
A Study on the Genetics of Primary Ciliary Dyskinesia


Abstract
:1. Introduction
2. Methods
2.1. Information from Public Databases
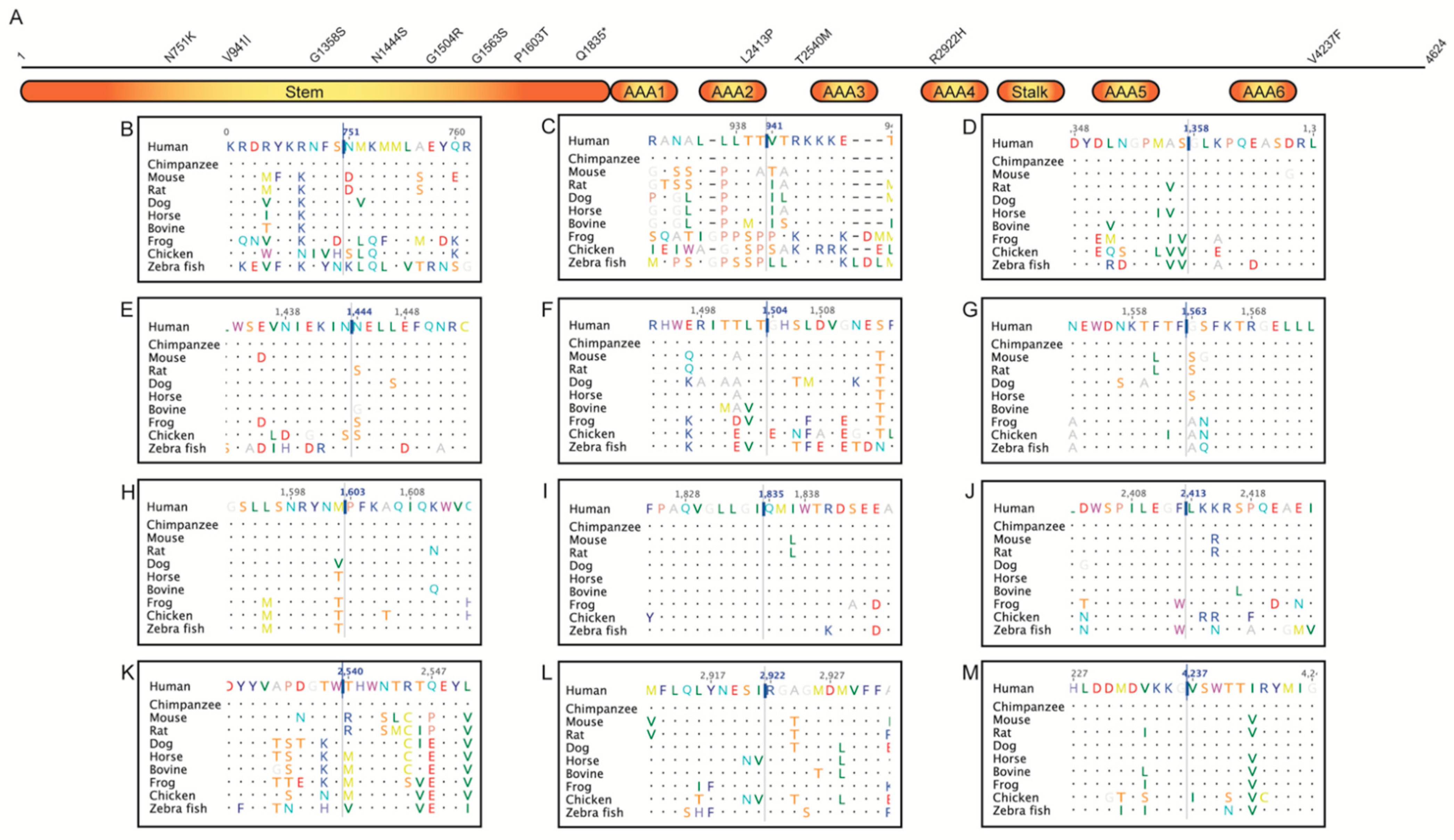
2.2. Multiple Sequence Alignment
2.3. Jensen–Shannon Divergence (JSD)
2.4. Clustering and Multidimensional Scaling
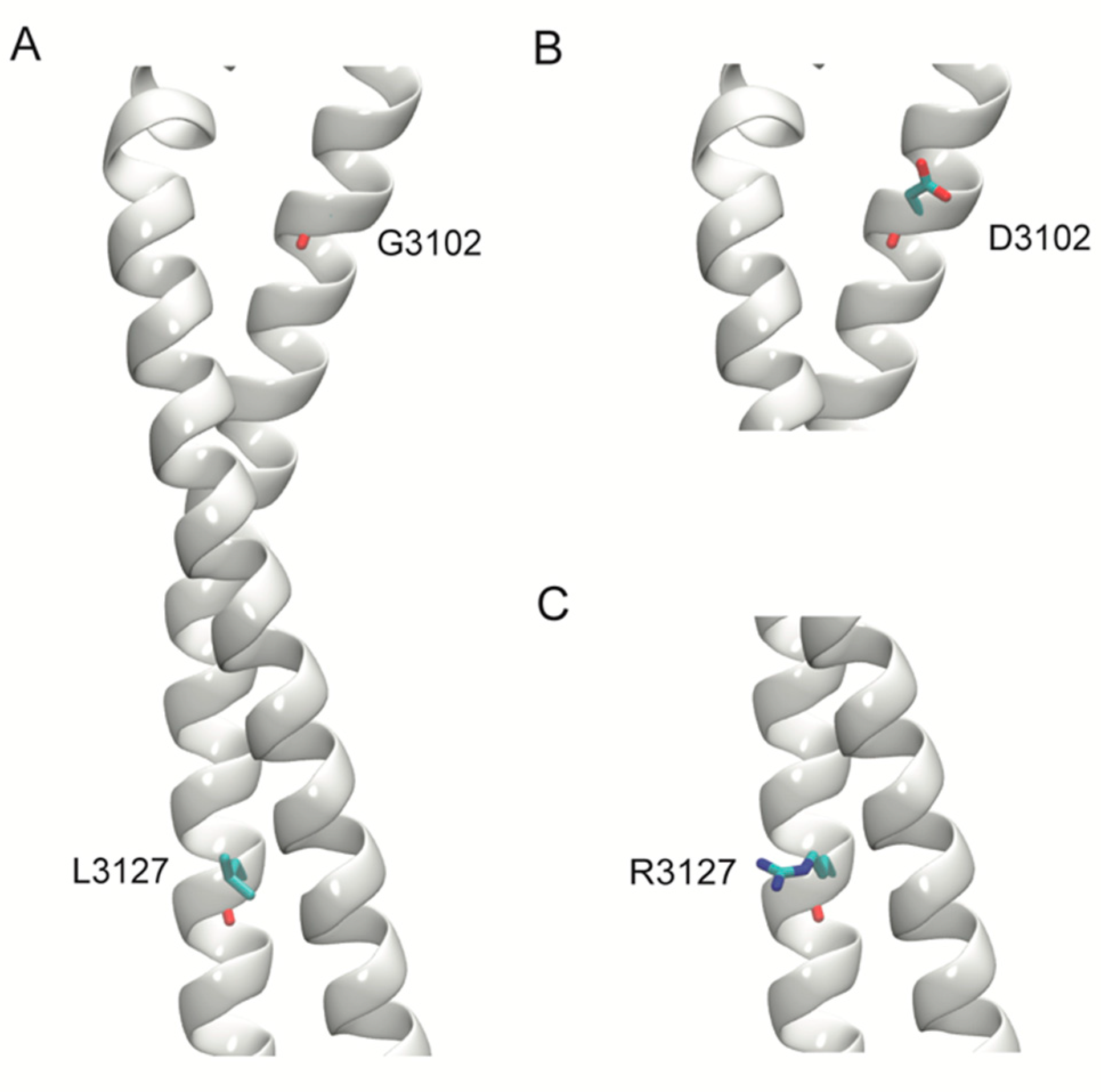
2.5. Structural Modeling
2.6. Statistics
3. Results
Novel Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Butterfield, R. Primary Ciliary Dyskinesia. Pediatr. Rev. 2017, 38, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Zariwala, M.; Leigh, M. Primary Ciliary Dyskinesia. Clin. Chest Med. 2016, 37, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosie, P.H.; Fitzgerald, D.A.; Jaffe, A.; Birman, C.S.; Rutland, J.; Morgan, L.C. Presentation of primary ciliary dyskinesia in children: 30 years’ experience. J. Paediatr. Child Health 2014, 51, 722–726. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.G.; Griffiths, A.; Iyer, N.P.; Shapiro, A.; Wilson, K.C.; Thomson, C.C. Summary for Clinicians: Diagnosis of Primary Ciliary Dyskinesia. Ann. Am. Thorac. Soc. 2019, 16, 171–174. [Google Scholar] [CrossRef]
- Lucas, J.S.; Davis, S.D.; Omran, H.; Shoemark, A. Primary ciliary dyskinesia in the genomics age. Lancet Respir. Med. 2020, 8, 202–216. [Google Scholar] [CrossRef]
- Horani, A.; Ferkol, T.W. Advances in the Genetics of Primary Ciliary Dyskinesia: Clinical Implications. Chest 2018, 154, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Ferkol, T.W.; Dutcher, S.K.; Brody, S.L. Genetics and biology of primary ciliary dyskinesia. Paediatr. Respir. Rev. 2016, 18, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Merveille, A.-C.; Davis, E.E.; Becker-Heck, A.; Legendre, M.; Amirav, I.; Bataille, G.; Belmont, J.W.; Beydon, N.; Billen, F.; Clément, A.; et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2010, 43, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Yanagisawa, H.; Kamiya, R.; Kikkawa, M. A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 2014, 346, 857–860. [Google Scholar] [CrossRef]
- Zariwala, M.A.; Knowles, M.R.; Leigh, M.W. Primary Ciliary Dyskinesia. In GeneReviews®; Adam, M.P., Ar-dinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1122/ (accessed on 25 October 2021).
- Antony, D.; Becker-Heck, A.; Zariwala, M.A.; Schmidts, M.; Onoufriadis, A.; Forouhan, M.; Wilson, R.; Taylor-Cox, T.; Dewar, A.; Jackson, C.; et al. Mutations inCCDC39andCCDC40are the Major Cause of Primary Ciliary Dyskinesia with Axonemal Disorganization and Absent Inner Dynein Arms. Hum. Mutat. 2013, 34, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Antony, D.; Brunner, H.G.; Schmidts, M. Ciliary Dyneins and Dynein Related Ciliopathies. Cells 2021, 10, 1885. [Google Scholar] [CrossRef] [PubMed]
