Abstract
Giant cell arteritis (GCA) is a granulomatous large-vessel vasculitis that affects adults above 50 years of age. In GCA, circulating monocytes are recruited to the inflamed arteries. With cues from the vascular microenvironment, they differentiate into macrophages and play important roles in the pathogenesis of GCA via pro-inflammatory cytokine production and vascular remodeling. However, a deeper understanding of macrophage heterogeneity in GCA pathogenesis is needed to assist the development of novel diagnostic tools and targeted therapies. Here, we review the current knowledge on macrophage heterogeneity and diverse functions of macrophage subsets in the pathogenesis of GCA. We next discuss the possibility to exploit their heterogeneity as a source of novel biomarkers and as targets for nuclear imaging. Finally, we discuss novel macrophage-targeted therapies and future directions for targeting these cells in GCA.
1. Introduction
Giant cell arteritis (GCA) is an auto-inflammatory disease that affects medium- and large-sized vessels in adults older than 50 years. GCA belongs to a disease spectrum that includes cranial GCA (C-GCA) and large-vessel GCA (LV-GCA), with or without overlapping polymyalgia rheumatica (PMR). The vasculitis in C-GCA mainly affects cranial arteries leading to symptoms such as headache, jaw claudication and vision loss [1]. Large-vessel (LV-GCA) GCA affects the aorta and its major branches leading mostly to symptoms such as weight loss, fatigue, night sweats and fever. Inflammation and remodeling in GCA can cause ischemic complications or aortic aneurysms and dissection [2]. C-GCA and LV-GCA may be often present together. Indeed, imaging studies reported a high percentage (up to 83%) of overlapping C-GCA and LV-GCA [3,4,5,6]. The American College of Rheumatology (ACR) 1990 classification criteria for GCA are exclusively based on the assessment of cranial features of GCA in addition to the age of the patient and an elevated erythrocyte sedimentation rate (ESR) and a positive temporal artery biopsy (TAB) [7]. However, there is room for improvement in the diagnosis of GCA. Even though a TAB has a high specificity, it is invasive with limited sensitivity. More recently, the European League Against Rheumatism (EULAR) proposed an expansion of the original classification criteria recommending the inclusion of clinical features of LV-GCA and PMR as well as the use of molecular imaging (ultrasound or magnetic resonance imaging (MRI), computerized tomography (CT), positron-emission-tomography (PET)-CT) [8,9]. However, the availability of imaging tools for GCA diagnosis remains challenging in daily care [10,11]. Therefore, reliable and disease-specific biomarkers are highly needed for GCA.
To date, glucocorticoids (GCs) are the mainstay of treatment for GCA. However, long-term GC usage causes serious adverse events such as the development of type II diabetes, hypertension, osteoporosis and increased risk of infection [12,13]. Additionally, the majority of patients experience relapses while on GCs treatment [14]. Although the acute-phase response is suppressed by GCs, there is proof of ongoing inflammation in the affected vascular tissues [15,16,17]. Therefore, alternative, more effective treatment options are highly needed in newly diagnosed and relapsing patients. Methotrexate has long been used with some GC-sparing effect in GCA [18]. More recently, the interleukin 6 receptor blocker, Tocilizumab, was shown to induce sustained GC-free remission in around 50% of the GCA patients after 52 weeks of treatment [19]. However, after 52 weeks of tocilizumab treatment, magnetic resonance angiography (MRA) revealed that vascular inflammation continues or reappears in two-third of GCA patients despite clinical remission [20]. Furthermore, an investigation of biomarkers in tocilizumab-treated patients documents immunological signs of subclinical disease activity in GCA patients especially in the early stages of treatment emphasizing the need for long-term treatment with tocilizumab [21]. Thus, these results pinpoint that current treatments do not abolish the local inflammation efficiently. As the persistent inflammation in the vessel wall is largely granulomatous [15], targeting macrophages may improve the effectiveness of immunosuppression and may induce lasting remission. Although the immunopathogenesis of GCA is unknown, it is most likely determined by the interaction of multiple factors including genetic susceptibility, environmental factors and aging of the immune system. Several studies showed an association between HLA-DRB1*04 and more recently also HLA-B*15:01 alleles with GCA, thereby suggesting the contribution of both MHC class II and class I genes to genetic susceptibility in GCA development [22,23]. Additionally, the role of activated dendritic cells (DCs), T cells, macrophages and type I IFN-related pathways in the pathogenesis of GCA points to a relationship between infection and the initiation of GCA [24]. However, the assessment of the microbiome in GCA-affected arteries has provided inconclusive and conflicting results leaving the question on the involvement of pathogens in the pathogenesis of GCA unresolved [25,26,27]. Besides, the aging of the immune system may also contribute to increased susceptibility to infections and, in combination with vascular ageing, accelerate the vasculopathy of GCA [24].
It is well-known that macrophages play critical roles in the pathogenesis of GCA, as they mediate inflammatory responses affecting processes like tissue remodeling and angiogenesis (Figure 1) [28]. Moreover, some of the macrophages in the vessel-wall fuse and form multinucleated giant cells, which is a hallmark of GCA [29]. Treatment with GCs cannot sufficiently suppress local inflammation which likely leads to relapse (Figure 1) [15]. Macrophages are highly plastic cells that can rapidly change their phenotypes upon cues from the tissue microenvironment. Recent research showed that a more comprehensive examination is needed for the characterization of macrophage heterogeneity both in phenotype and function in tissues [30,31,32]. Macrophage subsets involved in the vasculopathy of GCA could be exploited for diagnostic purposes, as a source of biomarkers, as targets for imaging and as a targets for treatment. In this review, we will discuss the current knowledge on the mechanisms underlying the distinct macrophage phenotypes and functions involved in the pathogenesis of GCA and the implications for improved diagnosis (biomarkers, imaging), monitoring (biomarkers, imaging), prognosis (biomarkers) and therapy in GCA.
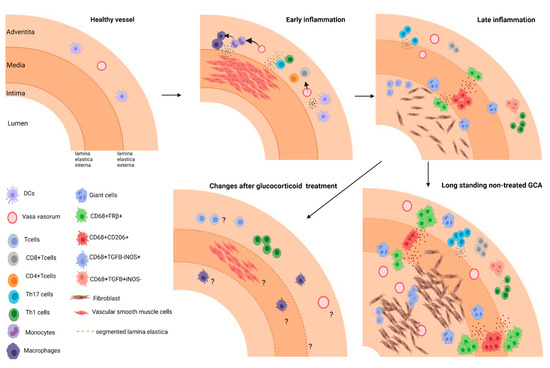
Figure 1.
Model of different stages in arterial inflammation in GCA. Early inflammation: Dendritic cell activation recruits T cells (CD4+ and CD8+) to the vessel wall and drives activation, and polarization of T cells. Secreted soluble factors lead to monocyte recruitment and their differentiation into macrophages. Late inflammation: Different macrophages subsets skewed by environmental cues contribute to tissue remodeling in GCA. CD206+ GM-CSF skewed macrophages producing MMPs mediate tissue destruction; FRβ+M-CSF skewed macrophages and fibroblast are activated and drive intimal proliferation. Long standing GCA: ultimately inflammation leads to vascular occlusion. Changes after glucocorticoid treatment: The effect of treatment on cellular infiltrates and vascular lesions/repair remain to be elucidated.
1.1. Pathogenesis of GCA
The initiation of the inflammatory response in the arterial vessel wall of GCA patients is not well understood. It is suggested that vascular dendritic cells are activated through Toll-like receptors (TLR) via unknown endogenous or exogenous ligands, leading to the production of chemokines (CCL18, CCL19, CCL20 and CCL21) and cytokines (IL-1, IL-6, IL-12, IL-18 and IL-33). These cytokines and chemokines recruit CD4+ T cells to the arterial wall and polarize them toward Th1 and Th17 cells. The infiltrating Th1 and Th17 cells produce proinflammatory cytokines including IFN-γ and IL-17, respectively. IFN-γ activates macrophages towards a proinflammatory phenotype that produces various proinflammatory cytokines and chemokines. Additionally, IFN-γ induces vascular endothelial growth factor (VEGF) and chemokine (CCL2, CXCL9, CXCL10 and CXCL11) expression by vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) leading to neoangiogenesis and recruitment of more monocytes and T cells to the site of inflammation [33] (Figure 1).
Studies on affected vessels of GCA patients point to a central role of macrophages in the vasculopathy. Once recruited, monocytes differentiate into macrophages and produce the pro-inflammatory cytokines IL-6, IL-1β and TNF-α that amplify the inflammatory response. These cytokines, particularly IL-6, initiate a systemic response of the body characterized by high levels of acute-phase markers in the blood giving rise to the systemic symptoms of GCA, such as fever, weight loss and malaise. In addition, macrophages at the site of inflammation play a critical role in vascular remodeling by promoting angiogenesis, intimal hyperplasia and tissue destruction. Macrophages are capable instigators of neoangiogenesis by the secretion of VEGF, IL-33 and YKL-40. Platelet-derived growth factor (PDGF), which is produced by macrophages, promotes VSMC and fibroblast migration and proliferation leading to intimal hyperplasia and eventually vessel-wall occlusion. (Figure 1) Furthermore, activated macrophages are the main contributors to tissue destruction by producing matrix metalloproteinases (MMPs) [33,34,35]. Therefore, gaining a deeper understanding of monocyte and macrophage involvement in GCA pathogenesis is vital.
1.2. Monocytes in GCA
Monocytes are the precursors of tissue macrophages and in the blood of GCA patients, altered dynamics and distribution of monocyte subsets have been documented [17]. At diagnosis, GCA patients present with elevated counts of circulating monocytes, and these elevated counts were found to associate with C reactive protein (CRP) levels [17]. During treatment with glucocorticoids, and even after termination of treatment, monocyte counts remained elevated in GCA patients. Monocytes display heterogeneity in their phenotype and function, and are currently subdivided into three subsets: classical monocytes (CD14brightCD16neg), intermediate monocytes (CD14brightCD16+) and non-classical monocytes (CD14dimCD16+). CD16+ monocytes were shown to be increased with age and associated with different inflammatory diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) [36,37]. The monocytosis observed in GCA patients was attributed to an expansion of the classical monocyte subset, although elevated intermediate monocyte counts have also been described [37,38]. Even though total monocyte counts remained high during treatment with glucocorticoids, substantially lower non-classical monocyte counts were noted, likely due to enhanced induction of apoptosis [37,39].
