Chromatin Remodeler CHD8 in Autism and Brain Development

Abstract
1. Introduction
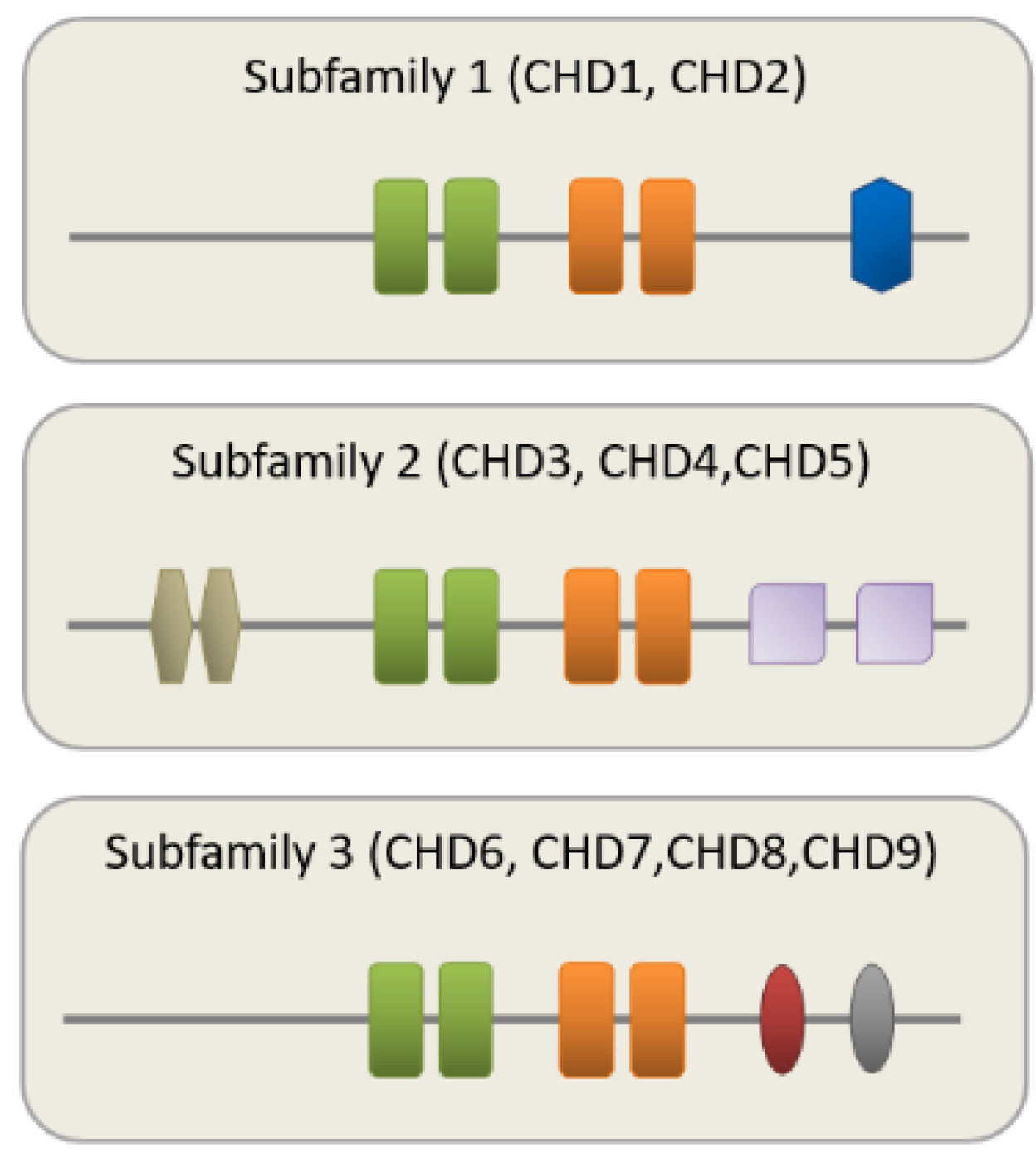
2. CHD8 Is a Member of the CHD Family of CRs
3. Mutations in CHD8 Associate with Autism and Macrocephaly
4. Chd8 Function in Neurodevelopment and Neuronal Function
4.1. A Role of Chd8 in Embryonic Development

{kind=link}
{kind=link}
{kind=link}
| Ref. | Model | System | Timepoint(s) | Tissue Type | Major Findings for Altered Chd8 Expression |
|---|---|---|---|---|---|
| [32] | Xenopus | Chd8 mRNA injection | Stage 40–41 | Whole embryo | Impaired β-catenin dependent axis duplication and head formation. |
| [20] | Zebrafish | Chd8 splice-block- ing anti-sense RNA | Day 4.3 post- fertilization | Embryo head | Enhanced proliferation of midbrain and forebrain progenitor cells. Increased interorbital distance. |
| [33,34] | Mice (C57BL/6J) | LoxP-neo cassette replacing all exons | E: 5.5, 6.5, 7.5 8.5, 9.5 | Embryonic growth retardation and lethality due to massive cell death. | |
| [35] | Mice (Swiss Webster) | E13 shRNA electroporation | E: 15 | GFP+ cortical cells | Impaired NPC proliferation and differentiation associates with reduced dendritic arborization and deficits in social behavior. |
| [36] | Mice (C57BL/6J) | Cre-LoxP Recombination Exon 11-13 | E: 10, 12, 14, 16, 18; P: 91 | Whole brain | Increased brain weight and ASD-like behavior. ASD risk genes and genes involved in synapse and ion channel function are downregulated. Up-regulation of the master regulator REST might underlie neuro- developmental delay. |
| [37] | Mice (C57BL/6J) | CRISPR/cas9 Exon 1 (7bp) | P: 70–84 | Ctx, Stria, NAc, VTA, Hipp, Amyg, Hyp | Macrocephaly. Expression changes in genes related to ASD, neuro- development, cell adhesion, histone and chromatin modification. Reduced local inhibitory signaling in the NAc leads to enhanced excitatory input on MSNs. |
| [38] | Mice (C57BL/6J) | CRISPR/cas9 Exon 5 (5bp) Both sexes | E: 12.5, 14.5, 17.5; P: 0, 58 | Bulk forebrain | Increased neocortical brain volume due to increased NPC proliferation. Cognitive deficits associate with expression changes in genes related to RNA processing chromatin remodeling, and cell cycle regulation. |
| [39] | Mice (C57BL/6J) | Chd8+/−, Exon 3 (Chd8flox/+ X β-actinCre+/−) | E: 12.5; P: 5, 26–27; W: 9–12, 15–18 | Ctx, Hipp, Cer | Increased cortical, hippocampal, and cerebellar areas. Hypertelorism. Cell adhesion and axon guidance genes are downregulated postnatally. Increased long-range connectivity in entorhinal, retrosplenial, auditory cortical, and posterior hippocampal areas. Hyperactive pups show delayed motor development, while hypoactive adults show heightened interest in social cues. |