- Baz-Redón, N.; Rovira-Amigo, S.; Fernández-Cancio, M.; Castillo-Corullón, S.; Cols, M.; Caballero-Rabasco, M.A.; Asensio, Ó.; de Vicente, C.M.; Martínez-Colls, M.D.M.; Torrent-Vernetta, A.; et al. Immunofluorescence Analysis as a Diagnostic Tool in a Spanish Cohort of Patients with Suspected Primary Ciliary Dyskinesia. J. Clin. Med. 2020, 9, 3603. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.G.; Horani, A.; Shapiro, A.J. Progress in Diagnosing Primary Ciliary Dyskinesia: The North American Perspective. Diagnostics 2021, 11, 1278. [Google Scholar] [CrossRef]
- Werner, C.; Onnebrink, J.G.; Omran, H. Diagnosis and management of primary ciliary dyskinesia. Cilia 2015, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shoemark, A.; Frost, E.; Dixon, M.; Ollosson, S.; Kilpin, K.; Patel, M.; Scully, J.; Rogers, A.V.; Mitchison, H.M.; Bush, A.; et al. Accuracy of Immunofluorescence in the Diagnosis of Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 2017, 196, 94–101. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Dell, S.D.; Gaston, B.; O’Connor, M.; Marozkina, N.; Manion, M.; Hazucha, M.J.; Leigh, M.W. Nasal Nitric Oxide Measurement in Primary Ciliary Dyskinesia. A Technical Paper on Standardized Testing Protocols. Ann. Am. Thorac. Soc. 2020, 17, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Ostrowski, L.E.; Leigh, M.W.; Sears, P.R.; Davis, S.D.; Wolf, W.E.; Hazucha, M.J.; Carson, J.L.; Olivier, K.N.; Sagel, S.D.; et al. Mutations inRSPH1Cause Primary Ciliary Dyskinesia with a Unique Clinical and Ciliary Phenotype. Am. J. Respir. Crit. Care Med. 2014, 189, 707–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horani, A.; Ferkol, T.W. Understanding Primary Ciliary Dyskinesia and Other Ciliopathies. J. Pediatr. 2021, 230, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.J.; Zariwala, M.A.; Ferkol, T.W.; Davis, S.D.; Sagel, S.D.; Dell, S.D.; Rosenfeld, M.; Olivier, K.N.; Milla, C.; Daniel, S.J.; et al. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr. Pulmonol. 2016, 51, 115–132. [Google Scholar] [CrossRef] [PubMed]
- AlSaadi, M.M.; Gaunt, T.R.; Boustred, C.R.; Guthrie, P.A.I.; Liu, X.; Lenzi, L.; Rainbow, L.; Hall, N.; Alharbi, K.K.; Day, I.N.M. From a Single Whole Exome Read to Notions of Clinical Screening: Primary Ciliary Dyskinesia and RSPH9 p.Lys268del in the Arabian Peninsula. Ann. Hum. Genet. 2012, 76, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United Arab Emirates Population (2021)—Worldometer. Available online: https://www.worldometers.info/world-population/united-arab-emirates-population/ (accessed on 9 October 2021).