Alterations in the functioning and migration of monocytes of GCA patients could play a role in initiating and fueling vascular inflammation. Monocyte subsets use different chemotaxis pathways to enter tissues, as classical monocytes mainly depend on the CCR2-CCL2 axis, and non-classical monocytes depend on the CX3CR1-CX3CL1 axis [40]. Further research on CCL2-CCR2 and CX3CR1-CX3CL1 pathways in tissue demonstrated that the majority of macrophages in TABs of GCA patients resemble non-classical monocytes with CD16 and CX3CR1 expression, but often lack CCR2 expression [37]. Albeit to a lower extent, influx of CCR2 expressing macrophages resembling the classical monocyte phenotype were also detected in the vessel wall of GCA patients [37,40]. Elevated CCL2 expression by VSMCs caused by the inflammatory microenvironment has been reported in GCA [40]. These reports suggest a central role for both classical and non-classical monocytes in the vasculopathy of GCA. In addition, tissue migrated monocytes/macrophages may aid the migration of T-cells to the vessel wall as well, through their production of MMP-9, which breaks down the extracellular matrix [41]. Further evidence indicates that the non-classical monocyte subset is the main source of MMP-9 and associated enzymes, in addition to pro-angiogenic factors such as YKL-40 [35,42,43]. Moreover, (classical) monocytes of GCA patients show upregulated CD64 expression but lowered expression of folate receptor β, which is likely a sign of an activated phenotype [41,44].
1.3. Macrophage Heterogeneity and Their Roles in the Vasculopathy of GCA
Upon migration of circulating monocytes to vascular tissue lesions, monocytes differentiate into macrophages. Although it is known that macrophages are highly plastic cells with the ability to adapt to the microenvironment, the knowledge regarding the phenotypic and functional diversity of macrophages in GCA tissues is steadily growing. Before, macrophages were classified into two major subtypes, widely known as the M1 and M2 macrophages. M1 type macrophages are induced by IFN-γ, and considered pro-inflammatory due to their capacity to produce proinflammatory cytokines (such as IL-1β, IL-6, IL-12 and IL-23), growth factors (VEGF, PDGFs), MMPs and reactive oxygen species (ROS). These factors play an important role in the immunopathology of GCA [45,46]. In contrast to M1 macrophages, M2 macrophages are defined as an anti-inflammatory and tissue repairing subtype. They are the source of IL-10 and express the macrophage mannose receptor (CD206) [45,46,47,48]. However, the distinction between M1 and M2 macrophage subsets is largely based on in vitro experiments under controlled conditions which is now regarded an oversimplification of the much more complex environment in tissues. Indeed, dedicated tissue studies have shown macrophage phenotypes with mixed traits of both M1 (CD64 expression) and M2 (CD206/FRβ) macrophages in pathological conditions including GCA [32,49,50,51,52].
A variety of important soluble mediators have been identified in GCA such as IFN-γ, PDGF, IL-17, IL-6 and GM-CSF that are secreted by different cell types in the vessel wall microenvironment and can modulate macrophage heterogeneity. As macrophages are highly plastic, it is no surprise that different phenotypes of macrophages with different functions may be present in GCA-affected vessel-walls. This notion of macrophage heterogeneity was first reported by Weyand et al. [53]. Immunohistochemical analysis revealed a functional heterogeneity of tissue-infiltrating macrophages where TGF-β1(+)iNOS (−) macrophages, localized in the adventitia in the vicinity of IFN(+) CD4+ T cells, contribute to IL-1β and IL-6 production. On the other hand, TGF-β1(−)iNOS(+) macrophages were found in the intimal layer of the inflamed artery and expressed MMP-2, a collagenase involved in tissue destruction [53,54].
Recently, our group reported a distinct spatial distribution of CD206+/YKL-40+/MMP-9+ macrophages and FRβ+/CD206- macrophages linked to tissue destruction and intimal hyperplasia, respectively, in GCA [35]. The observed macrophage phenotypic heterogeneity is likely caused by the local production of GM-CSF and M-CSF. The ability of GM-CSF and M-CSF to induce overexpression of CD206 and FRβ, respectively, has been reported previously [55,56,57]. CD206+YKL-40+MMP-9+ macrophages located in the media and media borders along the sites of elastic lamina degradation are likely skewed by local GM-CSF signals. Indeed, previous reports also revealed that MMP-2 and MMP-9 were expressed by macrophages and giant cells adjacent to the internal elastic lamina [58,59]. Both MMP-2 and MMP-9 play roles in the pathogenesis of GCA due to their ability to degrade elastin [41,60]. The overexpression of MMP-9 by macrophages in GCA vessels may be governed by YKL-40, acting as an upstream signal. YKL-40 also has the proangiogenic ability to induce vasa vasorum formation. Overall, these results showed that the spatial distribution of CD206+/YKL-40+/MMP-9+ macrophages in the media and media borders where elastic lamina degradation takes place matches with a tissue destructive and proangiogenic role of these macrophages.
In contrast to CD206+/MMP-9+/YKL-40+ macrophages, FRβ+ macrophages were mainly found in the adventitia and inner-intima, adjacent to CD206+MMP-9+ macrophages [44,61]. FRβ+ macrophages are likely skewed by local M-CSF signals as opposed to GM-CSF signals that have been reported to diminish FRβ expression by macrophages [55,56]. In inflamed GCA vessels, M-CSF expression was found to be highly localized in the area with CD206+/MMP-9+/YKL-40+ macrophages, suggesting that M-CSF produced by CD206+/MMP-9+/YKL-40+ macrophages primes adjacent macrophages to express FRβ. Intriguingly, significantly higher numbers of infiltrating FRβ+ macrophages were detected in the inner intima of vessels with massive intimal hyperplasia, suggesting their importance in promoting fibroblast proliferation. Indeed, M-CSF-primed macrophages, rather than GM-CSF-primed macrophages, secreted high levels of platelet-derived growth factor (PDGF)-AA, a factor known to promote fibroblast migration and proliferation. These data imply that spatial gradients of GM-CSF and M-CSF in the inflamed vessel wall might be responsible for the distinct macrophage subset distribution. Thus, these studies underline that the microenvironment shapes the phenotype and function of macrophages in the vasculopathy of GCA.
Th1 and Th17 polarized cells in the inflamed arteries also impact the phenotype and function of local macrophages. Th1 cells release IFN-γ, which primes macrophages to be more pro-inflammatory. IFN-γ activated macrophages produce PDGF. PDGF leads to activation, and proliferation of VSMCs, that subsequently migrate to the intima of the vessels, obstructing the lumen. Furthermore, the Th17 cytokine IL-17 induces macrophages to produce pro-inflammatory cytokines characteristic for “M1” like macrophages, but at the same time also stimulates CD163 expression, indicative for “M2” like polarization [62]. Moreover, T cells were identified as an essential source of GM-CSF in GCA TABs which may skew macrophages towards a CD206+ phenotypes in GCA lesions [63].
Recent findings regarding IL-6-producing B cells in GCA indicate a potential role of B cells in GCA pathogenesis. B cells have been described in the adventitia and media of GCA temporal artery and aorta, mostly colocalized with infiltrated CD3+ T cells [64]. Such lymphoid aggregates that show compartmentalization of T and B cells were defined as artery tertiary lymphoid organs (ATLOs) [65,66]. Investigation of ATLOs in GCA demonstrated an association with the ectopic expression of CXCL13 and B-cell activating factor (BAFF) which were shown to increase following in vitro stimulation of temporal arteries with IL-6 [66]. In vitro investigation on the interaction between B cells from GCA and macrophages revealed that soluble factors secreted by B cells enhanced pro-inflammatory cytokine production (IL-6, IL-1β andTNF-α) and induced higher expression of tissue-destructive factors (MMP-9, YKL-40) in macrophages [67]. Furthermore, GM-CSF producing B-cells that can efficiently activate myeloid cells were identified in multiple sclerosis, another autoimmune disease [68,69]. This suggests an interesting interaction between B-cells and macrophages, a notion that requires additional investigation in GCA. Overall, in GCA, different cell types and microenvironments direct functional and phenotypical diversity of macrophages (Figure 2). Novel high dimensional proteomic and transcriptomic methods such as GeoMXTM (Nanostring) and Visium Spatial gene expression platform (10× Genomics) may aid in identifying the macrophage phenotypes in GCA lesions in more detail and provide better insight on macrophage heterogeneity and their specific functions in the vasculopathy of GCA.
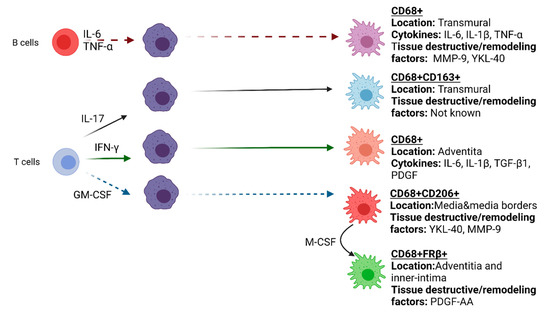
Figure 2.
Functional heterogeneity of macrophages in GCA affected vessel-wall.
1.4. Macrophage Metabolism in GCA
Activation of different metabolic pathways has been linked to different phenotypes of immune cells. Our increased knowledge on immune cell metabolism advocates its contribution to the pathogenesis of chronic inflammatory diseases. Therefore, understanding the metabolic pathways of macrophages at the cellular level is critical in elucidating their specific roles in the pathogenesis of chronic inflammatory diseases.
Environmental stimuli steer macrophages towards different metabolic pathways. The metabolism of “M1” (IFNγ-skewed) activated macrophages is characterized by a high rate of aerobic glycolysis, called Warburg metabolism, and impaired oxidative phosphorylation (OXPHOS). On the other hand, OXPHOS and fatty acid oxidation (FAO) are enhanced in “M2” macrophages after stimulation with IL-4 and IL-13 [70,71,72]. Possibly, activated macrophages skewed by LPS/IFNγ utilize glycolysis for rapid, short-term activation that is needed at the site of infection/inflammation, whereas IL-4-induced macrophages rely on OXPHOS that provides better energy support for prolonged cell survival and for prolonged responses to battle parasitic infections [72]. Additionally, it was shown that after macrophage polarization, reprogramming of metabolic pathways contributes to critical functional changes in macrophages [72,73,74].
Activated macrophages stimulated and skewed by IFNγ express iNOS to produce nitrous oxide (NO) from arginine and to form ROS [72]. Additionally, in activated macrophages, the Tricarboxylic acid (TCA) cycle (Krebs cycle) is broken leading to citrate and succinate accumulation. Citrate accumulation carries an important role in the production of NO, ROS and prostaglandins, thereby contributing to a pro-inflammatory and tissue-destructive macrophage phenotype [75,76]. iNOS(+) macrophages were detected in the TABs of GCA patients, thereby demonstrating the presence of metabolically active cells in the inflamed vessel. Besides citrate, succinate accumulation in activated macrophages results in HIF-1α activation that induces inflammatory mediators such as IL-1β [77]. IL-1β has been described as one of the important factors contributing to the pathogenesis of GCA [44,78]. The regulation of IL-1β via metabolic reprogramming in macrophages points to a possibility of metabolic activation via HIF-1α in GCA. Besides the pro-and anti-inflammatory stimulants, mitochondrial products could aid the identification of different subsets of macrophages. Gene expression analysis and immunohistochemical staining of inflamed and non-inflamed TABs revealed an overlap between mitochondrial activation and MMP-2 expression. Thus, the production of ROS and MMP-2 may distinguish a macrophage subset likely involved in tissue destruction [79].