| [40] | Mice | Knock-in | E: 15.5, 18.5; | Ctx, Hipp | Male-preponderant abnormalities in social communication in pups, anxiety-like behavior in juveniles, and isolation-induced grooming in adults. Opposite changes in excitatory and inhibitory neuronal firing in male and female hippocampi associate with gene expression changes enriched in matrix- and synapse-related gene sets. ASD- and glia gene sets are enriched in male hippocampi only. |
| (C57BL/6J) | Chd8+/N2373K | P: 0, 7, 14 | |||
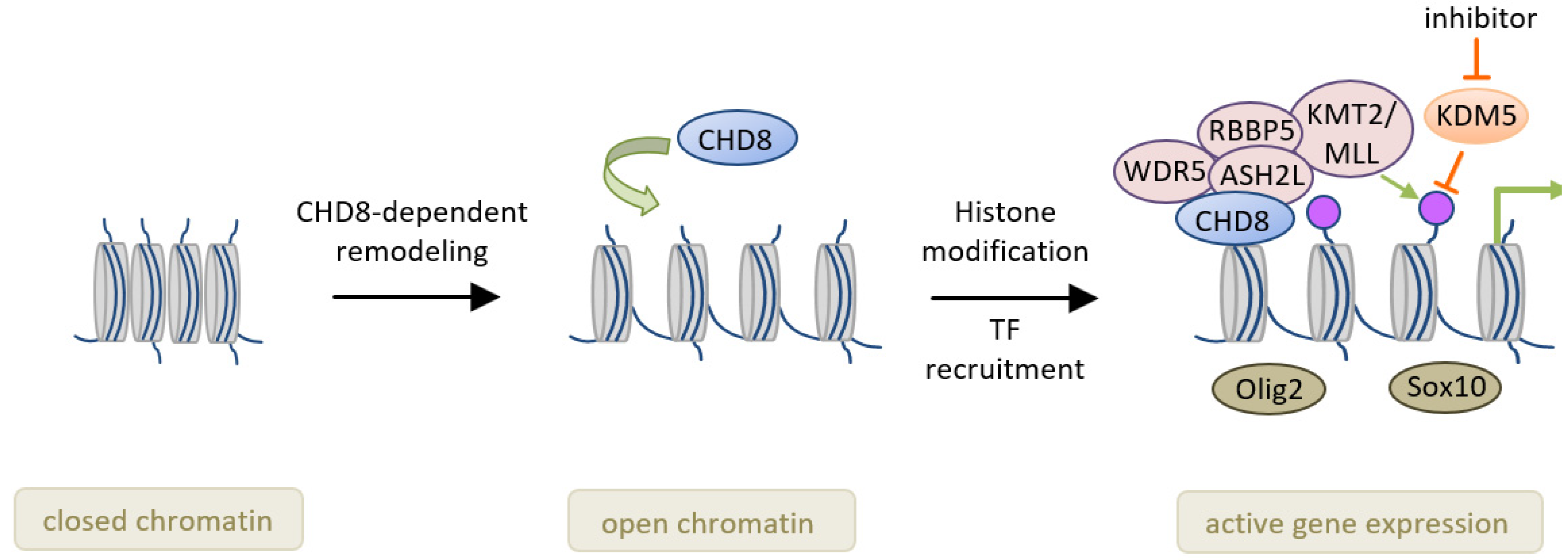
| [41] | Mice (mixed C57BL6;129Sc) | Chd8+/− and Chd8−/− Exon 4 (Chd8flox/+ X Olig1-Cre+/−) | E: 14.5 P: 1, 14 | CC, ON, VWM, SC | Chd8 chromatin binding prevail in early OLs and promotes expression of lineage specific genes through recruitment of the methytransferase KMT2. Null-mice die with severe myelination defects, whereas hetero- zygote mice show circumscribed myelination deficits. |
| [42] | Drosophila | Hypomorph kisk13460 and heteroallelic kisLM27/kisk13416 | Third instar larvae | NMJ, CNS | Under sustained stimulation. Kis promotes presynaptic endocytosis at GSEAthe NMJ. Kis-dependent transcription supports restoration of the recycling vesicle pool. |
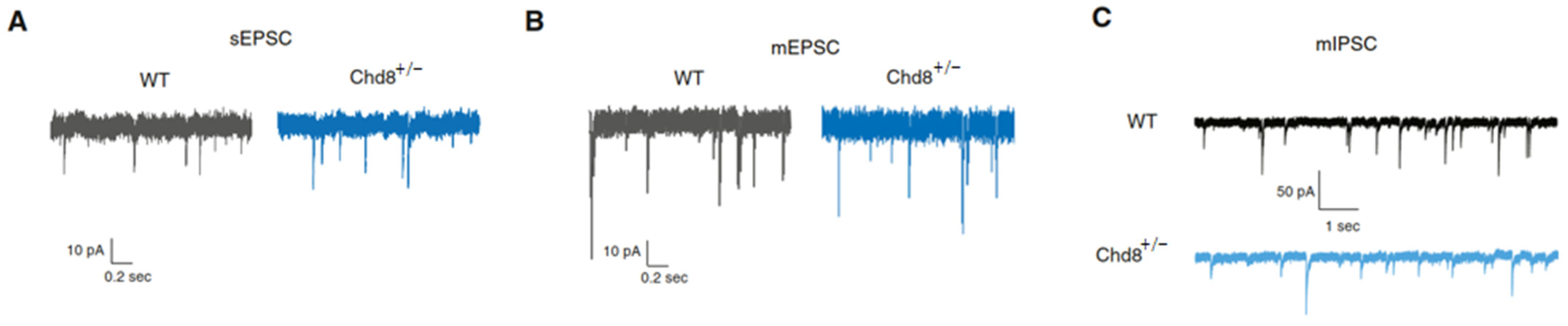
| [43] | Mice (C57BL/6J) | Chd8flox/+ X β-actin- Cre+/− or Nkx.1-Cre+/− or NEX-Cre+/− | P: 5, 13–15, 19- 22, 55–60 | Ctx (ex- vivo slices) | Impaired synaptic development of prefrontal pyramidal neurons in a stage-specific and cell-autonomous manner associates with contrasting changes in excitatory and inhibitory synaptic transmission. Homeostatic plasticity is perturbed in heterozygote neurons. |
| [44] | C. elegans (Bristol N2) | CHD8•chd-7(gk306) | 96 h posthatch | NA | Reduced synaptic vesicle recycling associates with reduced habituation of response probability indicating reduced plasticity of neuronal circuits. |
4.2. A Role of Chd8 for Neural Progenitors, Transcription, Neurotransmission, and RNA Splicing
4.3. A Complex Role of Sex for Chd8 Mutations
4.4. A Role of Chd8 in Oligodendrocytes
4.5. A Role of Chd8 in Synaptic Vesicle Recycling
4.6. A Role of Chd8 in Homeostatic Plasticity and Habituation
5. Discussion and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASD | autism spectrum disorder |
| ATP | adenosine triphosphate |
| CHD | Chromodomain Helicase DNA binding |
| ChIP-seq | chromatin immunoprecipitation sequencing |
| CR | chromatin remodeler |
| DEG | differentially expressed genes |
| ECM | extracellular matrix |
| ESC | embryonic stem cell |