- Alsamri, M.T.; Alabdouli, A.; Alkalbani, A.M.; Iram, D.; Antony, P.; Vijayan, R.; Souid, A.-K. Genetic variants in children with chronic respiratory diseases. Pediatr. Pulmonol. 2020, 55, 2389–2401. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Al Mogarri, I.; Alqwaiee, M.M.; Alharbi, A.S.; Baqais, K.; AlSaadi, M.; AlAnzi, T.; Alhashem, A.; Saghier, A.; Ameen, W.; et al. An exome-first approach to aid in the diagnosis of primary ciliary dyskinesia. Qual. Life Res. 2020, 139, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Sivadas, A.; Scaria, V. Pharmacogenomic survey of Qatari populations using whole-genome and exome sequences. Pharm. J. 2018, 18, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Hammoudeh, S.; Gadelhak, W.; Janahi, I.A. Primary ciliary dyskinesia among Arabs: Where do we go from here? Paediatr. Respir. Rev. 2018, 29, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Reish, O.; Slatkin, M.; Chapman-Shimshoni, D.; Elizur, A.; Chioza, B.; Castleman, V.; Mitchison, H.M. Founder Mutation(s) in theRSPH9Gene Leading to Primary Ciliary Dyskinesia in Two Inbred Bedouin Families. Ann. Hum. Genet. 2010, 74, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stannard, W.; Rutman, A.; Wallis, C.; O’Callaghan, C. Central Microtubular Agenesis Causing Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 2004, 169, 634–637. [Google Scholar] [CrossRef]
- Castleman, V.H.; Romio, L.; Chodhari, R.; Hirst, R.A.; de Castro, S.C.P.; Parker, K.A.; Ybot-Gonzalez, P.; Emes, R.D.; Wilson, S.W.; Wallis, C.; et al. Mutations in Radial Spoke Head Protein Genes RSPH9 and RSPH4A Cause Primary Ciliary Dyskinesia with Central-Microtubular-Pair Abnormalities. Am. J. Hum. Genet. 2009, 84, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, C.G.; Cox, J.; Springell, K.; Hampshire, D.J.; Mohamed, M.D.; McKibbin, M.; Stern, R.; Raymond, F.L.; Sandford, R.; Sharif, S.M.; et al. Quantification of Homozygosity in Consanguineous Individuals with Autosomal Recessive Disease. Am. J. Hum. Genet. 2006, 78, 889–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunning, A.C.; Fryer, V.; Fasham, J.; Crosby, A.H.; Ellard, S.; Baple, E.L.; Wright, C.F. Assessing performance of pathogenicity predictors using clinically relevant variant datasets. J. Med. Genet. 2021, 58, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Ba, W.; Yan, Y.; Reijnders, M.R.F.; Schuurs-Hoeijmakers, J.H.M.; Feenstra, I.; Bongers, E.M.H.F.; Bosch, D.G.M.; de Leeuw, N.; Pfundt, R.; Gilissen, C.; et al. TRIOloss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet. 2016, 25, 892–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citti, A.; Peca, D.; Petrini, S.; Cutrera, R.; Biban, P.; Haass, C.; Boldrini, R.; Danhaive, O. Ultrastructural Characterization of Genetic Diffuse Lung Diseases in Infants and Children: A Cohort Study and Review. Ultrastruct. Pathol. 2013, 37, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Kurkowiak, M.; Zietkiewicz, E.; Witt, M. Recent advances in primary ciliary dyskinesia genetics. J. Med. Genet. 2015, 52, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comprehensive Pulmonary Disease Panel | Whole (Diagnostic) Exome Sequencing (WES) | Chromosomal Microarray (CMA) | Targeted Deletion/Duplication Analysis | Single Gene Sequencing | |
|---|---|---|---|---|---|
| Tests done | 48 | 16 | 15 | 5 | 8 |
| PCD genetic defect detected | 48 | 13 | 5 | 0 | 8 |
| PCD genetic defect not detected | 0 | 3 (Three children had negative WES; thereafter, a variant was detected by the Panel in two of them (Patients 47 and 48; Supplementary Table S2) and duplication in exons 1–47 of DNAH5 was detected by CMA in another child (Patient 50)). | 10 | 5 | 0 |
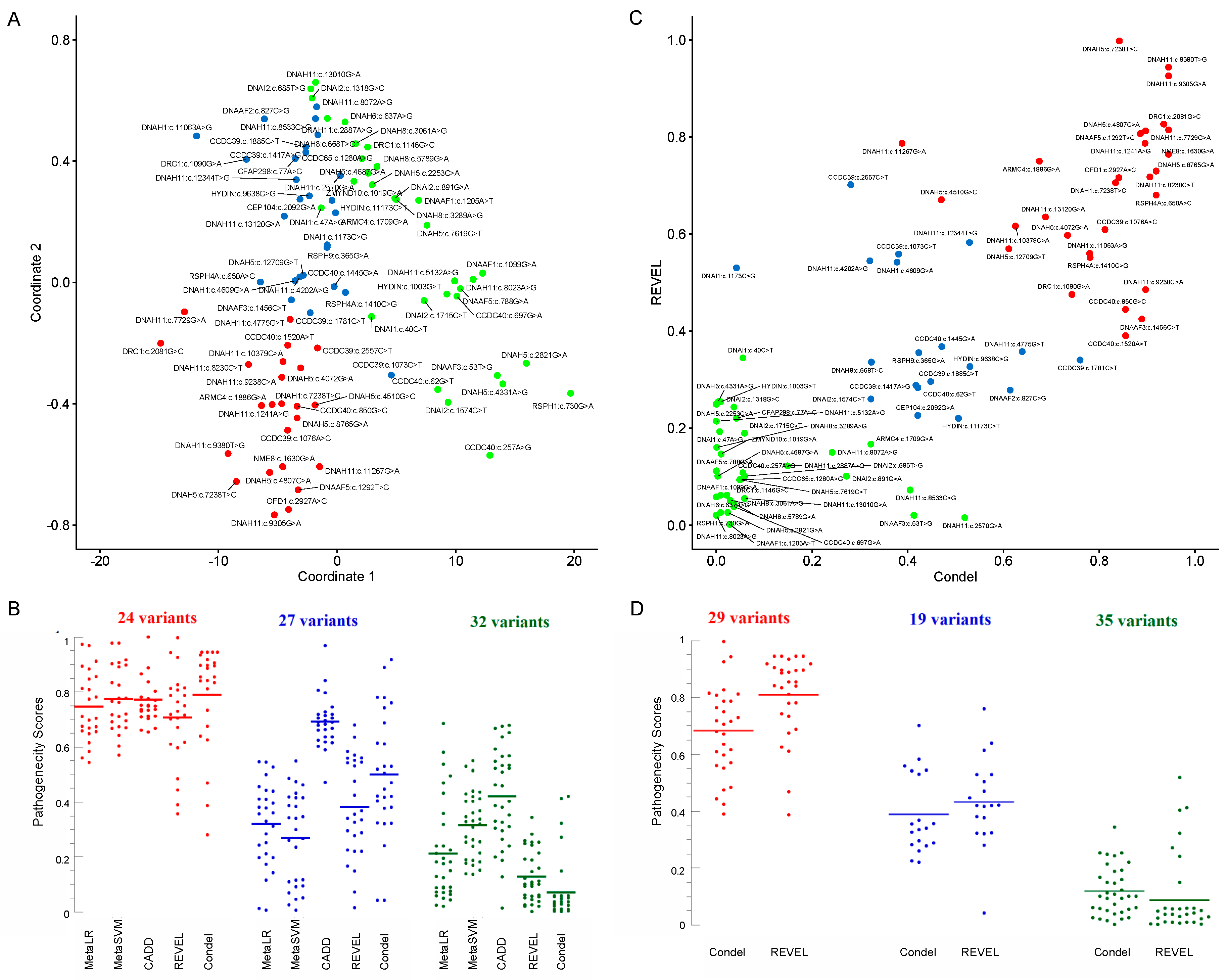
| Variant | Frequency | JSD | MetaLR | MetaSVM | CADD | REVEL | Condel | Scatter | MDS | ACMG | Clinical Assessment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ARMC4(NM_018076.4):c.1709G>A [cGg/cAg] (p.Arg570Gln) Missense, rs140569195 | 0.0004742 | 0.82265 Conserved Figure S5K | 0.41008 T | 0.26729 T | 20.3 | 0.16698 | 0.323 N | Green | Blue | Likely benign | - |
| ARMC4(NM_018076.4):c.1886G>A [cGc/cAc] (p.Arg629His) Missense, rs200127444 | 0.00007571 * | 0.82467 Conserved Figure S5L | 0.774 T | 0.7778 T | 25.6 | 0.75005 | 0.675 D | Red | Red | Likely benign | - |
| C1orf127(NM_001170754.1):c.337C>T [Cga/Tga] (p.Arg113Ter) Nonsense, rs558323413 | 0.00002156 | 0.80196 Conserved Figure S5Q | - | - | 28.1 | - | - | - | - | Uncertain significance | Patient 2 |
| CCDC39(NM_181426.2):c.1073C>T [aCa/aTa] (p.Thr358Ile) Missense, rs183413880 | 0.003822 * | 0.63091 Figure S2A | 0.44228 T | 0.41582 T | 15.58 | 0.55883 | 0.381 N | Blue | Blue | Benign | Patient 3 |
| CCDC39(NM_181426):c.1076A>C [aAa/aCa] (p.Lys359Thr) Missense, rs956532574 | - | 0.7388 Conserved Figure S2B | 0.81295 D | 0.77653 T | 24.3 | 0.60952 | 0.812 D | Red | Red | Uncertain significance | Patient 3 |