More recently, our group studied the roles of GM-CSF and M-CSF in skewing different macrophage populations in GCA. Reports have shown that stimulation of macrophages with M-CSF, demonstrated increased expression of both glycolytic and TCA cycle enzymes resulting in increased glycolysis and OXPHOS [73,80]. On the other hand, stimulation of macrophages by GM-CSF affects glucose and lipid metabolism. Inhibition of LPS-stimulated glycolysis by 2-deoxyglucose decreased GM-CSF-mediated TNF-α, IL-1β and IL-6 production levels [81]. All in all, these data suggest possible alteration of metabolic activities in the macrophages found in GCA affected vessels which may be exploited to target specific macrophage populations for diagnostics and therapeutic purposes.
1.5. Aging Macrophages
GCA is a disease that exclusively affects people over 50 years of age. With aging, systemic low-grade inflammation increases which is activated by damage associated molecular patterns (DAMPs) in the absence of acute inflammatory stimuli and/or pathogen-associated molecular patterns (PAMPs). This constant low-grade inflammation drives tissue and organ damage over time and was coined inflammaging [49,82,83,84,85]. Thus, the inflammaging process and the involvement of cellular aging monocytes and macrophages in the pathogenesis of GCA cannot be ignored.
Cellular senescence is an essential process in aging. The number of senescent cells at the pathologic sites in chronic diseases increases with age [86,87]. Senescent cells secrete pro-inflammatory cytokines, chemokines and tissue-destructive proteins referred to as the senescence-associated secretory phenotype (SASP). The SASP is also able to induce senescence of the surrounding healthy cells [86]. Cellular senescence has not been assessed in vascular tissues of GCA patients; however, the literature indicates its possible role in GCA pathology. Counts of aged non-classical CD14dimCD16+ monocytes significantly increase with age. Non-classical monocytes have short telomeres, demonstrate a distinct pro-inflammatory phenotype and have characteristics of senescent cells [88,89]. Interestingly, non-classical monocyte proportions were found decreased in the circulation of GCA patients [37,90]. The relative decrease of non-classical monocytes in GCA may imply tissue migration of these cells. Moreover, absent in melanoma (AIM)2, a DNA damage sensor important in initiating the senescent phenotype, is upregulated in inflamed arteries of GCA patients [91].
Another effect of aging becomes apparent when analyzing macrophage responses to TLR stimulation, which have been implicated in GCA pathogenesis as well. TLRs are pathogen recognition receptors (PRRs) that enable macrophages to detect PAMPs, produce pro-inflammatory cytokines and trigger inflammation. Studies on peripheral blood monocytes from young and elderly participants showed that TLR4 expression was elevated in elderly participants in response to TLR stimulation and associated with elevated IL-8 levels [92]. TLR gene expression analysis in six different human vessel types showed vessel-specific expression of TLRs 1 to 9. TLR2 and TLR4 were abundantly expressed in medium- and large-vessels, while TLR7 and TLR9 were detected at low levels [93,94]. Interestingly, Rodriguez et al. investigated the expression and function of TLRs in GCA patients and revealed an increased TLR7 expression on circulating monocytes from GCA patients with active disease compared to healthy controls. However, despite this higher expression of TLR7, patient monocytes responded with a dampened pro-inflammatory cytokine secretion after stimulation with TLR7 agonists [95]. Investigating possible factors that could affect the TLR7 response showed that neither genetic defects nor amino acid substitutions in TLR7 were responsible for the observed effects. More-in depth research is needed to identify the cause of decreased TLR7 responses, whether due to inflammatory processes or overstimulation by ligands such as single-stranded RNA viruses.
1.6. Macrophage Related Biomarkers as a Tool for GCA Diagnosis and Disease Monitoring
Although the gold standard for GCA diagnosis is a TAB with evidence of vascular inflammation, the invasiveness of the procedure, the lack of sensitivity, as well as the risk of false-negative assessments due to patchy inflammation hampers the utility of a TAB as a diagnostic tool [96]. Recently, imaging techniques such as ultrasonography, detecting the “halo” sign in inflamed cranial and axillary arteries of GCA patients, and 18F-Fluorodeoxyglucose PET/CT (FDG-PET/CT) are emerging as more sensitive and specific diagnostic tools for GCA [97,98,99]. Although vasculitic lesions as detected by these imaging methods often improve upon treatment-induced remission, vascular abnormalities may still persist despite clinical remission [100,101]. This observation could reflect ongoing smoldering inflammation in the vessel or perhaps post-inflammatory vascular remodeling. Besides imaging techniques, current blood markers for GCA diagnosis are not disease-specific, have low diagnostic accuracy and cannot predict a non-favorable disease course. Therefore, more specific macrophage-targeted imaging tools and novel macrophage-derived serum markers may potentially be exploited to improve diagnostic accuracy and monitoring of disease activity in GCA patients, including patients on GC treatment [102].
1.6.1. A. Serum Markers
GCA is a heterogeneous disease, and the extent of the local and systemic inflammatory response may differ among patients [103]. Early detection is important due to the danger of serious ischemic events, leading to blindness or stroke, requiring timely start of treatment. So far, several non-specific biomarkers (CRP, ESR, Serum amyloid A (SAA), IL-6) have been identified that can help to distinguish GCA from healthy controls [102]. However, the identification of new biomarkers to further characterize patient heterogeneity (disease subsets) can help to implement improved personalized medications for GCA patients [103].
As novel biomarker candidates, proteins released by macrophages important in the pathology of GCA may provide a source of biomarkers for diagnosis and tracking disease activity. Indeed, levels of macrophage products such as soluble CD163, YKL-40, Angiopoietin-2 (Angpt-2), IL-33, VEGF, MMP-9, calprotectin and osteopontin were found elevated in the serum of GCA patients compared to healthy donors [51,104,105,106,107,108,109,110,111,112] (Figure 3).
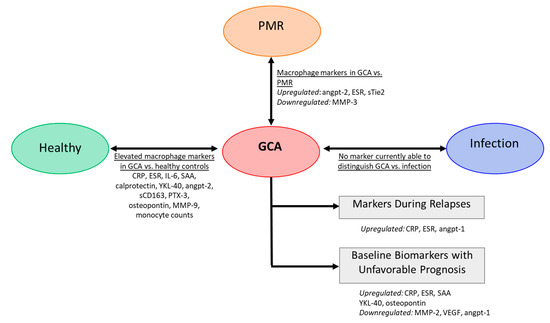
Figure 3.
Serum markers in GCA. Several markers are altered in GCA compared to healthy controls. Some studies reported markers to differentiate GCA patients from patients with isolated polymyalgia rheumatica (PMR). There is currently no macrophage markers to discriminate between GCA and infection (e.g., respiratory and urinary infection) [51,104,105,106,107,108,109,110,111,112].
Ideally, a biomarker or combination of biomarkers should be disease-specific. Investigations on biomarkers to distinguish concurrent arterial inflammation in patients phenotypically presenting as PMR demonstrated that high levels of angiopoietin-2, ESR and low MMP-3 levels at baseline may aid the diagnosis of concurrent vasculitis in PMR [113,114]. Although differences in macrophage-related markers may potentially distinguish GCA patients from patients with isolated PMR, distinguishing GCA patients from patients with infections is still a challenge, as high-level acute phase protein (CRP) is also elevated in patients with active infection. Infection triggers an acute inflammatory response, whereas GCA leads to more chronic granulomatous inflammation. As different inflammatory microenvironment may skew macrophages into different subtypes, macrophage products may outperform CRP, thereby improving the diagnostic process. However, such markers have yet to be identified (Figure 3).
Markers reflecting monocyte/macrophage activity may also serve as prognostic biomarkers. High levels of VEGF and Angiopoietin-1 (Angpt-1) and low levels of YKL-40 at baseline predicted a short time to GC-free remission in GCA patients and thus may identify easy to treat patients [104]. Serum levels of osteopontin may also have prognostic value as they were found to predict a relapsing disease course [110,111]. Additionally, MMP-2 was found to be negatively associated with relapse in GCA patients, while SAA, CRP, ESR were positively associated [102]. Other studies, however, failed to confirm the prognostic value of acute-phase markers CRP, ESR and SAA [104,115] (Figure 3).
Lastly, in addition to their utility in the diagnosis and prognosis of GCA, several serum biomarkers are gaining importance in monitoring disease activity. Currently, CRP, ESR and other inflammatory markers are used for monitoring patients. However, the specificity/sensitivity of CRP and ESR in predicting relapses in GCA was shown to be poor [17,116] (Figure 3). Although GC-sparing agents are developed, CRP and ESR remain unreliable as prediction markers for GCA flares. Recently, combined treatment with tocilizumab (IL-6R blockade) and GCs was shown to be beneficial for GCA patients. However, in tocilizumab-treated patients CRP levels and ESR, which are highly dependent on IL-6, are completely suppressed making it even more difficult for physicians to monitor disease activity [116]. Therefore, other markers that can flag relapses are needed and macrophage-derived markers might be good candidates. There is evidence that GCA symptoms can reappear as a result of smoldering inflammation in the tissue, despite suppression of systemic inflammation. Ultrasound imaging showed that vessel-wall thickening continues in GC-treated GCA patients [11]. In follow-up, temporal artery biopsies in GCA patients showed ongoing vascular inflammation with macrophages and T cells [15], which underscores the potential of macrophages or macrophage-derived factors in disease monitoring. As an example, our group recently demonstrated that serum levels of two macrophage-derived proteins, Calprotectin and YKL-40, remained elevated in GCA patients during the first year of treatment. It is tempting to speculate that the persistence of elevated calprotectin and YKL-40 levels reflects persisting vessel wall inflammation although this was not formally proven [104].
1.6.2. B. Macrophage Targeted Imaging
Apart from blood biomarkers, positron emission tomography combined with computed tomography (PET/CT) is emerging as a potent diagnostic tool for GCA. Visualization of inflammation via [18F]FDG-PET imaging is now a useful tool for diagnosis and monitoring of treatment in GCA [101]. However, [18F]FDG-PET imaging still shows a number of disadvantages. Firstly, the diagnostic utility of [18F]FDG-PET in glucocorticoid-treated patients is significantly reduced only after 10 days of glucocorticoid treatment [117,118]. Yet, it is not always feasible to perform diagnostic and disease monitoring imaging of the patients within the narrow timeframe due to limited hospital capacities. [18F]FDG-PET scan relies on glucose uptake of metabolically active immune cells and stromal cells. Treatment with GCs was shown to reduce glycolysis in immune cells and therefore may downmodulate the vascular wall uptake of FGD [101]. Higher [18F]FDG uptake in aging vessels due to changes in metabolic activity, persistent vessel wall remodeling, and atherosclerotic calcifications may also pose a problem in the diagnosis of an aging disease such as GCA [119,120]. Therefore, recently discovered radiotracers for visualizing specific macrophage subsets may improve the diagnosis as well as the monitoring of treatment efficacy and disease progression during follow-up [121,122].