| GEP | gene expression profiling |
| GSEA | gene set enrichment analysis |
| GWAS | genome-wide association studies |
| ID | intellectual disability |
| iPSC | induced pluripotent stem cell |
| Kis | Kismet |
| NMJ | neuromuscular junction |
| MRI | magnetic resonance imaging |
| NAc | nucleus accumbens |
| NPC | neural progenitor cell |
| OPC | oligodendrocyte precursor cell |
| OL | oligodendrocyte |
| p53 | tumor suppressor gene p53 |
| PcG | Polycomb group complex |
| TF | transcription factor |
| WES | whole exome sequencing |
| Wnt | wingless |
References
- Tyagi, M.; Imam, N.; Verma, K.; Patel, A.K. Chromatin remodelers: We are the drivers! Nucleus 2016, 7, 388–404. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-Y.; Meaney, M.J. Epigenetics and the environmental regulation of the genome and its function. Annu. Rev. Psychol. 2010, 61, 439–466. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Wu, Y.; Bockmühl, Y.; Spengler, D. Genes learn from stress: How infantile trauma programs us for depression. Epigenetics 2010, 5, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Spengler, D. Genetic variation in the epigenetic machinery and mental health. Curr. Psychiatry Rep. 2012, 14, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; O’Roak, B.J.; Shendure, J.; Eichler, E.E. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014, 37, 95–105. [Google Scholar] [CrossRef]
- Barnard, R.A.; Pomaville, M.B.; O’Roak, B.J. Mutations and Modeling of the Chromatin Remodeler CHD8 Define an Emerging Autism Etiology. Front. Neurosci. 2015, 9, 477. [Google Scholar] [CrossRef]
- Wade, A.A.; Lim, K.; Catta-Preta, R.; Nord, A.S. Common CHD8 Genomic Targets Contrast With Model-Specific Transcriptional Impacts of CHD8 Haploinsufficiency. Front. Mol. Neurosci. 2018, 11, 481. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef]
- Hu, Y.; Lai, Y.; Zhu, D. Transcription regulation by CHD proteins to control plant development. Front. Plant Sci. 2014, 5, 223. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Spengler, D. Chromatin Remodeling Complex NuRD in Neurodevelopment and Neurodevelopmental Disorders. Front. Genet. 2019, 10, 682. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; State, M.W. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 2015, 14, 1109–1120. [Google Scholar] [CrossRef]