| CCDC39(NM_181426.1):c.1167+1261A>G, Splice donor, rs577069249 | - | - | - | - | - | - | - | - | - | Uncertain significance | Pathogenic, Patient 4 |
| CCDC39(NM_181426.1):c.1363-3delC, Splice acceptor, rs551191744 | - | - | - | - | - | - | - | - | - | Uncertain significance | Patient 5 |
| CCDC39(NM_181426.1):c.1417A>G [Aat/Gat] (p.Asn473Asp) Missense, rs1241950069 | 0.00000811 | 0.81432 Conserved Figure S2C | 0.28445 T | 0.06946 T | 22.8 | 0.28867 | 0.417 N | Blue | Blue | Likely benign | - |
| CCDC39(NM_181426.1):c.1528-11_1528-10delCT; Splice acceptor, rs765966793 | 0.00005236 | - | - | - | - | - | - | - | - | Likely benign | - |
| CCDC39(NM_181426.2):c.1781C>T [aCa/aTa] (p.Thr594Ile) Missense, rs140505857 | 0.0006629 | 0.73478 Figure S2D | 0.5455 T | 0.41927 T | 22.4 | 0.34042 | 0.760 D | Blue | Blue | Likely benign | Patient 5 |
| CCDC39(NM_181426.1):c.1792G>T [Gag/Tag] (p.Glu598Ter) Nonsense, Novel | - | 0.79808 Figure S2E | - | - | 37 | - | - | - | - | - | - |
| CCDC39(NM_181426.1):c.1885C>T [Cgc/Tgc] (p.Arg629Cys) Missense, rs199526690 | 0.0001162 | 0.77558 Figure S2F | 0.198 T | 0.09238 T | 22.8 | 0.29614 | 0.448 N | Blue | Blue | Likely benign | - |
| CCDC39(NM_181426.2):c.2557C>T [Cgt/Tgt] (p.Arg853Cys) Missense, rs201097154 | 0.0003535 | 0.60342 Figure S2G | 0.65899 T | 0.57055 T | 21.8 | 0.70179 | 0.281 N | Blue | Red | Likely benign | - |
| CCDC39(NM_181426.1):c.2660dupT [cta/ctTa] (p.Ser888fs) Frameshift, rs200353947 | 0.007916 * | - | - | - | - | - | - | - | - | Benign | - |
| CCDC40(NM_017950.3):c.62G>T [gGa/gTa] (p.Gly21Val) Missense, Novel | - | 0.48751 Figure S2H | 0.43298 T | 0.34655 T | 11.66 | 0.28361 | 0.421 N | Blue | Green | - | Pathogenic, Patient 8 |
| CCDC40(NM_017950.3):c.257A>G [tAt/tGt] (p.Tyr86Cys) Missense, rs202220442 | 0.0000728 | 0.41972 Figure S2I | 0.68522 T | 0.55015 T | 7.262 | 0.10822 | 0.056 N | Green | Green | Likely benign | Pathogenic, Patient 6 |
| CCDC40(NM_017950.3):c.697G>A [Gat/Aat] (p.Asp233Asn Missense, rs201815496 | 0.0002182 | 0.47098 Figure S2J | 0.2398 T | 0.36059 T | 10.07 | 0.0383 | 0.037 N | Green | Green | Likely benign | Patient 7 |
| CCDC40(NM_017950.3):c.850G>C [Gac/Cac] (p.Asp284His) Missense, rs201042940 | 0.002833 * | 0.79987 Figure S2K | 0.71147 T | 0.76216 T | 23.5 | 0.44471 | 0.855 D | Red | Red | Benign | Likely pathogenic or uncertain significance, Patient 7 |
| CCDC40(NM_017950.3):c.1097delT [cTg/cg], Frameshift, Novel | - | - | - | - | - | - | - | - | - | - | - |
| CCDC40(NM_017950.3):c.1445G>A [tGc/tAc] (p.Cys482Tyr) Missense, rs367601192 | 0.0000483 | 0.74673 Figure S2L | 0.45678 T | 0.38742 T | 20.4 | 0.36828 | 0.471 D | Blue | Blue | Likely benign | Patient 6 |
| CCDC40(NM_017950.3):c.1520A>T [aAg/aTg] (p.Lys507Met) Missense, rs563467821 | 0.0003944 | 0.76562 Figure S2M | 0.61518 T | 0.60299 T | 24.3 | 0.39053 | 0.855 D | Red | Red | Uncertain significance | - |
| CCDC40(NM_017950.4):c.2440C>T [Cga/Tga] (p.Arg814Ter) Nonsense, rs747233125 | 0.00000877 | 0.79007 Figure S2N | - | - | 42.0 | - | - | - | - | Pathogenic | - |
| CCDC65(NM_033124.4):c.1280A>G [gAt/gGt] (p.Asp427Gly) Missense, rs866658813 | 0.00000795 | 0.66012 Figure S2O | 0.02439 T | 0.42009 T | 18.07 | 0.0939 | 0.051 N | Green | Green | Likely benign | - |
| CCDC114(NM_144577.3):c.747G>C [ggG/ggC] (p.Gly249=) Synonymous, rs745962113 | 0.00002048 | - | - | - | - | - | - | - | - | Likely benign | - |
| CCDC114(NM_144577.3):c.1032G>A [aaG/aaA] (p.Lys344=) Synonymous, rs753921661 | 0.00003589 | - | - | - | - | - | - | - | - | Likely benign | - |
| CEP104(NM_014704.3):c.2092G>A [Gaa/Aaa] (p.Glu698Lys) Missense, Novel | - | 0.79226 Conserved Figure S5M | 0.39747 T | 0.34116 T | 23.3 | 0.22627 | 0.421 N | Blue | Blue | - | Pathogenic, Patient 9 |
| CFAP298(NM_021254.4):c.77A>C [gAg/gCg] (p.Glu26Ala) Missense, rs138178722 | 0.002517 * | 0.70416 Figure S5N | 0.44009 T | 0.41632 T | 23.7 | 0.220036 | 0.042 N | Green | Blue | Benign | - |
| DNAAF1(NM_178452.4):c.241_242delAG, [AGg/g], Frameshift, rs761836563 | 0.00000398 | - | - | - | - | - | - | - | - | Likely pathogenic | - |
| DNAAF1(NM_178452.5):c.1099G>A [Ggg/Agg] (p.Gly367Arg) Missense, rs763129355 | 0.00008352 | 0.55445 Figure S3A | 0.12497 T | 0.13655 T | 7.909 | 0.06188 | 0.009 N | Green | Green | Likely benign | - |
| DNAAF1(NM_178452.5):c.1205A>T [gAg/gTg] (p.Glu402Val) Missense, rs144034147 | 0.0001472 | 0.32593 Figure S3B | 0.19559 T | 0.15188 T | 13.3 | 0.00175 | 0.028 N | Green | Green | Likely benign | - |
| DNAAF1(NM_178452.4):c.1698+1G>A, Splice donor, rs139519641 | 0.0004176 | - | - | - | - | - | - | - | - | Likely pathogenic | - |
| DNAAF2(NM_018139.2):c.827C>G [cCg/cGg] (p.Pro276Arg) Missense, rs562712293 | 0.00001243 | 0.7484 Figure S3C | 0.19839 T | 0.11656 T | 26.3 | 0.27849 | 0.614 D | Blue | Blue | Likely benign | - |
| DNAAF3(NM_001256716):c.53T>G [aTt/aGt] (p.Ile18Ser) Missense, rs537635826 | 0.0008102 | - | 0.06425 T | 0.40327 T | 6.642 | 0.02051 | 0.413 N | Green | Green | Likely benign | - |