The fact that macrophages play key roles in various inflammatory diseases has encouraged many researchers to extensively explore the viability of targeting macrophages for diagnostic purposes. Several of these radiotracers are still in preclinical development but some radiotracers are already being studied in clinical trials (Table 1). One of the potential macrophage targeted radiotracers for the diagnosis and treatment monitoring of GCA is [18F]NOS, a radiopharmaceutical targeting iNOS. [18F]NOS was utilized in a study to quantify iNOS expression from endotoxin-induced lung inflammation in healthy volunteers. In this study, they showed that imaging iNOS activity is efficient in acute lung inflammation with abundant iNOS+ macrophage infiltration [123]. Considering the expression of iNOS in metabolically active macrophages and the presence of CD68(+)iNOS(+) macrophages in inflamed arteries of GCA patients, iNOS could be a candidate tracer to image macrophages in GCA.

Table 1.
Radiotracers targeting macrophage subsets in the GCA affected vessel-wall.
Apart from iNOS+ macrophages, our recent observations highlighting the presence of CD206+ and FRβ+ macrophages in GCA have sparked interest in exploring novel radiotracers specifically targeting these macrophage subsets in GCA. In a first clinical study, [18F]fluoro-PEG-folate, a novel radiotracer that targets the FRβ was recently shown to be a potent tool in imaging FRβ macrophages at the site of inflammation in rheumatoid arthritis [124]. More recently, a clinical grade gallium-68-tagged antibody fragment targeting CD206 ([68Ga]NOTA-anti-MMR-sdAb) has been developed for phase I clinical studies in humans [136]. In addition, a ligand for the translocator protein (TSPO) on activated macrophages, 11C-(R)-PK11195, was shown to improve imaging of macrophage infiltration to vessel wall in large- vessel vasculitis patients [137]. Apart from the aforementioned radiotracers, several other radiotracers currently in preclinical development may also potentially be useful for disease monitoring in GCA. Targeting CD163 by 68Ga labeled anti-CD163-antibody in rats with acute collagen-induced arthritis (CIA) displayed a significant uptake at the site of inflammation [129]. Likewise, MMP-2/9 targeting [68Ga]DOTA-TCTP-1 showed a specific uptake in inflamed atherosclerotic lesions in mice [130]. To conclude, a variety of macrophage targeted imaging tracers are currently being developed and evaluated. These novel radiotracers may potentially be useful for diagnostic and disease monitoring purposes in GCA patients.
1.7. Targeting Macrophages as Alternative Therapeutic Strategies for the Treatment of GCA
Glucocorticoids are currently the cornerstone in the treatment of GCA. GC treatment may resolve systemic inflammation rapidly but is not able to completely suppress vascular inflammation, leading to a relapsing/chronic disease. Moreover, long-term high doses of GCs come with serious adverse events in GCA patients [13,138,139]. Therefore, identification of GC-sparing agents for GCA treatment is imperative. Studies on temporal arteries engrafted into SCID mice demonstrated that after administration of dexamethasone for one week, T cells and macrophage functions were partially suppressed via blockage of the nuclear localization of NFkappaB which markedly reduced IL-6, IL-2, IL-1β and iNOS mRNA levels [140]. Chronic steroid therapy was shown to deplete some T cell products such as IL-17 but not IFN-γ, whereas, in macrophages, TGF-β1 synthesis was not affected [140,141]. This implies that T-cells and macrophages are only partially suppressed by GCs at the vascular site which may underlie the chronicity of the disease and thus emphasizes the need for alternative therapy in GCA. Studies revealed that increased CD68+ cells in TABs are linked to relapses with patients eventually requiring Disease-modifying antirheumatic drugs (DMARDs) [142]. Furthermore, persistence of macrophage infiltration in the vessels of GCA patients while on treatment, indicates that current treatments do not sufficiently suppress the local inflammatory response [15]. Therefore, targeting macrophages or macrophage-related pathways may prove to be more effective for suppressing chronic ongoing vascular inflammation and may provide lasting remission.
Indeed, IL-6 signaling, a cytokine released also by macrophages at the inflammatory site, has an essential role in systemic inflammation in GCA, as its serum levels correlate with CRP and other acute-phase proteins [19]. In the Giant-Cell Arteritis Actemra (GiACTA) trial, blocking the IL-6 receptor with tocilizumab was found to induce higher rates of sustained GC-free remission in GCA patients compared to placebo [19]. However, despite the clear efficacy of tocilizumab in GCA, up to 44% of patients treated with tocilizumab did not achieve sustained GC-free remission after 1 year of treatment. This underlines the heterogeneity among patients with GCA and the need for other targets for more personalized GCA treatment [19]. Furthermore, there is ongoing debate over the efficacy of tocilizumab in resolving vascular inflammation in GCA despite normalized inflammatory markers [143,144,145]. Additional to targeting IL-6 receptor with tocilizumab, sirukumab (IL-6 neutralization) and inhibition of the Janus kinase–signal transducers and activators of transcription (JAK–STAT) pathway [2] are being investigated as possible treatment options to resolve inflammation in GCA patients. The JAK-STAT pathway acts downstream of IL-6, IFN-γ and GM-CSF [146,147]. Therefore, targeting IL-6, IFNγ, GM-CSF or downstream cytokine signal transduction pathways may provide efficient treatment options in GCA.
Besides the IL-6 signaling pathway, the GM-CSF signaling pathway may be an important target for treatment in GCA. GM-CSF transcripts and protein were detected in vascular lesions and reported to play a major role in the pathogenesis of GCA [44,148]. In line with these reports, Mavrilimumab, a GM-CSF receptor antagonist is currently being evaluated as a therapeutic option for GCA patients. The phase 2 trial (NCT03827018) has demonstrated that 83% of Mavrilimumab treated patients were in sustained remission at week 26 compared to 50% of placebo-treated patients, when added to a 26-week prednisolone taper [149,150].
2. Future Perspectives
Investigating macrophage phenotypes and functions in GCA pathogenesis may facilitate the development of novel therapeutics and monitoring tools. Apart from intervening in cytokine-related pathways, targeting metabolic activity in macrophages may be an interesting alternative. Targeting glucose metabolism with anti-diabetic drugs in three PMR patients with concurrent type II diabetes improved the PMR symptoms and laboratory measurements like CRP [151]. Furthermore, small molecules such as DASA-58, TEPP-46, or shikonin, which target glucose metabolism led to a shift in macrophage phenotype by suppression of pro-inflammatory cytokine production in vitro [152,153]. Thus, targeting the glucose metabolism in GCA with small molecules may provide a novel option for GCA treatment. Clearly, this remains to be investigated in dedicated clinical trials. Lastly, considering the involvement of senescent cells in the production of pro-inflammatory mediators, ablation of senescent cells by senolytics is another therapeutically interesting concept. However, to be considered as a possible targeted therapy in GCA, the senescent cells contributing to the SASP in affected tissues needs first to be investigated [154].
3. Conclusions
In this review, we underlined the central role of monocytes and macrophages in the pathogenesis of GCA. In inflamed arteries, infiltrated and differentiated macrophages are key in shaping cellular and molecular processes involved in tissue destruction and tissue remodeling. Reversely, different cell types and their products such as metabolites, cytokines and chemokines in the vascular microenvironment influence differentiation, function, and heterogeneity of macrophages in GCA. Further research into the role of these constantly changing macrophages and their products in vasculitis lesions in GCA may eventually lead to discovery of new relevant targets for diagnosis, prognosis, monitoring disease activity, imaging and therapeutic intervention in GCA.
Author Contributions
Writing original draft: I.E., writing—review & editing: W.F.J., Y.v.S., A.M.H.B., P.H., M.S., K.S.M.v.d.G., E.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Dutch Arthritis Foundation (Reumafonds [grant number RF 14-3-401]) and European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement 754425.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
van der Geest and Brouwer receive a speaker/consulting fee from Roche paid to the University Medical Center Groningen. All other authors declare no conflict of interest.