- Diagnostic and statistical manual of mental disorders: DSM-5. In Development, 5th ed.; American Psychiatric Association, Ed.; American Psychiatric Publishing: Washington, DC, USA, 2013; ISBN 978-0-89042-555-8. [Google Scholar]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.-F.; Stevens, C.; Wang, L.-S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Stessman, H.A.; Boyle, E.A.; Witherspoon, K.T.; Martin, B.; Lee, C.; Vives, L.; Baker, C.; Hiatt, J.B.; Nickerson, D.A.; et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun. 2014, 5, 5595. [Google Scholar] [CrossRef]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, H.; Xiong, B.; Stessman, H.A.F.; Wu, H.; Coe, B.P.; Turner, T.N.; Liu, Y.; Zhao, W.; Hoekzema, K.; et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 2016, 7, 13316. [Google Scholar] [CrossRef]
- Stessman, H.A.F.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012, 149, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Yasin, H.; Gibson, W.T.; Langlois, S.; Stowe, R.M.; Tsang, E.S.; Lee, L.; Poon, J.; Tran, G.; Tyson, C.; Wong, C.K.; et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J. Hum. Genet. 2019, 64, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, P.J.; Zachariou, A.; Loveday, C.; Beleza-Meireles, A.; Bertoli, M.; Dean, J.; Douglas, A.G.L.; Ellis, I.; Foster, A.; Graham, J.M.; et al. The CHD8 overgrowth syndrome: A detailed evaluation of an emerging overgrowth phenotype in 27 patients. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 557–564. [Google Scholar] [CrossRef]
- An, Y.; Zhang, L.; Liu, W.; Jiang, Y.; Chen, X.; Lan, X.; Li, G.; Hang, Q.; Wang, J.; Gusella, J.F.; et al. De novo variants in the Helicase-C domain of CHD8 are associated with severe phenotypes including autism, language disability and overgrowth. Hum. Genet. 2020, 139, 499–512. [Google Scholar] [CrossRef]
- Wu, H.; Li, H.; Bai, T.; Han, L.; Ou, J.; Xun, G.; Zhang, Y.; Wang, Y.; Duan, G.; Zhao, N.; et al. Phenotype-to-genotype approach reveals head-circumference-associated genes in an autism spectrum disorder cohort. Clin. Genet. 2020, 97, 338–346. [Google Scholar] [CrossRef]
- Beighley, J.S.; Hudac, C.M.; Arnett, A.B.; Peterson, J.L.; Gerdts, J.; Wallace, A.S.; Mefford, H.C.; Hoekzema, K.; Turner, T.N.; O’Roak, B.J.; et al. Clinical Phenotypes of Carriers of Mutations in CHD8 or Its Conserved Target Genes. Biol. Psychiatry 2020, 87, 123–131. [Google Scholar] [CrossRef]
- Chenn, A. Wnt/beta-catenin signaling in cerebral cortical development. Organogenesis 2008, 4, 76–80. [Google Scholar] [CrossRef]