| DNAAF3(NM_001256714.1):c.1116+5G>C Splice donor, rs1037483400 | - | - | - | - | - | - | - | - | - | Uncertain significance | Patient 18; an aberrant effect on splicing is likely. |
| DNAAF3(NM_001256714.1):c.1456C>T, [Cgg/Tgg] (p.Arg486Trp) Missense, rs201929981 | 0.0002648 * | 0.72226 Figure S3D | 0.38193 T | 0.47481 T | 24 | 0.42485 | 0.889 D | Red | Blue | Likely benign | - |
| DNAAF5(NM_017802.3):c.788G>A [cGg/cAg] (p.Arg263Gln) Missense, rs201059622 | 0.0001155 * | 0.75769 Figure S3E | 0.06998 T | 0.42963 T | 9.761 | 0.11179 | 0.000 N | Green | Green | Likely benign | - |
| DNAAF5(NM_017802.3):c.1131G>T [gtG/gtT] (p.Val377=) Synonymous, rs151119269 | 0.00001593 | 0.69489 | - | - | - | - | - | - | - | Likely benign | - |
| DNAAF5(NM_017802.4):c.1292T>C [gTc/gCc] (p.Val431Ala) Missense, Novel | - | 0.79355 Conserved Figure S3F | 0.80849 T | 0.83461 D | 23.4 | 0.80713 | 0.886 D | Red | Red | - | - |
| DNAH1(NM_015512.5):c.4609G>A [Gtg/Atg] (p.Val1537Met) Missense, rs768532151 | 0.00001204 | 0.81432 Conserved Figure S1A | 0.50078 T | 0.54831 T | 23.7 | 0.54201 | 0.378 N | Blue | Blue | Uncertain significance | - |
| DNAH1(NM_015512.5):c.7238T>C [cTt/cCt] (p.Val2413Ala) Missense, rs1164570685 | 0.000004013 | 0.7822 Conserved Figure S1B | 0.65577 T | 0.72023 T | 24.8 | 0.70589 | 0.834 D | Red | Red | Uncertain significance | - |
| DNAH1(NM_015512.4):c.11063A>G [tAc/tGc] (p.Tyr3688Cys) Missense, rs369995851 | 0.0000241 | 0.85784 Conserved Figure S1C | 0.33071 T | 0.23649 T | 32 | 0.5602 | 0.780 D | Red | Blue | Uncertain significance | - |
| DNAH5(NM_001369.2):c.278-3T>C Splice acceptor, rs1244727714 | 0.00001593 | - | - | - | - | - | - | - | - | Uncertain significance | - |
| DNAH5(NM_001369.2):c.2053-23A>C Splice acceptor, rs114717951 | 0.009576 * | - | - | - | - | - | - | - | - | Benign | - |
| DNAH5(NM_001369.2):c.2253C>A [aaC/aaA] (p.Asn751Lys) Missense, rs115004914 | 0.009721 * | 0.68547 Figure 2B | 0.17657 T | 0.25336 T | 17.21 | 0.24907 | 0.000 N | Green | Green | Benign | - |
| DNAH5(NM_001369.2):c.2821G>A [Gtc/Atc] (p.Val941Ile) Missense, rs370080157 | 0.00009554 | 0.56602 Figure 2C | 0.09101 T | 0.40345 T | 4.202 | 0.02609 | 0.024 N | Green | Green | Likely benign | - |
| DNAH5(NM_001369.2):c.3471G>A [aaG/aaA] (p.Lys1157=) Synonymous, rs865979045 | - | - | - | - | - | - | - | - | - | Likely benign | - |
| DNAH5(NM_001369.2):c.4072G>A [Ggc/Agc] (p.Gly1358Ser) Missense, rs752638332 | 0.00002807 | 0.75801 Conserved Figure 2D | 0.54345 T | 0.64213 T | 23.2 | 0.5976 | 0.734 D | Red | Red | Uncertain significance | - |
| DNAH5(NM_001369.2):c.4331A>G [aAt/aGt] (p.Asn1444Ser) Missense, rs567013299 | 0.0003152 * | 0.7356 Figure 2E | 0.35818 T | 0.26259 T | 6.187 | 0.25457 | 0.004 N | Green | Green | Benign | - |
| DNAH5(NM_001369.2):c.4510G>C [Ggg/Cgg] (p.Gly1504Arg) Missense, rs143567667 | 0.000728 | 0.71857 Figure 2F | 0.69826 T | 0.76696 T | 22 | 0.67108 | 0.470 D | Red | Red | Uncertain significance | - |
| DNAH5(NM_001369.2):c.4680C>T [ttC/ttT] (p.Phe1560=) Synonymous, rs1283006383 | 0.000003978 | 0.76067 | - | - | - | - | - | - | - | Likely benign | - |
| DNAH5(NM_001369.2):c.4687G>A [Ggc/Agc] (p.Gly1563Ser) Missense, rs147567352 | 0.00005569 | 0.68677 Figure 2G | 0.23942 T | 0.27354 T | 17.56 | 0.10108 | 0.003 N | Green | Green | Likely benign | - |
| DNAH5(NM_001369.2):c.4807C>A [Cca/Aca] (p.Pro1603Thr) Missense, rs369137751 | 0.002391 * | 0.84684 Conserved Figure 2H | 0.84694 D | 0.89805 D | 25.8 | 0.8124 | 0.897 D | Red | Red | Benign | Pathogenic, Patient 10 |
| DNAH5(NM_001369.2):c.5503C>T [Cag/Tag] (p.Gln1835Ter) Nonsense, rs761622153 | 0.00000799 | 0.8610 Conserved Figure 2I | - | - | 50 | - | - | - | - | Pathogenic | Pathogenic, Patient 11 |
| DNAH5(NM_001369.2):c.7238T>C [cTt/cCt] (p.Leu2413Pro) Missense, Novel | - | 0.78526 Conserved Figure 2J | 0.96992 D | 0.97908 D | 28.6 | 0.99806 | 0.842 D | Red | Red | - | - |
| DNAH5(NM_001369.2):c.7619C>T [aCg/aTg] (p.Thr2540Met) Missense, rs144428526 | 0.00001591 | 0.74254 Figure 2K | 0.08732 T | 0.25497 T | 12.59 | 0.0939 | 0.049 N | Green | Green | Likely benign | - |
| DNAH5(NM_001369.2):c.8765G>A [cGt/cAt] (p.Arg2922His) Missense, rs148539877 | 0.00009165 | 0.80492 Conserved v 2L | 0.58306 T | 0.66715 T | 23.5 | 0.73 | 0.919 D | Red | Red | Uncertain significance | - |
| DNAH5(NM_001369.2):c.12709G>T [Gtc/Ttc] (p.Val4237Phe) Missense, rs138045391 | 0.0001117 | 0.77942 Conserved Figure 2M | 0.37894 T | 0.34678 T | 23 | 0.56961 | 0.611 D | Red | Blue | Uncertain significance | - |
| DNAH5(NM_001369.2):c.13492-15T>C, Splice acceptor, rs192514899 | 0.0003582 * | - | - | - | - | - | - | - | - | Likely benign | - |
| DNAH5(NM_001369.2): Duplication of exons 1-47, Duplication, Novel | - | - | - | - | - | - | - | - | - | - | - |