References
- van der Geest, K.S.M.; Sandovici, M.; Brouwer, E.; Mackie, S.L. Diagnostic Accuracy of Symptoms, Physical Signs, and Laboratory Tests for Giant Cell Arteritis: A Systematic Review and Meta-analysis. JAMA Intern. Med. 2020, 180, 1295–1304. [Google Scholar] [CrossRef]
- Dejaco, C.; Brouwer, E.; Mason, J.C.; Buttgereit, F.; Matteson, E.L.; Dasgupta, B. Giant cell arteritis and polymyalgia rheumatica: Current challenges and opportunities. Nat. Rev. Rheumatol. 2017, 13, 578–592. [Google Scholar] [CrossRef]
- Agard, C.; Barrier, J.-H.; Dupas, B.; Ponge, T.; Mahr, A.; Fradet, G.; Chevalet, P.; Masseau, A.; Batard, E.; Pottier, P.; et al. Aortic involvement in recent-onset giant cell (temporal) arteritis: A case-control prospective study using helical aortic computed tomodensitometric scan. Arthritis Rheum. 2008, 59, 670–676. [Google Scholar] [CrossRef]
- Koster, M.J.; Matteson, E.L.; Warrington, K.J. Large-vessel giant cell arteritis: Diagnosis, monitoring and management. Rheumatology 2018, 57, ii32–ii42. [Google Scholar] [CrossRef]
- Prieto-González, S.; Arguis, P.; García-Martínez, A.; Espígol-Frigolé, G.; Tavera-Bahillo, I.; Butjosa, M.; Sánchez, M.; Hernández-Rodríguez, J.; Grau, J.M.; Cid, M.C. Large vessel involvement in biopsy-proven giant cell arteritis: Prospective study in 40 newly diagnosed patients using CT angiography. Ann. Rheum. Dis. 2012, 71, 1170–1176. [Google Scholar] [CrossRef]
- Schmidt, W.A.; Seifert, A.; Gromnica-Ihle, E.; Krause, A.; Natusch, A. Ultrasound of proximal upper extremity arteries to increase the diagnostic yield in large-vessel giant cell arteritis. Rheumatology 2008, 47, 96–101. [Google Scholar] [CrossRef]
- Hunder, G.G.; Bloch, D.A.; Michel, B.A.; Stevens, M.B.; Arend, W.P.; Calabrese, L.H.; Edworthy, S.M.; Fauci, A.S.; Leavitt, R.Y.; Lie, J.T. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990, 33, 1122–1128. [Google Scholar] [CrossRef]
- Dejaco, C.; Duftner, C.; Buttgereit, F.; Matteson, E.L.; Dasgupta, B. The spectrum of giant cell arteritis and polymyalgia rheumatica: Revisiting the concept of the disease. Rheumatology 2017, 56, 506–515. [Google Scholar] [CrossRef]
- Hellmich, B.; Agueda, A.; Monti, S.; Buttgereit, F.; De Boysson, H.; Brouwer, E.; Cassie, R.; Cid, M.C.; Dasgupta, B.; Dejaco, C.; et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann. Rheum. Dis. 2020, 79, 19–130. [Google Scholar] [CrossRef]
- Luqmani, R.; Lee, E.; Singh, S.; Gillett, M.; Schmidt, W.A.; Bradburn, M.; Dasgupta, B.; Diamantopoulos, A.P.; Forrester-Barker, W.; Hamilton, W.; et al. The Role of Ultrasound Compared to Biopsy of Temporal Arteries in the Diagnosis and Treatment of Giant Cell Arteritis (TABUL): A diagnostic accuracy and cost-effectiveness study. Health Technol. Assess. 2016, 20, 1–238. [Google Scholar] [CrossRef]
- Schmidt, W.A. Ultrasound in the diagnosis and management of giant cell arteritis. Rheumatology 2018, 57, ii22–ii31. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Matteson, E.L.; Dejaco, C.; Dasgupta, B. Prevention of glucocorticoid morbidity in giant cell arteritis. Rheumatology 2018, 57, ii11–ii21. [Google Scholar] [CrossRef] [PubMed]
- Broder, M.S.; Sarsour, K.; Chang, E.; Collinson, N.; Tuckwell, K.; Napalkov, P.; Klearman, M. Corticosteroid-related adverse events in patients with giant cell arteritis: A claims-based analysis. Semin. Arthritis Rheum. 2016, 46, 246–252. [Google Scholar] [CrossRef]
- Restuccia, G.; Boiardi, L.; Cavazza, A.; Catanoso, M.; Macchioni, P.; Muratore, F.; Cimino, L.; Aldigeri, R.; Crescentini, F.; Pipitone, N.; et al. Flares in Biopsy-Proven Giant Cell Arteritis in Northern Italy: Characteristics and Predictors in a Long-Term Follow-Up Study. Medicine 2016, 95, e3524. [Google Scholar] [CrossRef]
- Maleszewski, J.J.; Younge, B.R.; Fritzlen, J.T.; Hunder, G.G.; Goronzy, J.J.; Warrington, K.J.; Weyand, C.M. Clinical and pathological evolution of giant cell arteritis: A prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod. Pathol. 2017, 30, 788–796. [Google Scholar] [CrossRef]
- Schmidt, W.A.; Moll, A.; Seifert, A.; Schicke, B.; Gromnica-Ihle, E.; Krause, A. Prognosis of large-vessel giant cell arteritis. Rheumatology 2008, 47, 1406–1408. [Google Scholar] [CrossRef] [PubMed]
- van Sleen, Y.; Graver, J.C.; Abdulahad, W.H.; van der Geest, K.S.M.; Boots, A.M.H.; Sandovici, M.; Brouwer, E. Leukocyte Dynamics Reveal a Persistent Myeloid Dominance in Giant Cell Arteritis and Polymyalgia Rheumatica. Front. Immunol. 2019, 10, 1981. [Google Scholar] [CrossRef]
- Jover, J.A.; Hernández-García, C.; Morado, I.C.; Vargas, E.; Bañares, A.; Fernández-Gutiérrez, B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2001, 134, 106–114. [Google Scholar] [CrossRef]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; et al. Trial of Tocilizumab in Giant-Cell Arteritis. N. Engl. J. Med. 2017, 377, 317–328. [Google Scholar] [CrossRef]
- Reichenbach, S.; Adler, S.; Bonel, H.; Cullmann, J.L.; Kuchen, S.; Bütikofer, L.; Seitz, M.; Villiger, P.M. Magnetic resonance angiography in giant cell arteritis: Results of a randomized controlled trial of tocilizumab in giant cell arteritis. Rheumatology 2018, 57, 982–986. [Google Scholar] [CrossRef]
- Gloor, A.D.; Yerly, D.; Adler, S.; Reichenbach, S.; Kuchen, S.; Seitz, M.; Villiger, P.M. Immuno-monitoring reveals an extended subclinical disease activity in tocilizumab-treated giant cell arteritis. Rheumatology 2018, 57, 1795–1801. [Google Scholar] [CrossRef]
- González-Gay, M.A.; Amoli, M.M.; Garcia-Porrua, C.; Ollier, W.E. r Genetic markers of disease susceptibility and severity in giant cell arteritis and polymyalgia rheumatica. Semin. Arthritis Rheum. 2003, 33, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Peña, D.; Remuzgo-Martínez, S.; Ocejo-Vinyals, J.G.; Atienza-Mateo, B.; Genre, F.; Muñoz-Jimenez, A.; Ortiz-Sanjuán, F.; Romero-Yuste, S.; Moriano, C.; Galindez-Agirregoikoa, E.; et al. The presence of both HLA-DRB1[*]04:01 and HLA-B[*]15:01 increases the susceptibility to cranial and extracranial giant cell arteritis. Clin. Exp. Rheumatol. 2021, 39, 21–26. [Google Scholar]
- Rhee, R.L.; Grayson, P.C.; Merkel, P.A.; Tomasson, G. Infections and the risk of incident giant cell arteritis: A population-based, case-control study. Ann. Rheum. Dis. 2017, 76, 1031–1035. [Google Scholar] [CrossRef]
- Hoffman, G.S.; Getz, T.M.; Padmanabhan, R.; Villa-Forte, A.; Clifford, A.H.; Funchain, P.; Sankunny, M.; Perry, J.D.; Blandford, A.; Kosmorsky, G.; et al. The Microbiome of Temporal Arteries. Pathog. Immun. 2019, 4, 21–38. [Google Scholar] [CrossRef]
- Getz, T.M.; Hoffman, G.S.; Padmanabhan, R.; Villa-Forte, A.; Roselli, E.E.; Blackstone, E.; Johnston, D.; Pettersson, G.; Soltesz, E.; Svensson, L.G.; et al. Microbiomes of Inflammatory Thoracic Aortic Aneurysms Due to Giant Cell Arteritis and Clinically Isolated Aortitis Differ From Those of Non-Inflammatory Aneurysms. Pathog. Immun. 2019, 4, 105–123. [Google Scholar] [CrossRef][Green Version]
- Bhatt, A.S.; Manzo, V.E.; Pedamallu, C.S.; Duke, F.; Cai, D.; Bienfang, D.C.; Padera, R.F.; Meyerson, M.; Docken, W.P. In search of a candidate pathogen for giant cell arteritis: Sequencing-based characterization of the giant cell arteritis microbiome. Arthritis Rheumatol. 2014, 66, 1939–1944. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Watanabe, R.; Zhang, H.; Akiyama, M.; Berry, G.J.; Goronzy, J.J. Cytokines, growth factors and proteases in medium and large vessel vasculitis. Clin. Immunol. 2019, 206, 33–41. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, J.; Kurban, M.; Abbas, O. Elastophagocytosis: Underlying mechanisms and associated cutaneous entities. J. Am. Acad. Dermatol. 2014, 70, 934–944. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Bosco, M.C. Macrophage polarization: Reaching across the aisle? J. Allergy Clin. Immunol. 2019, 143, 1348–1350. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Corbera-Bellalta, M.; Audia, S.; Planas-Rigol, E.; Martin, L.; Cid, M.C.; Bonnotte, B. Recent advances in our understanding of giant cell arteritis pathogenesis. Autoimmun. Rev. 2017, 16, 833–844. [Google Scholar] [CrossRef]
- Ciccia, F.; Rizzo, A.; Ferrante, A.; Guggino, G.; Croci, S.; Cavazza, A.; Salvarani, C.; Triolo, G. New insights into the pathogenesis of giant cell arteritis. Autoimmun. Rev. 2017, 16, 675–683. [Google Scholar] [CrossRef] [PubMed]
- van Sleen, Y.; Jiemy, W.F.; Pringle, S.; van der Geest, K.S.M.; Abdulahad, W.H.; Sandovici, M.; Brouwer, E.; Heeringa, P.; Boots, A.M.H. A Distinct Macrophage Subset Mediating Tissue Destruction and Neovascularization in Giant Cell Arteritis: Implication of the YKL-40-IL-13 Receptor α2 Axis. Arthritis Rheumatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Hofer, T.P.J. Toward a refined definition of monocyte subsets. Front. Immunol. 2013, 4, 23. [Google Scholar] [CrossRef]
- van Sleen, Y.; Wang, Q.; van der Geest, K.S.M.; Westra, J.; Abdulahad, W.H.; Heeringa, P.; Boots, A.M.H.; Brouwer, E. Involvement of Monocyte Subsets in the Immunopathology of Giant Cell Arteritis. Sci. Rep. 2017, 7, 6553. [Google Scholar] [CrossRef]
- Matsumoto, K.; Suzuki, K.; Yoshimoto, K.; Seki, N.; Tsujimoto, H.; Chiba, K.; Takeuchi, T. Significant association between clinical characteristics and changes in peripheral immuno-phenotype in large vessel vasculitis. Arthritis Res. Ther. 2019, 21, 304. [Google Scholar] [CrossRef]