- Sakamoto, I.; Kishida, S.; Fukui, A.; Kishida, M.; Yamamoto, H.; Hino, S.; Michiue, T.; Takada, S.; Asashima, M.; Kikuchi, A. A novel beta-catenin-binding protein inhibits beta-catenin-dependent Tcf activation and axis formation. J. Biol. Chem. 2000, 275, 32871–32878. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, M.; Nakayama, K.; Tsunematsu, R.; Tsukiyama, T.; Kikuchi, A.; Nakayama, K.I. Early embryonic death in mice lacking the beta-catenin-binding protein Duplin. Mol. Cell. Biol. 2004, 24, 8386–8394. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, M.; Oshikawa, K.; Tsukada, Y.; Nakagawa, T.; Iemura, S.; Natsume, T.; Fan, Y.; Kikuchi, A.; Skoultchi, A.I.; Nakayama, K.I. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol. 2009, 11, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Durak, O.; Gao, F.; Kaeser-Woo, Y.J.; Rueda, R.; Martorell, A.J.; Nott, A.; Liu, C.Y.; Watson, L.A.; Tsai, L.-H. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 2016, 19, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Nishiyama, M.; Shoji, H.; Ohkawa, Y.; Kawamura, A.; Sato, T.; Suyama, M.; Takumi, T.; Miyakawa, T.; Nakayama, K.I. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 2016, 537, 675–679. [Google Scholar] [CrossRef]
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.-A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep. 2017, 19, 335–350. [Google Scholar] [CrossRef]
- Gompers, A.L.; Su-Feher, L.; Ellegood, J.; Copping, N.A.; Riyadh, M.A.; Stradleigh, T.W.; Pride, M.C.; Schaffler, M.D.; Wade, A.A.; Catta-Preta, R.; et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci. 2017, 20, 1062–1073. [Google Scholar] [CrossRef]
- Suetterlin, P.; Hurley, S.; Mohan, C.; Riegman, K.L.H.; Pagani, M.; Caruso, A.; Ellegood, J.; Galbusera, A.; Crespo-Enriquez, I.; Michetti, C.; et al. Altered Neocortical Gene Expression, Brain Overgrowth and Functional Over-Connectivity in Chd8 Haploinsufficient Mice. Cereb. Cortex 2018, 28, 2192–2206. [Google Scholar] [CrossRef]
- Jung, H.; Park, H.; Choi, Y.; Kang, H.; Lee, E.; Kweon, H.; Roh, J.D.; Ellegood, J.; Choi, W.; Kang, J.; et al. Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat. Neurosci. 2018, 21, 1218–1228. [Google Scholar] [CrossRef]
- Zhao, C.; Dong, C.; Frah, M.; Deng, Y.; Marie, C.; Zhang, F.; Xu, L.; Ma, Z.; Dong, X.; Lin, Y.; et al. Dual Requirement of CHD8 for Chromatin Landscape Establishment and Histone Methyltransferase Recruitment to Promote CNS Myelination and Repair. Dev. Cell 2018, 45, 753–768.e8. [Google Scholar] [CrossRef]
- Latcheva, N.K.; Delaney, T.L.; Viveiros, J.M.; Smith, R.A.; Bernard, K.M.; Harsin, B.; Marenda, D.R.; Liebl, F.L.W. The CHD Protein, Kismet, is Important for the Recycling of Synaptic Vesicles during Endocytosis. Sci. Rep. 2019, 9, 19368. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, R.A.; de Meritens, E.R.; Shaunak, R.; Naybour, L.; Basson, M.A.; Andreae, L.C. Cell-type-specific synaptic imbalance and disrupted homeostatic plasticity in cortical circuits of ASD-associated Chd8 haploinsufficient mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- McDiarmid, T.A.; Belmadani, M.; Liang, J.; Meili, F.; Mathews, E.A.; Mullen, G.P.; Hendi, A.; Wong, W.-R.; Rand, J.B.; Mizumoto, K.; et al. Systematic phenomics analysis of autism-associated genes reveals parallel networks underlying reversible impairments in habituation. Proc. Natl. Acad. Sci. USA 2020, 117, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Paredes, M.; Ceballos-Chávez, M.; Esteller, M.; García-Domínguez, M.; Reyes, J.C. The chromatin remodeling factor CHD8 interacts with elongating RNA polymerase II and controls expression of the cyclin E2 gene. Nucleic Acids Res. 2009, 37, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Menon, T.; Yates, J.A.; Bochar, D.A. Regulation of androgen-responsive transcription by the chromatin remodeling factor CHD8. Mol. Endocrinol. 2010, 24, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Subtil-Rodríguez, A.; Vázquez-Chávez, E.; Ceballos-Chávez, M.; Rodríguez-Paredes, M.; Martín-Subero, J.I.; Esteller, M.; Reyes, J.C. The chromatin remodeller CHD8 is required for E2F-dependent transcription activation of S-phase genes. Nucleic Acids Res. 2014, 42, 2185–2196. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Chávez, M.; Subtil-Rodríguez, A.; Giannopoulou, E.G.; Soronellas, D.; Vázquez-Chávez, E.; Vicent, G.P.; Elemento, O.; Beato, M.; Reyes, J.C. The chromatin Remodeler CHD8 is required for activation of progesterone receptor-dependent enhancers. PLoS Genet. 2015, 11, e1005174. [Google Scholar] [CrossRef]
- Hoffmann, A.; Zimmermann, C.A.; Spengler, D. Molecular epigenetic switches in neurodevelopment in health and disease. Front. Behav. Neurosci. 2015, 9, 120. [Google Scholar] [CrossRef]
- Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration. Epigenomes 2017, 1, 21. [Google Scholar] [CrossRef]
- Elsabbagh, M.; Divan, G.; Koh, Y.-J.; Kim, Y.S.; Kauchali, S.; Marcín, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef]
- Menger, Y.; Bettscheider, M.; Murgatroyd, C.; Spengler, D. Sex differences in brain epigenetics. Epigenomics 2010, 2, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Kandel, E.R.; Schwartz, J.H.; Jessel, T.M.; Siegelbaum, S.A.; Hudspeth, A.J.; Mack, S. (Eds.) Principles of Neural Science, 5th ed.; McGraw-Hill Medical: New York, NY, USA; Lisbon, Portugal; London, UK, 2013; ISBN 978-0-07-139011-8. [Google Scholar]
- Belmonte, M.K.; Allen, G.; Beckel-Mitchener, A.; Boulanger, L.M.; Carper, R.A.; Webb, S.J. Autism and abnormal development of brain connectivity. J. Neurosci. 2004, 24, 9228–9231. [Google Scholar] [CrossRef] [PubMed]