| DNAH6(NM_001370.1):c.637A>G [Att/Gtt] (p.Ile213Val) Missense, rs774899113 | 0.0002001 | 0.82579 Conserved Figure S1D | 0.18202 T | 0.23812 T | 19.52 | 0.0584 | 0.000 N | Green | Green | Likely benign | - |
| DNAH8(NM_001206927.1):c.668T>C [aTa/aCa] (p.Ile223Thr) Missense, rs1554195443 | - | 0.73642 Figure S1E | 0.14334 T | 0.05175 T | 21.8 | 0.33598 | 0.324 N | Blue | Blue | Uncertain significance | - |
| DNAH8(NM_001206927.1):c.3061A>G, [Agt/Ggt] (p.Ser1021Gly) Missense, rs865933270 | - | 0.72356 Figure S1F | 0.02018 T | 0.44006 T | 18.63 | 0.05153 | 0.029 N | Green | Green | Likely benign | - |
| DNAH8(NM_001206927.1):c.3289A>G, [Att/Gtt] (p.Ile1097Val) Missense, rs147941001 | 0.0000963 | 0.74036 Figure S1G | 0.07329 T | 0.33784 T | 15.18 | 0.16033 | 0.001 N | Green | Green | Likely benign | Patient 12 |
| DNAH8(NM_001206927.2):c.5789G>A, [cGt/cAt] (p.Arg1930His) Missense, rs758923038 | 0.00004782 | 0.74754 Figure S1H | 0.05849 T | 0.36883 T | 16.81 | 0.04481 | 0.037 N | Green | Green | Likely benign | Patient 12 |
| DNAH11(NM_001277115):c.1241A>G [gAa/gGa] (p.Glu414Gly) Missense, Novel | - | 0.77167 Conserved Figure 3B | 0.70404 T | 0.71692 T | 26.5 | 0.7872 | 0.896 D | Red | Red | - | - |
| DNAH11(NM_001277115.1):c.2570G>A [cGa/cAa] (p.Arg857Gln) Missense, rs376572966 | 0.0000444 | 0.72811 Figure 3C | 0.11574 T | 0.26196 T | 19.89 | 0.01542 | 0.519 D | Green | Blue | Likely benign | Patient 13 |
| DNAH11(NM_001277115.1):c.2887A>G [Aga/Gga] (p.Arg963Gly) Missense, rs185803317 | 0.000008103 | 0.74574 Figure 3D | 0.18285 T | 0.16981 T | 21 | 0.12243 | 0.149 N | Green | Green | Likely benign | - |
| DNAH11(NM_001277115.2):c.4202A>G [cAg/cGg] (p.Gln1401Arg) Missense, rs199629774 | 0.002357 * | 0.77106 Figure 3E | 0.54477 T | 0.48696 T | 23.3 | 0.54486 | 0.321 N | Blue | Blue | Benign | - |
| DNAH11(NM_001277115.1):c.4775G>T [tGc/tTc] (p.Cys1592Phe) Missense, rs72657327 | 0.0001981 | 0.79927 Figure 3F | 0.56045 T | 0.69537 T | 24.1 | 0.35779 | 0.639 D | Blue | Red | Uncertain significance | - |
| DNAH11(NM_001277115.1):c.4945-12T>C, Splice acceptor, rs141572016 | 0.0004917 | - | - | - | - | - | - | - | - | Uncertain significance | Patient 14 |
| DNAH11(NM_001277115.1):c.5132A>G [cAa/cGa] (p.Gln1711Arg) Missense, rs189432084 | 0.0005305 | 0.74319 Figure 3G | 0.22617 T | 0.17802 T | 10.26 | 0.21439 | 0.000 N | Green | Green | Likely benign | - |
| DNAH11(NM_001277115.1):c.7729G>A [Gac/Aac] (p.Asp2577Asn) Missense, rs770532527 | 0.00008837 | 0.85404 Conserved Figure 3H | 0.67362 T | 0.71014 T | 33 | 0.81472 | 0.945 D | Red | Red | Uncertain significance | - |
| DNAH11(NM_001277115.1):c.8023A>G [Att/Gtt] (p.Ile2675Val) Missense, rs72657364 | 0.001758 | 0.717 Figure 3I | 0.08778 T | 0.33151 T | 8.696 | 0.02051 | 0.000 N | Green | Green | Likely benign | - |
| DNAH11(NM_001277115.2):c.8072A>G [cAg/cGg] (p.Gln2691Arg) Missense, rs183682756 | 0.0002329 | 0.72089 Figure 3J | 0.21222 T | 0.04597 T | 21.9 | 0.1502 | 0.242 N | Green | Blue | Likely benign | - |
| DNAH11(NM_001277115.1):c.8230C>T [Cgt/Tgt] (p.Arg2744Cys) Missense, rs374826188 | 0.00008435 | 0.81402 Conserved Figure 3K | 0.64824 T | 0.67368 T | 27.6 | 0.71783 | 0.906 D | Red | Red | Uncertain significance | Patient 15 |
| DNAH11(NM_001277115.1):c.8533C>G [Cga/Gga] (p.Arg2845Gly) Missense, rs121908854 | 0.001171 | 0.70209 Figure 3L | 0.17319 T | 0.09501 T | 22 | 0.07246 | 0.405 N | Green | Blue | Likely benign | - |
| DNAH11(NM_001277115.1):c.9238C>A [Ctg/Atg] (p.Leu3080Met) Missense, Novel | - | 0.77741 Conserved Figure 3M | 0.67455 T | 0.67005 T | 24.8 | 0.48544 | 0.897 D | Red | Red | - | - |
| DNAH11(NM_001277115.1):c.9305G>A [gGc/gAc] (p.Gly3102Asp) Missense, rs774083447 | 0.00001615 | 0.77066 Conserved Figure 3N | 0.91256 D | 0.91719 D | 25.4 | 0.92601 | 0.945 D | Red | Red | Uncertain significance | Patient 13 |
| DNAH11(NM_001277115.1):c.9380T>G [cTg/cGg] (p.Leu3127Arg) Missense, rs755885697 | - | 0.76849 Conserved Figure 3O | 0.8943 D | 0.9065 D | 29.3 | 0.94394 | 0.945 D | Red | Red | Uncertain significance | - |
| DNAH11(NM_001277115.2):c.10379C>A [aCg/aAg] (p.Thr3460Lys) Missense, rs573384750 | 0.0001832 | 0.81088 Conserved Figure 3P | 0.66914 T | 0.69202 T | 24.7 | 0.61646 | 0.625 D | Red | Red | Likely benign | - |
| DNAH11(NM_001277115.1):c.11267G>A [cGc/cAc] (p.Arg3756His) Missense, rs554657293 | 0.00006853 | 0.73608 Figure 3Q | 0.88418 D | 0.89161 D | 21.6 | 0.7872 | 0.388 N | Red | Red | Uncertain significance | - |
| DNAH11(NM_001277115.1):c.11839+1G>A Splice donor, Novel | - | - | - | - | - | - | - | - | - | - | Pathogenic, Patient 15 |
| DNAH11(NM_001277115.1):c.12344T>G [aTt/aGt] (p.Ile4115Ser) Missense, rs371418299 | 0.0005882 * | 0.80147 Figure 3R | 0.24391 T | 0.00625 T | 23.6 | 0.58263 | 0.529 D | Blue | Blue | Likely benign | - |
| DNAH11(NM_001277115.1):c.13010G>A [aGc/aAc] (p.Ser4337Asn) Missense, rs759646661 | 0.00002809 | 0.7162 Figure 3S | 0.04369 T | 0.32012 T | 22 | 0.05495 | 0.059 N | Green | Green | Likely benign | Patient 13 |