- Dayyani, F.; Belge, K.-U.; Frankenberger, M.; Mack, M.; Berki, T.; Ziegler-Heitbrock, L. Mechanism of glucocorticoid-induced depletion of human CD14+ CD16+ monocytes. J. Leukoc. Biol. 2003, 74, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.C.; Hoffman, M.P.; Hernández-Rodríguez, J.; Segarra, M.; Elkin, M.; Sánchez, M.; Vilardell, C.; García-Martínez, A.; Pla-Campo, M.; Grau, J.M.; et al. Association between increased CCL2 (MCP-1) expression in lesions and persistence of disease activity in giant-cell arteritis. Rheumatology 2006, 45, 1356–1363. [Google Scholar] [CrossRef]
- Watanabe, R.; Maeda, T.; Zhang, H.; Berry, G.J.; Zeisbrich, M.; Brockett, R.; Greenstein, A.E.; Tian, L.; Goronzy, J.J.; Weyand, C.M. MMP (Matrix Metalloprotease)-9-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ. Res. 2018, 123, 700–715. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Boots, A.M.H.; Steenbakkers, P.G.A.; Elewaut, D.; Bos, E.; Verheijden, G.F.M.; Verbruggen, G.; Miltenburg, A.M.M.; Rijnders, A.W.M.; Veys, E.M.; et al. Human cartilage gp-39, CD16+ monocytes in peripheral blood and synovium: Correlation with joint destruction in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1233–1243. [Google Scholar] [CrossRef]
- Campos, T.M.; Passos, S.T.; Novais, F.O.; Beiting, D.P.; Costa, R.S.; Queiroz, A.; Mosser, D.; Scott, P.; Carvalho, E.M.; Carvalho, L.P. Matrix metalloproteinase 9 production by monocytes is enhanced by TNF and participates in the pathology of human cutaneous Leishmaniasis. PLoS Negl. Trop. Dis. 2014, 8, e3282. [Google Scholar] [CrossRef]
- Jiemy, W.F.; van Sleen, Y.; van der Geest, K.S.; Ten Berge, H.A.; Abdulahad, W.H.; Sandovici, M.; Boots, A.M.; Heeringa, P.; Brouwer, E. Distinct macrophage phenotypes skewed by local granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) are associated with tissue destruction and intimal hyperplasia in giant cell arteritis. Clin. Transl. Immunol. 2020, 9, e1164. [Google Scholar] [CrossRef]
- Shirai, T.; Hilhorst, M.; Harrison, D.G.; Goronzy, J.J.; Weyand, C.M. Macrophages in vascular inflammation--From atherosclerosis to vasculitis. Autoimmunity 2015, 48, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Laria, A.; Lurati, A.; Marrazza, M.; Mazzocchi, D.; Re, K.A.; Scarpellini, M. The macrophages in rheumatic diseases. J. Inflamm. Res. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Di Benedetto, P.; Ruscitti, P.; Vadasz, Z.; Toubi, E.; Giacomelli, R. Macrophages with regulatory functions, a possible new therapeutic perspective in autoimmune diseases. Autoimmun. Rev. 2019, 18, 102369. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Anders, H.J. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 989–997. [Google Scholar] [CrossRef] [PubMed]
- De Maeyer, R.P.H.; Chambers, E.S. The impact of ageing on monocytes and macrophages. Immunol. Lett. 2021, 230, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.D.; Björnsson, J.; Bartley, G.B.; Goronzy, J.J.; Weyand, C.M. Interferon-gamma-producing T cells in giant cell vasculitis represent a minority of tissue-infiltrating cells and are located distant from the site of pathology. Am. J. Pathol. 1996, 148, 1925–1933. [Google Scholar]
- Ciccia, F.; Alessandro, R.; Rizzo, A.; Raimondo, S.; Giardina, A.; Raiata, F.; Boiardi, L.; Cavazza, A.; Guggino, G.; De Leo, G.; et al. IL-33 is overexpressed in the inflamed arteries of patients with giant cell arteritis. Ann. Rheum. Dis. 2013, 72, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. Immune mechanisms in medium and large-vessel vasculitis. Nat. Rev. Rheumatol. 2013, 9, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Wagner, A.D.; Björnsson, J.; Goronzy, J.J. Correlation of the topographical arrangement and the functional pattern of tissue-infiltrating macrophages in giant cell arteritis. J. Clin. Investig. 1996, 98, 1642–1649. [Google Scholar] [CrossRef]
- Wagner, A.D.; Goronzy, J.J.; Weyand, C.M. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. Evidence for two components of the disease. J. Clin. Investig. 1994, 94, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Samaniego, R.; Palacios, B.S.; Domiguez-Soto, Á.; Vidal, C.; Salas, A.; Matsuyama, T.; Sánchez-Torres, C.; de la Torre, I.; Miranda-Carús, M.E.; Sánchez-Mateos, P.; et al. Macrophage uptake and accumulation of folates are polarization-dependent in vitro and in vivo and are regulated by activin A. J. Leukoc. Biol. 2014, 95, 797–808. [Google Scholar] [CrossRef]
- Puig-Kröger, A.; Sierra-Filardi, E.; Domínguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gómez-Aguado, F.; Ratnam, M.; Sánchez-Mateos, P.; Corbí, A.L. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef]
- Kittan, N.A.; Allen, R.M.; Dhaliwal, A.; Cavassani, K.A.; Schaller, M.; Gallagher, K.A.; Carson, W.F., 4th; Mukherjee, S.; Grembecka, J.; Cierpicki, T.; et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE 2013, 8, e78045. [Google Scholar] [CrossRef]
- Tomita, T.; Imakawa, K. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in giant cell arteritis: An immunocytochemical study. Pathology 1998, 30, 40–50. [Google Scholar] [CrossRef]
- Rodríguez-Pla, A.; Bosch-Gil, J.A.; Rosselló-Urgell, J.; Huguet-Redecilla, P.; Stone, J.H.; Vilardell-Tarres, M. Metalloproteinase-2 and -9 in giant cell arteritis: Involvement in vascular remodeling. Circulation 2005, 112, 264–269. [Google Scholar] [CrossRef]
- Nikkari, S.T.; Höyhtyä, M.; Isola, J.; Nikkari, T. Macrophages contain 92-kd gelatinase (MMP-9) at the site of degenerated internal elastic lamina in temporal arteritis. Am. J. Pathol. 1996, 149, 1427–1433. [Google Scholar]
- Albano-Aluquin, S.; Malysz, J.; Aluquin, V.R.; Ratnam, M.; Olsen, N. An immunohistochemical analysis of folate receptor beta expression and distribution in giant cell arteritis—A pilot study. Am. J. Clin. Exp. Immunol. 2017, 6, 107–114. [Google Scholar] [PubMed]
- Erbel, C.; Akhavanpoor, M.; Okuyucu, D.; Wangler, S.; Dietz, A.; Zhao, L.; Stellos, K.; Little, K.M.; Lasitschka, F.; Doesch, A.; et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J. Immunol. 2014, 193, 4344–4355. [Google Scholar] [CrossRef]
- Jiemy, W.F.; Reitsema, R.; Kwant, A.; Abdulahad, W.; Boots, A.; Heeringa, P.; Brouwer, E. POS0112 CD8+ T-CELL infiltration is associated with lesional GM-CSF overexpression in GCA. Ann. Rheum. Dis. 2021, 80, 267. [Google Scholar] [CrossRef]
- Van Der Geest, K.S.M.; Abdulahad, W.H.; Chalan, P.; Rutgers, A.; Horst, G.; Huitema, M.G.; Roffel, M.P.; Roozendaal, C.; Kluin, P.M.; Bos, N.A.; et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol. 2014, 66, 1927–1938. [Google Scholar] [CrossRef] [PubMed]
- Graver, J.C.; Sandovici, M.; Diepstra, A.; Boots, A.M.H.; Brouwer, E. Artery tertiary lymphoid organs in giant cell arteritis are not exclusively located in the media of temporal arteries. Ann. Rheum. Dis. 2018, 77, 2–3. [Google Scholar] [CrossRef]
- Ciccia, F.; Rizzo, A.; Maugeri, R.; Alessandro, R.; Croci, S.; Guggino, G.; Cavazza, A.; Raimondo, S.; Cannizzaro, A.; Iacopino, D.G.; et al. Ectopic expression of CXCL13, BAFF, April and LT-β is associated with artery tertiary lymphoid organs in giant cell arteritis. Ann. Rheum. Dis. 2017, 76, 235–243. [Google Scholar] [CrossRef]
- Graver, J.C.; Jiemy, W.F.; Altulea, D.; Boots, A.; Heeringa, P.; Abdulahad, W.; Brouwer, E.; Sandovici, M. OP0062 Cytokine producing B cells skew macrophages towards a pro-inflammatory phenotype in giant cell arteritis. Ann. Rheum. Dis. 2021, 80, 33–34. [Google Scholar] [CrossRef]
- Kostic, M.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I. Granulocyte-macrophage colony-stimulating factor as a mediator of autoimmunity in multiple sclerosis. J. Neuroimmunol. 2018, 323, 1–9. [Google Scholar] [CrossRef]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef] [PubMed]
- Artyomov, M.N.; Sergushichev, A.; Schilling, J.D. Integrating immunometabolism and macrophage diversity. Semin. Immunol. 2016, 28, 417–424. [Google Scholar] [CrossRef]
- Batista-Gonzalez, A.; Vidal, R.; Criollo, A.; Carreño, L.J. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front. Immunol. 2020, 10, 2993. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Saraber, D.L. Metabolic regulation of macrophages in tissues. Cell. Immunol. 2018, 330, 54–59. [Google Scholar] [CrossRef]
- Kang, S.; Kumanogoh, A. The spectrum of macrophage activation by immunometabolism. Int. Immunol. 2020, 32, 467–473. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J. A critical role for citrate metabolism in LPS signalling. Biochem. J. 2011, 438, e5–e6. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, J.; Segarra, M.; Vilardell, C.; Sánchez, M.; García-Martínez, A.; Esteban, M.J.; Queralt, C.; Grau, J.M.; Urbano-Márquez, A.; Palacín, A.; et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology 2004, 43, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Rittner, H.L.; Kaiser, M.; Brack, A.; Szweda, L.I.; Goronzy, J.J.; Weyand, C.M. Tissue-destructive macrophages in giant cell arteritis. Circ. Res. 1999, 84, 1050–1058. [Google Scholar] [CrossRef]
- Tavakoli, S.; Short, J.D.; Downs, K.; Nguyen, H.N.; Lai, Y.; Zhang, W.; Jerabek, P.; Goins, B.; Sadeghi, M.M.; Asmis, R. Differential Regulation of Macrophage Glucose Metabolism by Macrophage Colony-stimulating Factor and Granulocyte-Macrophage Colony-stimulating Factor: Implications for (18)F FDG PET Imaging of Vessel Wall Inflammation. Radiology 2017, 283, 87–97. [Google Scholar] [CrossRef]
