 (−26.0–−4.0) | −7.0 (−17.0–3.0) | −11.0 (−22.0–−5.0) | −8.0 (−16.0–−3.0) | −10.0 (−17.0–−5.0) | 0.319 18 |
| GLS septal middle, median (range) | −13.0 (−29.0–−4.0) | −10.0 (−27.0–1.0) | −8.0 (−27.0–−4.0) | −7.0 (−17.0–−4.0) | −12.0 (−25.0–−4.0) | 0.687 18 |
| GLS septal apex, median (range) | −18.0 (−32.0–−5.0) | −15.0(−39.0–−8.0) | −14.0 (−33.0–−7.0) | −13.0 (−13.0–−9.0) | −16.0 (−33.0–10.0) | 0.849 18 |
| GLS lateral basal, median (range) | −20.5 (−38.0–7.0) | −13.0 (−27.0–−5.0) | −18.0 (−33.0–−7.0) | −1.5 (−8.0–5.0) | −14.0 (−27.0–−6.0) | 0.304 18 |
| GLS lateral middle, median (range) | −15.0 (−28.0–3.0) | −18.5 (−32.0–1.0) | −9.5 (−21.0–−6.0) | N/A 20 | −16.0 (−18.0–−12.0) | 0.626 18 |
| GLS lateral apex, median (range) | −15.0 (−30.0–−5.0) | −34.0 (−34–−34.0) | −12.0 (−29.0–−10.0) | −8.0 (−8.0–−8.0) | −16.0 (−20.0–−12.0) | 0.360 18 |
| Diastolic function | ||||||
| MV 16 E/A Ratio, median (range) | 1.4 (0.7–2.2) | 1.8 (1.4–3.0) | 1.7 (1.0–2.5) | 2.0 (1.1–2.0) | 1.5 (1.0–12.8) | 0.420 18 |
| MV E Deceleration (m/s), median (range) | 2.0 (1.1–2.5) | 2.0 (1.5–2.9) | 2.1 (1.7–2.8) | 2.2 (2.0–2.6) | 2.0 (1.6–2.9) | 0.610 18 |
| MV E maximum (m/s), median (range) | 0.8 (0.4–1.3) | 0.8 (0.7–1.5) | 0.7 (0.6–1.1) | 0.6 (0.5–0.8) | 0.8 (0.6–1.8) | 0.504 18 |
| MV E’ septal (m/s), median (range) | 9.5 (5.0–13.0) | 5.7 (5.0–7.0) | 11.0 (3.0–15.0) | 5.5 (4.0–7.0) | 7.5 (3.0–12.0) | 0.176 18 |
| MV E’ lateral (m/s), median (range) | 10.0 (5.0–12.0) | 9.0 (7.0–11.0) | 7.0 (5.0–18.0) | 7.0 (5.0–9.0) | 11.5 (7.0–14.0) | 0.459 18 |
| E/E’ septal, median (range) | 7.9 (5.2–21.8) | 12.3 (1.7–14.7) | 7.5 (6.1–20.0) | 10.5 (8.6–12.5) | 11.6 (6.9–19.8) | 0.507 18 |
| E/E’ lateral, median (range) | 8.0 (5.8–11.9) | 8.1 (7.0–9.4) | 8.1 (−5.1–12.0) | 8.3 (6.7–10.0) | 7.3 (5.2–14.1) | 0.895 18 |
| LA 17 Diameter (cm), median (range) | 3.3 (1.7–5.2) | 3.2 (2.0–4.8) | 2.5 (1.9–4.9) | 4.6 (4.3–5.1) | 2.6 (2.0–3.8) | 0.106 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pollmann, K.; Kaltenecker, E.; Schleihauf, J.; Ewert, P.; Görlach, A.; Wolf, C.M. Compound Mutation in Cardiac Sarcomere Proteins Is Associated with Increased Risk for Major Arrhythmic Events in Pediatric Onset Hypertrophic Cardiomyopathy. J. Clin. Med. 2021, 10, 5256. https://doi.org/10.3390/jcm10225256
Pollmann K, Kaltenecker E, Schleihauf J, Ewert P, Görlach A, Wolf CM. Compound Mutation in Cardiac Sarcomere Proteins Is Associated with Increased Risk for Major Arrhythmic Events in Pediatric Onset Hypertrophic Cardiomyopathy. Journal of Clinical Medicine. 2021; 10(22):5256. https://doi.org/10.3390/jcm10225256
Chicago/Turabian StylePollmann, Kathrin, Emanuel Kaltenecker, Julia Schleihauf, Peter Ewert, Agnes Görlach, and Cordula M. Wolf. 2021. "Compound Mutation in Cardiac Sarcomere Proteins Is Associated with Increased Risk for Major Arrhythmic Events in Pediatric Onset Hypertrophic Cardiomyopathy" Journal of Clinical Medicine 10, no. 22: 5256. https://doi.org/10.3390/jcm10225256
APA StylePollmann, K., Kaltenecker, E., Schleihauf, J., Ewert, P., Görlach, A., & Wolf, C. M. (2021). Compound Mutation in Cardiac Sarcomere Proteins Is Associated with Increased Risk for Major Arrhythmic Events in Pediatric Onset Hypertrophic Cardiomyopathy. Journal of Clinical Medicine, 10(22), 5256. https://doi.org/10.3390/jcm10225256