| DNAH11(NM_001277115.1):c.13120G>A [Gtg/Atg] (p.Val4374Met) Missense, rs560018723 | - | 0.81781 Figure 3T | 0.35791 T | 0.02566 T | 24.6 | 0.63525 | 0.688 D | Red | Blue | Uncertain significance | Patient 14 |
| DNAI1(NM_012144.3):c.40C>T [Cat/Tat] (p.His14Tyr) Missense, rs146501326 | 0.0001551 | 0.49721 Figure S4A | 0.58033 T | 0.52983 T | 17.25 | 0.34483 | 0.056 N | Green | Green | Likely benign | - |
| DNAI1(NM_012144.3):c.47A>G [cAg/cGg] (p.Gln16Arg) Missense, rs148701985 | 0.0003422 | 0.68698 Figure S4B | 0.49514 T | 0.5142 T | 21.5 | 0.19284 | 0.007 N | Green | Green | Likely benign | - |
| DNAI1(NM_012144.4):c.81+20T>C Splice donor, rs572257884 | 0.000249 | - | - | - | - | - | - | - | - | Uncertain significance | - |
| DNAI1(NM_012144.4):c.274_281delAAGCCTAT (p.Lys92Trpfs) Frameshift, Novel | - | - | - | - | - | - | - | - | - | - | - |
| DNAI1(NM_012144.3):c.1173C>G [atC/atG] (p.Ile391Met) Missense, rs151097256 | 0.00009949 | 0.7792 Figure S4C | 0.52782 T | 0.34281 T | 21 | 0.5304 | 0.042 N | Blue | Blue | Likely benign | - |
| DNAI1(NM_012144.3):c.1265_1267del [tTCTgc/tgc] (p.Phe422del) Inframe deletion, rs567346433 | 0.0006057 | - | - | - | - | - | - | - | - | Likely benign | - |
| DNAI2(NM_023036.4):c.685T>G [Tcc/Gcc] (p.Ser229Ala) Missense, rs576683556 | 0.0002585 * | 0.83226 Conserved Figure S4D | 0.213 T | 0.18233 T | 22.4 | 0.10108 | 0.059 N | Green | Green | Likely benign | - |
| DNAI2(NM_023036.4):c.891G>A [atG/atA] (p.Met297Ile) Missense, rs750750518 | 0.00003181 | 0.75677 Figure S4E | 0.09009 | 0.13899 T | 15.3 | 0.10108 | 0.272 N | Green | Green | Likely benign | - |
| DNAI2(NM_023036.4):c.1318G>C [Gag/Cag] (p.Glu440Gln) Missense, rs182986650 | 0.00002387 | 0.74919 Figure S4F | 0.12935 T | 0.21396 T | 22.3 | 0.24349 | 0.037 N | Green | Green | Likely benign | - |
| DNAI2(NM_023036.4):c.1574C>T [gCg/gTg] (p.Ala525Val) Missense, rs145602856 | 0.0007551 | 0.61318 Figure S4G | 0.47021 T | 0.41194 T | 10.79 | 0.26002 | 0.323 N | Blue | Green | Likely benign | - |
| DNAI2(NM_023036.4):c.1715C>T [cCa/cTa] (p.Pro572Leu) Missense, rs151241589 | 0.001541 * | 0.46808 Figure S4H | 0.53676 T | 0.18762 T | 12.81 | 0.18967 | 0.059 N | Green | Green | Likely benign | - |
| DRC1(NM_145038.4):c.1090G>A [Gag/Aag] (p.Glu364Lys) Missense, rs184506507 | 0.00000398 | 0.73269 Figure S5A | 0.31284 T | 0.10997 T | 27.8 | 0.47558 | 0.743 D | Red | Blue | Likely benign | Likely pathogenic, Patient 16 |
| DRC1(NM_145038.4):c.1146G>C [gaG/gaC] (p.Glu382Asp) Missense, Novel | - | 0.76693 Figure S5B | 0.04985 T | 0.32928 T | 17.61 | 0.06188 | 0.022 N | Green | Green | - | - |
| DRC1(NM_145038.5):c.2081G>C [aGg/aCg] (p.Arg694Thr) Missense, rs372797665 | 0.0001233 | 0.77074 Conserved Figure S5C | 0.85587 D | 0.90334 D | 35 | 0.82668 | 0.935 D | Red | Red | Uncertain significance | Likely pathogenic, Patient 16 |
| HYDIN(NM_001270974.1):c.1003G>T [Gta/Tta] (p.Val335Leu) Missense, rs755584531 | 0.000076450 | 0.63033 Figure S5D | 0.07565 T | 0.43605 T | 10.91 | 0.25457 | 0.009 N | Green | Green | Likely benign | Patients 17-18 |
| HYDIN(NM_001270974.1):c.9638C>G [cCc/cGc] (p.Pro3213Arg) Missense, Novel | - | 0.60436 Figure S5E | 0.0133 T | 0.46351 T | 22.5 | 0.32697 | 0.530 D | Blue | Blue | - | Likely pathogenic, Patients 17-18 |
| HYDIN(NM_001270974.2):c.11173C>T [Cgg/Tgg] (p.Arg3725Trp) Missense, rs79417681 | 0.00008865 | 0.6387 Figure S5F | 0.00646 T | 0.43195 T | 20.6 | 0.22036 | 0.506 D | Blue | Blue | Likely benign | - |
| NME8(NM_016616.4):c.1630G>A [Gca/Aca] (p.Ala544Thr) Missense, rs140494494 | 0.0005613 * | 0.68484 Figure S5O | 0.78209 T | 0.82492 D | 24.7 | 0.76421 | 0.945 D | Red | Red | Likely benign | - |
| NME8(NM_016616.4):c.271-27C>T Splice acceptor, rs117149381 | 0.01787 * | - | - | - | - | - | - | - | - | Benign | - |
| OFD1(NM_003611.3):c.2927A>C [aAg/aCg ](p.Lys976Thr) Missense, rs1458317780 | 0.000005470 | 0.68731 Figure S5R | 0.97362 D | 0.97841 D | 24.2 | 0.71627 | 0.841 D | Red | Red | Uncertain significance | See Results |
| RSPH1(NM_080860.4):c.730G>A [Gca/Aca] (p.Ala244Thr) Missense, rs150400022 | 0.0007761 * | 0.60579 Figure S5G | 0.15074 T | 0.19078 T | 0.478 | 0.02609 | 0.009 N | Green | Green | Likely benign | - |
| RSPH4A(NM_001010892.3):c.650A>C [tAc/tCc] (p.Tyr217Ser) Missense, rs762313827 | 0.00006861 | 0.83409 Conserved Figure S5H | 0.36055 T | 0.3837 T | 26.6 | 0.68023 | 0.919 D | Red | Blue | Uncertain significance | - |
| RSPH4A(NM_001010892):c.1410C>G [atC/atG] (p.Ile470Met) Missense, rs775326896 | 0.000027850 | 0.7634 Figure S5I | 0.25237 T | 0.11706 T | 19.44 | 0.5519 | 0.781 D | Red | Blue | Likely benign | - |
| RSPH9(NM_001193341.1):c.365G>A [gGt/gAt] (p.Gly122Asp) Missense, rs1195999841 | 0.00000398 | 0.66683 Figure S5J | 0.41053 T | 0.29917 T | 21 | 0.35566 | 0.423 N | Blue | Blue | Uncertain significance | - |