- Na, Y.R.; Gu, G.J.; Jung, D.; Kim, Y.W.; Na, J.; Woo, J.S.; Cho, J.Y.; Youn, H.; Seok, S.H. GM-CSF Induces Inflammatory Macrophages by Regulating Glycolysis and Lipid Metabolism. J. Immunol. 2016, 197, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- van Beek, A.A.; Van den Bossche, J.; Mastroberardino, P.G.; de Winther, M.P.J.; Leenen, P.J.M. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Appay, V.; Campisi, J.; Frasca, D.; Fülöp, T.; Sauce, D.; Larbi, A.; Weinberger, B.; Cossarizza, A. Aging of the immune system: Focus on inflammation and vaccination. Eur. J. Immunol. 2016, 46, 2286–2301. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Mohan, S.V.; Liao, Y.J.; Kim, J.W.; Goronzy, J.J.; Weyand, C.M. Giant cell arteritis: Immune and vascular aging as disease risk factors. Arthritis Res. Ther. 2011, 13, 231. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, J.; Ramirez, R.; Soriano, S.; Alvarez de Lara, M.A.; Rodriguez, M.; Martin-Malo, A.; Aljama, P. Monocytes from dialysis patients exhibit characteristics of senescent cells: Does it really mean inflammation? Contrib. Nephrol. 2005, 149, 208–218. [Google Scholar] [CrossRef]
- Merino, A.; Buendia, P.; Martin-Malo, A.; Aljama, P.; Ramirez, R.; Carracedo, J. Senescent CD14+CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J. Immunol. 2011, 186, 1809–1815. [Google Scholar] [CrossRef]
- Seidler, S.; Zimmermann, H.W.; Bartneck, M.; Trautwein, C.; Tacke, F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 2010, 11, 30. [Google Scholar] [CrossRef]
- Coit, P.; De Lott, L.B.; Nan, B.; Elner, V.M.; Sawalha, A.H. DNA methylation analysis of the temporal artery microenvironment in giant cell arteritis. Ann. Rheum. Dis. 2016, 75, 1196–1202. [Google Scholar] [CrossRef]
- Wang, Q.; Westra, J.; van der Geest, K.S.M.; Moser, J.; Bijzet, J.; Kuiper, T.; Lorencetti, P.G.; Joosten, L.A.B.; Netea, M.G.; Heeringa, P.; et al. Reduced levels of cytosolic DNA sensor AIM2 are associated with impaired cytokine responses in healthy elderly. Exp. Gerontol. 2016, 78, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Pryshchep, O.; Ma-Krupa, W.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation 2008, 118, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.; Molloy, E.S. The role of toll like receptors in giant cell arteritis. Rheumatology 2016, 55, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, L.Á.; López-Hoyos, M.; Mata, C.; Fontalba, A.; Alen, J.C.; Marín, M.J.; Fernández-Luna, J.L.; Balbín, J.A.; Zaldunbide, M.A.; Blanco, R.; et al. Expression and function of toll-like receptors in peripheral blood mononuclear cells of patients with polymyalgia rheumatica and giant cell arteritis. Ann. Rheum. Dis. 2011, 70, 1677–1683. [Google Scholar] [CrossRef] [PubMed]
- Bardi, M.; Diamantopoulos, A.P. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice summary. Radiol. Med. 2019, 124, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Van Der Geest, K.S.M.; Borg, F.; Kayani, A.; Paap, D.; Gondo, P.; Schmidt, W.; Luqmani, R.A.; Dasgupta, B. Novel ultrasonographic Halo Score for giant cell arteritis: Assessment of diagnostic accuracy and association with ocular ischaemia. Ann. Rheum. Dis. 2020, 79, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Prieto-González, S.; Depetris, M.; García-Martínez, A.; Espígol-Frigolé, G.; Tavera-Bahillo, I.; Corbera-Bellata, M.; Planas-Rigol, E.; Alba, M.A.; Hernández-Rodríguez, J.; Grau, J.M.; et al. Positron emission tomography assessment of large vessel inflammation in patients with newly diagnosed, biopsy-proven giant cell arteritis: A prospective, case-control study. Ann. Rheum. Dis. 2014, 73, 1388–1392. [Google Scholar] [CrossRef]
- Nienhuis, P.H.; Sandovici, M.; Glaudemans, A.W.; Slart, R.H.; Brouwer, E. Visual and semiquantitative assessment of cranial artery inflammation with FDG-PET/CT in giant cell arteritis. Semin. Arthritis Rheum. 2020, 50, 616–623. [Google Scholar] [CrossRef]
- Aschwanden, M.; Schegk, E.; Imfeld, S.; Staub, D.; Rottenburger, C.; Berger, C.T.; Daikeler, T. Vessel wall plasticity in large vessel giant cell arteritis: An ultrasound follow-up study. Rheumatology 2019, 58, 792–797. [Google Scholar] [CrossRef]
- van der Geest, K.S.M.; Treglia, G.; Glaudemans, A.W.J.M.; Brouwer, E.; Sandovici, M.; Jamar, F.; Gheysens, O.; Slart, R.H.J.A. Diagnostic value of [18F]FDG-PET/CT for treatment monitoring in large vessel vasculitis: A systematic review and meta-analysis. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3886–3902. [Google Scholar] [CrossRef] [PubMed]
- Burja, B.; Feichtinger, J.; Lakota, K.; Thallinger, G.G.; Sodin-Semrl, S.; Kuret, T.; Rotar, Ž.; Ješe, R.; Žigon, P.; Čučnik, S.; et al. Utility of serological biomarkers for giant cell arteritis in a large cohort of treatment-naïve patients. Clin. Rheumatol. 2019, 38, 317–329. [Google Scholar] [CrossRef] [PubMed]
- van der Geest, K.S.M.; Sandovici, M.; van Sleen, Y.; Sanders, J.-S.; Bos, N.A.; Abdulahad, W.H.; Stegeman, C.A.; Heeringa, P.; Rutgers, A.; Kallenberg, C.G.M.; et al. Review: What Is the Current Evidence for Disease Subsets in Giant Cell Arteritis? Arthritis Rheumatol. 2018, 70, 1366–1376. [Google Scholar] [CrossRef]
- van Sleen, Y.; Sandovici, M.; Abdulahad, W.H.; Bijzet, J.; van der Geest, K.S.M.; Boots, A.M.H.; Brouwer, E. Markers of angiogenesis and macrophage products for predicting disease course and monitoring vascular inflammation in giant cell arteritis. Rheumatology 2019, 58, 1383–1392. [Google Scholar] [CrossRef]
- Tombetti, E.; Hysa, E.; Mason, J.C.; Cimmino, M.A.; Camellino, D. Blood Biomarkers for Monitoring and Prognosis of Large Vessel Vasculitides. Curr. Rheumatol. Rep. 2021, 23, 17. [Google Scholar] [CrossRef]
- Johansen, J.S.; Baslund, B.; Garbarsch, C.; Hansen, M.; Stoltenberg, M.; Lorenzen, I.; Price, P.A. YKL-40 in giant cells and macrophages from patients with giant cell arteritis. Arthritis Rheum. 1999, 42, 2624–2630. [Google Scholar] [CrossRef]
- Baldini, M.; Maugeri, N.; Ramirez, G.A.; Giacomassi, C.; Castiglioni, A.; Prieto-González, S.; Corbera-Bellalta, M.; Di Comite, G.; Papa, I.; Dell’antonio, G.; et al. Selective up-regulation of the soluble pattern-recognition receptor pentraxin 3 and of vascular endothelial growth factor in giant cell arteritis: Relevance for recent optic nerve ischemia. Arthritis Rheum. 2012, 64, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Younge, B.; Björnsson, J.; Goronzy, J.J.; Weyand, C.M. Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells. Am. J. Pathol. 1999, 155, 765–774. [Google Scholar] [CrossRef]
- Sorbi, D.; French, D.L.; Nuovo, G.J.; Kew, R.R.; Arbeit, L.A.; Gruber, B.L. Elevated levels of 92-kd type IV collagenase (matrix metalloproteinase 9) in giant cell arteritis. Arthritis Rheum. 1996, 39, 1747–1753. [Google Scholar] [CrossRef]
- Grossman, C.; Yassin, N.; Avivi, C.; Bornstein, G.; Ben-Zvi, I.; Barshack, I. Cytokine expression in temporal arteries: Comparative analysis between patients with biopsy-positive giant cell arteritis, biopsy-negative giant cell arteritis and biopsy-negative without arteritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 1), 122–129. [Google Scholar] [PubMed]
- Prieto-González, S.; Terrades-García, N.; Corbera-Bellalta, M.; Planas-Rigol, E.; Miyabe, C.; Alba, M.A.; Ponce, A.; Tavera-Bahillo, I.; Murgia, G.; Espígol-Frigolé, G.; et al. Serum osteopontin: A biomarker of disease activity and predictor of relapsing course in patients with giant cell arteritis. Potential clinical usefulness in tocilizumab-treated patients. RMD Open 2017, 3, e000570. [Google Scholar] [CrossRef]
- Desbois, A.-C.; Cacoub, P.; Leroyer, A.S.; Tellier, E.; Garrido, M.; Maciejewski-Duval, A.; Comarmond, C.; Barete, S.; Arock, M.; Bruneval, P.; et al. Immunomodulatory role of Interleukin-33 in large vessel vasculitis. Sci. Rep. 2020, 10, 6405. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Nunokawa, T.; Kobayashi, S.; Kamei, S.; Yokogawa, N.; Takizawa, Y.; Shimada, K.; Sugii, S.; Setoguchi, K. MMP-3 can distinguish isolated PMR from PMR with GCA: A retrospective study regarding PMR and GCA in Japan. Mod. Rheumatol. 2016, 26, 259–264. [Google Scholar] [CrossRef] [PubMed]
- van Sleen, Y.; Boots, A.M.H.; Abdulahad, W.H.; Bijzet, J.; Sandovici, M.; van der Geest, K.S.M.; Brouwer, E. High angiopoietin-2 levels associate with arterial inflammation and long-term glucocorticoid requirement in polymyalgia rheumatica. Rheumatology 2020, 59, 176–184. [Google Scholar] [CrossRef]
- Muratore, F.; Boiardi, L.; Restuccia, G.; Cavazza, A.; Catanoso, M.; Macchioni, P.; Spaggiari, L.; Cimino, L.; Aldigeri, R.; Pipitone, N.; et al. Relapses and long-term remission in large vessel giant cell arteritis in northern Italy: Characteristics and predictors in a long-term follow-up study. Semin. Arthritis Rheum. 2020, 50, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; et al. Glucocorticoid Dosages and Acute-Phase Reactant Levels at Giant Cell Arteritis Flare in a Randomized Trial of Tocilizumab. Arthritis Rheumatol. 2019, 71, 1329–1338. [Google Scholar] [CrossRef]
- Stellingwerff, M.D.; Brouwer, E.; Lensen, K.-J.D.F.; Rutgers, A.; Arends, S.; van der Geest, K.S.M.; Glaudemans, A.W.J.M.; Slart, R.H.J.A. Different Scoring Methods of FDG PET/CT in Giant Cell Arteritis: Need for Standardization. Medicine 2015, 94, e1542. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, B.D.; Gormsen, L.C.; Hansen, I.T.; Keller, K.K.; Therkildsen, P.; Hauge, E.-M. Three days of high-dose glucocorticoid treatment attenuates large-vessel 18F-FDG uptake in large-vessel giant cell arteritis but with a limited impact on diagnostic accuracy. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1119–1128. [Google Scholar] [CrossRef]
- Dunphy, M.P.S.; Freiman, A.; Larson, S.M.; Strauss, H.W. Association of vascular 18F-FDG uptake with vascular calcification. J. Nucl. Med. 2005, 46, 1278–1284. [Google Scholar]
- Bural, G.G.; Torigian, D.A.; Basu, S.; Bozkurt, M.F.; Houseni, M.; Alavi, A. Atherosclerotic inflammatory activity in the aorta and its correlation with aging and gender as assessed by 18F-FDG-PET. Hell. J. Nucl. Med. 2013, 16, 164–168. [Google Scholar] [CrossRef]
- Jiemy, W.F.; Heeringa, P.; Kamps, J.A.A.M.; van der Laken, C.J.; Slart, R.H.J.A.; Brouwer, E. Positron emission tomography (PET) and single photon emission computed tomography (SPECT) imaging of macrophages in large vessel vasculitis: Current status and future prospects. Autoimmun. Rev. 2018, 17, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Farrah, T.E.; Basu, N.; Dweck, M.; Calcagno, C.; Fayad, Z.A.; Dhaun, N. Advances in Therapies and Imaging for Systemic Vasculitis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1520–1541. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.J.; Isakow, W.; Byers, D.E.; Engle, J.T.; Griffin, E.A.; Kemp, D.; Brody, S.L.; Gropler, R.J.; Miller, J.P.; Chu, W.; et al. Imaging pulmonary inducible nitric oxide synthase expression with PET. J. Nucl. Med. 2015, 56, 76–81. [Google Scholar] [CrossRef]
- Verweij, N.J.F.; Yaqub, M.; Bruijnen, S.T.G.; Pieplenbosch, S.; ter Wee, M.M.; Jansen, G.; Chen, Q.; Low, P.S.; Windhorst, A.D.; Lammertsma, A.A.; et al. First in man study of [18F]fluoro-PEG-folate PET: A novel macrophage imaging technique to visualize rheumatoid arthritis. Sci. Rep. 2020, 10, 1047. [Google Scholar] [CrossRef] [PubMed]
- Betzel, T.; Müller, C.; Groehn, V.; Müller, A.; Reber, J.; Fischer, C.R.; Krämer, S.D.; Schibli, R.; Ametamey, S.M. Radiosynthesis and preclinical evaluation of 3′-Aza-2′-[(18)F]fluorofolic acid: A novel PET radiotracer for folate receptor targeting. Bioconjug. Chem. 2013, 24, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Moisio, O.; Palani, S.; Virta, J.; Elo, P.; Liljenbäck, H.; Tolvanen, T.; Käkelä, M.; Miner, M.G.; Herre, E.A.; Marjamäki, P.; et al. Radiosynthesis and preclinical evaluation of [(68)Ga]Ga-NOTA-folate for PET imaging of folate receptor β-positive macrophages. Sci. Rep. 2020, 10, 13593. [Google Scholar] [CrossRef]
- Schniering, J.; Benešová, M.; Brunner, M.; Haller, S.; Cohrs, S.; Frauenfelder, T.; Vrugt, B.; Feghali-Bostwick, C.; Schibli, R.; Distler, O.; et al. (18)F-AzaFol for Detection of Folate Receptor-β Positive Macrophages in Experimental Interstitial Lung Disease-A Proof-of-Concept Study. Front. Immunol. 2019, 10, 2724. [Google Scholar] [CrossRef] [PubMed]
- Silvola, J.M.U.; Li, X.-G.; Virta, J.; Marjamäki, P.; Liljenbäck, H.; Hytönen, J.P.; Tarkia, M.; Saunavaara, V.; Hurme, S.; Palani, S.; et al. Aluminum fluoride-18 labeled folate enables in vivo detection of atherosclerotic plaque inflammation by positron emission tomography. Sci. Rep. 2018, 8, 9720. [Google Scholar] [CrossRef] [PubMed]
- Eichendorff, S.; Svendsen, P.; Bender, D.; Keiding, S.; Christensen, E.I.; Deleuran, B.; Moestrup, S.K. Biodistribution and PET imaging of a novel [68Ga]-anti-CD163-antibody conjugate in rats with collagen-induced arthritis and in controls. Mol. Imaging Biol. 2015, 17, 87–93. [Google Scholar] [CrossRef]
- Kiugel, M.; Hellberg, S.; Käkelä, M.; Liljenbäck, H.; Saanijoki, T.; Li, X.-G.; Tuomela, J.; Knuuti, J.; Saraste, A.; Roivainen, A. Evaluation of [(68)Ga]Ga-DOTA-TCTP-1 for the Detection of Metalloproteinase 2/9 Expression in Mouse Atherosclerotic Plaques. Molecules 2018, 23, 3168. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Pan, D.; Cheng, C.; Zhang, A.; Ma, C.; Wang, L.; Zhang, D.; Liu, H.; Jiang, H.; Wang, T.; et al. Targeting of MMP2 activity in malignant tumors with a 68Ga-labeled gelatinase inhibitor cyclic peptide. Nucl. Med. Biol. 2015, 42, 939–944. [Google Scholar] [CrossRef]
- Tahara, N.; Mukherjee, J.; de Haas, H.J.; Petrov, A.D.; Tawakol, A.; Haider, N.; Tahara, A.; Constantinescu, C.C.; Zhou, J.; Boersma, H.H.; et al. 2-deoxy-2-[18F]fluoro-D-mannose positron emission tomography imaging in atherosclerosis. Nat. Med. 2014, 20, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kim, S.; Seo, H.S.; Lee, Y.J.; Eo, J.S.; Jeong, J.M.; Lee, B.; Kim, J.Y.; Park, Y.M.; Jeong, M. Novel PET Imaging of Atherosclerosis with 68Ga-Labeled NOTA-Neomannosylated Human Serum Albumin. J. Nucl. Med. 2016, 57, 1792–1797. [Google Scholar] [CrossRef]
- Blykers, A.; Schoonooghe, S.; Xavier, C.; D’hoe, K.; Laoui, D.; D’Huyvetter, M.; Vaneycken, I.; Cleeren, F.; Bormans, G.; Heemskerk, J.; et al. PET Imaging of Macrophage Mannose Receptor-Expressing Macrophages in Tumor Stroma Using 18F-Radiolabeled Camelid Single-Domain Antibody Fragments. J. Nucl. Med. 2015, 56, 1265–1271. [Google Scholar] [CrossRef]
- Locke, L.W.; Mayo, M.W.; Yoo, A.D.; Williams, M.B.; Berr, S.S. PET imaging of tumor associated macrophages using mannose coated 64Cu liposomes. Biomaterials 2012, 33, 7785–7793. [Google Scholar] [CrossRef]
- Xavier, C.; Blykers, A.; Laoui, D.; Bolli, E.; Vaneyken, I.; Bridoux, J.; Baudhuin, H.; Raes, G.; Everaert, H.; Movahedi, K.; et al. Clinical Translation of [(68)Ga]Ga-NOTA-anti-MMR-sdAb for PET/CT Imaging of Protumorigenic Macrophages. Mol. Imaging Biol. 2019, 21, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Lamare, F.; Hinz, R.; Gaemperli, O.; Pugliese, F.; Mason, J.C.; Spinks, T.; Camici, P.G.; Rimoldi, O.E. Detection and quantification of large-vessel inflammation with 11C-(R)-PK11195 PET/CT. J. Nucl. Med. 2011, 52, 33–39. [Google Scholar] [CrossRef]
- Wilson, J.C.; Sarsour, K.; Collinson, N.; Tuckwell, K.; Musselman, D.; Klearman, M.; Napalkov, P.; Jick, S.S.; Stone, J.H.; Meier, C.R. Incidence of outcomes potentially associated with corticosteroid therapy in patients with giant cell arteritis. Semin. Arthritis Rheum. 2017, 46, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.C.; Sarsour, K.; Collinson, N.; Tuckwell, K.; Musselman, D.; Klearman, M.; Napalkov, P.; Jick, S.S.; Stone, J.H.; Meier, C.R. Serious adverse effects associated with glucocorticoid therapy in patients with giant cell arteritis (GCA): A nested case–control analysis. Semin. Arthritis Rheum. 2017, 46, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Brack, A.; Rittner, H.L.; Younge, B.R.; Kaltschmidt, C.; Weyand, C.M.; Goronzy, J.J. Glucocorticoid-mediated repression of cytokine gene transcription in human arteritis-SCID chimeras. J. Clin. Investig. 1997, 99, 2842–2850. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Younge, B.R.; Olshen, R.A.; Goronzy, J.J.; Weyand, C.M. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation 2010, 121, 906–915. [Google Scholar] [CrossRef]
- Sultan, H.; Smith, S.V.; Lee, A.G.; Chévez-Barrios, P. Pathologic Markers Determining Prognosis in Patients With Treated or Healing Giant Cell Arteritis. Am. J. Ophthalmol. 2018, 193, 45–53. [Google Scholar] [CrossRef]
- Schönau, V.; Roth, J.; Tascilar, K.; Corte, G.; Manger, B.; Rech, J.; Schmidt, D.; Cavallaro, A.; Uder, M.; Crescentini, F.; et al. Resolution of vascular inflammation in patients with new-onset giant cell arteritis: Data from the RIGA study. Rheumatology 2021, 60, 3851–3861. [Google Scholar] [CrossRef]
- Slart, R.H.J.A.; Glaudemans, A.W.J.M.; Brouwer, E.; van der Geest, K.S.M. Therapy response evaluation in large-vessel vasculitis: A new role for [18F]FDG-PET/CT? Rheumatology 2021, 60, 3494–3495. [Google Scholar] [CrossRef]
- Unizony, S.; Arias-Urdaneta, L.; Miloslavsky, E.; Arvikar, S.; Khosroshahi, A.; Keroack, B.; Stone, J.R.; Stone, J.H. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res. 2012, 64, 1720–1729. [Google Scholar] [CrossRef]
- Monin, L.; Gaffen, S.L. Interleukin 17 family cytokines: Signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb. Perspect. Biol. 2018, 10, a028522. [Google Scholar] [CrossRef]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.C.; Muralidharan, S.; Corbera-Bellalta, M.; Espigol-Frigole, G.; Marco Hernandez, J.; Denuc, A.; Rios-Garces, R.; Terrades-Garcia, N.; Paolini, J.F.; D’andrea, A. FRI0010 GM-CSFR pathway is implicated in pathogenic inflammatory mechanisms in giant cell arteritis. Ann. Rheum. Dis. 2020, 79, 576. [Google Scholar] [CrossRef]
- Cid, M.C.; Unizony, S.; Pupim, L.; Fang, F.; Pirrello, J.; Ren, A.; Samant, M.; Zhou, T.; Paolini, J.F. OP0059 mavrilimumab (anti GM-CSF receptor α monoclonal antibody) reduces risk of flare and increases sustained remission in a phase 2 trial of patients with giant cell arteritis. Ann. Rheum. Dis. 2021, 80, 31–32. [Google Scholar] [CrossRef]
- Harrington, R.; Al Nokhatha, S.A.; Conway, R. Biologic Therapies for Giant Cell Arteritis. Biol. Targets Ther. 2021, 15, 17–29. [Google Scholar] [CrossRef]
- Yoshida, K.; Sakamoto, N.; Kurosaka, D. Improvement in Polymyalgia Rheumatica Associated With Improved Control of Diabetes Mellitus: A Case Series. Ann. Intern. Med. 2021, 174, 274–276. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 13280. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).