| SPAG1(NM_172218.2):c.957T>A [gtT/gtA] (p.Val319=) Synonymous, rs146528350 | 0.0008908 * | - | - | - | - | - | - | - | - | Likely benign | - |
| SPAG1(NM_172218.2):c.1435+16C>T Splice donor, rs148767962 | 0.0002789 | - | - | - | - | - | - | - | - | Likely benign | - |
| ZMYND10(NM_015896.4):c.1019G>A [cGg/cAg] (p.Arg340Gln) Missense, rs148328402 | 0.002811 * | 0.66864 Figure S5P | 0.31943 T | 0.30892 T | 18.76 | 0.14679 | 0.010 N | Green | Green | Benign | - |
| Patient | First | Second | Third | Clinical Assessment |
|---|---|---|---|---|
| 1 Homo | C1orf127:c.337C>T (p.Arg113Ter) | C1orf127:c.337C>T (p.Arg113Ter) | - | Heterotaxy, asplenia, midline liver, pulmonary stenosis, interrupted inferior vena cava, bilateral superior vena cava, and right aortic arch. |
| 2 Homo | C1orf127:c.337C> (p.Arg113Ter) | C1orf127:c.337C> (p.Arg113Ter) | - | Dextrocardia, pulmonary stenosis, respiratory infections. Parents are heterozygous for EP400 and asymptomatic. Family members with congenital heart anomalies. |
| EP400:c.323C>T (p.Ala108Val) rs762116055-Likely benign | EP400:c.323C>T (p.Ala108Val) rs762116055 - Likely benign | - | ||
| 3 | CCDC39:c.1073C>T (p.Thr358Ile) | CCDC39:c.1076A>C (p.Lys359Thr) | - | Prematurity (32 weeks’ gestation) with persistent atelectasis. Parental studies revealed the variants are in cis phase. |
| 4 Homo | CCDC39:c.1167+1261A>G | CCDC39:c.1167+1261A>G | - | Two cousins with homozygosity and diagnostic features of PCD. § |
| 5 | CCDC39:c.1363-3delC | CCDC39:c.1781C>T (p.Thr594Ile) | - | Clinical features of PCD. § |
| 6 | CCDC40:c.1445G>A (p.Cys482Tyr) | CCDC40:c.257A>G (p.Tyr86Cys) | - | Heterotaxy syndrome (isomerism). |
| 7 | CCDC40:c.850G>C (p.Asp284His) | CCDC40:c.697G>A (p.Asp233Asn) | - | Recurrent sinusitis. |
| 8 Homo | CCDC40:c.62G>T (p.Gly21Val) | CCDC40:c.62G>T (p.Gly21Val) | - | Two siblings with sinopulmonary infections (including chronic otorrhea) from early infancy with ultrastructural defects in the cilia (significant microtubular disorganizations, including distorted dynein arms and absent inner dynein arms). |
| 9 Homo | CEP104:c.2092G>A (p.Glu698Lys) | CEP104:c.2092G>A (p.Glu698Lys) | - | Joubert syndrome 25 (MIM#616781). Parents are asymptomatic carriers. |
| 10 Homo | DNAH5:c.4807C>A (p.Pro1603Tyr) | DNAH5:c.4807C>A (p.Pro1603Tyr) | - | Two sisters with bronchiectasis and chronic sinusitis. Parents are heterozygous and asymptomatic. Mother had recurrent abortions and an ectopic pregnancy. |
| 11 Homo | DNAH5:c.5503C>T (p.Gln1835Ter) | DNAH5:c.5503C>T (p.Gln1835Ter) | - | Two sisters with chronic respiratory infections and ultrastructural defects in the cilia (significant microtubular disorganizations, including distorted outer dynein arms). |
| 12 | DNAH8:c.3289A>G (p.Ile1097Val) | DNAH8:c.5789G>A (p.Arg1930His) | - | Clinical features of PCD. |
| 13 | DNAH11:c.2570G>A (p.Arg857Gln) | DNAH11:c.9305G>A (p.Gly3102Asp) | DNAH11:c.13010G>A (p.Ser4337Asn) | Recurrent respiratory infections from childhood and non-motile sperms. Since parents were not tested, phasing of variants could not be performed. |
| 14 | DNAH11:c.13120G>A (p.Val4374Met) | DNAH11:c.4945-12T>C | DNAH11:c.5132A>G (p.Gln1711Arg) | Chronic sinusitis. Since parents were not tested, phasing of variants could not be performed. |
| 15 | DNAH11:c.8230C>T (p.Arg2744Cys) | DNAH11:c.11839+1G>A | - | Situs inversus totalis with dextrocardia. |
| 16 | DRC1:c.1090G>A (p.Glu364Lys) | DRC1:c.2081G>C (p.Arg694Thr) | - | Clinical features of PCD. Parents are first cousins. The mother also has clinical features of PCD with the same two variants. The father is not tested. |
| 17 | HYDIN:c.1003G>T (p.Val335Leu) | HYDIN:c.9638C>G (p.Pro3213Arg) | - | Clinical features of PCD. |
| 18 | HYDIN:c.1003G>T (p.Val335Leu) | HYDIN:c.9638C>G (p.Pro3213Arg) | - | Prematurity (33 weeks’ gestation) with clinical features of PCD. |
| DNAAF3:c.1053+5G>C | DNAAF3:c.1116+5G>C | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsamri, M.T.; Alabdouli, A.; Iram, D.; Alkalbani, A.M.; Almarzooqi, A.S.; Souid, A.-K.; Vijayan, R. A Study on the Genetics of Primary Ciliary Dyskinesia. J. Clin. Med. 2021, 10, 5102. https://doi.org/10.3390/jcm10215102
Alsamri MT, Alabdouli A, Iram D, Alkalbani AM, Almarzooqi AS, Souid A-K, Vijayan R. A Study on the Genetics of Primary Ciliary Dyskinesia. Journal of Clinical Medicine. 2021; 10(21):5102. https://doi.org/10.3390/jcm10215102
Chicago/Turabian StyleAlsamri, Mohammed T., Amnah Alabdouli, Durdana Iram, Alia M. Alkalbani, Ayesha S. Almarzooqi, Abdul-Kader Souid, and Ranjit Vijayan. 2021. "A Study on the Genetics of Primary Ciliary Dyskinesia" Journal of Clinical Medicine 10, no. 21: 5102. https://doi.org/10.3390/jcm10215102
APA StyleAlsamri, M. T., Alabdouli, A., Iram, D., Alkalbani, A. M., Almarzooqi, A. S., Souid, A.-K., & Vijayan, R. (2021). A Study on the Genetics of Primary Ciliary Dyskinesia. Journal of Clinical Medicine, 10(21), 5102. https://doi.org/10.3390/jcm10215102