- Picci, G.; Gotts, S.J.; Scherf, K.S. A theoretical rut: Revisiting and critically evaluating the generalized under/over-connectivity hypothesis of autism. Dev. Sci. 2016, 19, 524–549. [Google Scholar] [CrossRef] [PubMed]
- Bedford, R.; Pickles, A.; Lord, C. Early gross motor skills predict the subsequent development of language in children with autism spectrum disorder. Autism Res. 2016, 9, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.; Forgeot d’Arc, B.; Bell, S.C.; Maussion, G.; Peng, H.; Gauthier, J.; Crapper, L.; Hamdan, F.F.; Michaud, J.L.; Mottron, L.; et al. A de novo frameshift mutation in chromodomain helicase DNA-binding domain 8 (CHD8): A case report and literature review. Am. J. Med. Genet. A 2016, 170A, 1225–1235. [Google Scholar] [CrossRef]
- Werling, D.M.; Parikshak, N.N.; Geschwind, D.H. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat. Commun. 2016, 7, 10717. [Google Scholar] [CrossRef] [PubMed]
- Barak, B.; Feng, G. Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat. Neurosci. 2016, 19, 647–655. [Google Scholar] [CrossRef]
- Kim, Y.; Yang, G.R.; Pradhan, K.; Venkataraju, K.U.; Bota, M.; García Del Molino, L.C.; Fitzgerald, G.; Ram, K.; He, M.; Levine, J.M.; et al. Brain-wide Maps Reveal Stereotyped Cell-Type-Based Cortical Architecture and Subcortical Sexual Dimorphism. Cell 2017, 171, 456–469.e22. [Google Scholar] [CrossRef]
- Hardan, A.Y.; Fung, L.K.; Frazier, T.; Berquist, S.W.; Minshew, N.J.; Keshavan, M.S.; Stanley, J.A. A proton spectroscopy study of white matter in children with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 66, 48–53. [Google Scholar] [CrossRef]
- Deoni, S.; Dean, D.; Joelson, S.; O’Regan, J.; Schneider, N. Early nutrition influences developmental myelination and cognition in infants and young children. Neuroimage 2018, 178, 649–659. [Google Scholar] [CrossRef]
- Douzgou, S.; Liang, H.W.; Metcalfe, K.; Somarathi, S.; Tischkowitz, M.; Mohamed, W.; Kini, U.; McKee, S.; Yates, L.; Bertoli, M.; et al. The clinical presentation caused by truncating CHD8 variants. Clin. Genet. 2019, 96, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, B.; Popko, B. Molecular Control of Oligodendrocyte Development. Trends Neurosci. 2019, 42, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Marie, C.; Clavairoly, A.; Frah, M.; Hmidan, H.; Yan, J.; Zhao, C.; Van Steenwinckel, J.; Daveau, R.; Zalc, B.; Hassan, B.; et al. Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc. Natl. Acad. Sci. USA 2018, 115, E8246–E8255. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Marie, C.; Zhao, C.; Kim, B.; Wang, J.; Deng, Y.; Clavairoly, A.; Frah, M.; Wang, H.; He, X.; et al. Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat. Neurosci. 2016, 19, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Batsukh, T.; Pieper, L.; Koszucka, A.M.; von Velsen, N.; Hoyer-Fender, S.; Elbracht, M.; Bergman, J.E.H.; Hoefsloot, L.H.; Pauli, S. CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum. Mol. Genet. 2010, 19, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef]
- Turrigiano, G.G. Homeostatic plasticity in neuronal networks: The more things change, the more they stay the same. Trends Neurosci. 1999, 22, 221–227. [Google Scholar] [CrossRef]
- Antoine, M.W.; Langberg, T.; Schnepel, P.; Feldman, D.E. Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron 2019, 101, 648–661.e4. [Google Scholar] [CrossRef]
- Tatavarty, V.; Torrado Pacheco, A.; Groves Kuhnle, C.; Lin, H.; Koundinya, P.; Miska, N.J.; Hengen, K.B.; Wagner, F.F.; Van Hooser, S.D.; Turrigiano, G.G. Autism-Associated Shank3 Is Essential for Homeostatic Compensation in Rodent V1. Neuron 2020, 106, 769–777.e4. [Google Scholar] [CrossRef]
- Carney, D.S.; Davies, B.A.; Horazdovsky, B.F. Vps9 domain-containing proteins: Activators of Rab5 GTPases from yeast to neurons. Trends Cell Biol. 2006, 16, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef] [PubMed]
- McDiarmid, T.A.; Bernardos, A.C.; Rankin, C.H. Habituation is altered in neuropsychiatric disorders-A comprehensive review with recommendations for experimental design and analysis. Neurosci. Biobehav. Rev. 2017, 80, 286–305. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.E.; Cheroni, C.; López-Tóbon, A.; Dotter, C.P.; Oliveira, B.; Sacco, R.; Yahya, A.C.; Morandell, J.; Gabriele, M.; Sommer, C.; et al. CHD8 haploinsufficiency alters the developmental trajectories of human excitatory and inhibitory neurons linking autism phenotypes with transient cellular defects. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jin, X.; Simmons, S.K.; Guo, A.; Shetty, A.S.; Ko, M.; Nguyen, L.; Jokhi, V.; Robinson, E.; Oyler, P.; Curry, N.; et al. In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 2020, 370. [Google Scholar] [CrossRef]
- Treutlein, B.; Camp, J.G. Sequencing perturbed cortex development. Science 2020, 370, 1038–1039. [Google Scholar] [CrossRef]
- Gandal, M.J.; Leppa, V.; Won, H.; Parikshak, N.N.; Geschwind, D.H. The road to precision psychiatry: Translating genetics into disease mechanisms. Nat. Neurosci. 2016, 19, 1397–1407. [Google Scholar] [CrossRef]
- Tsankova, N.; Renthal, W.; Kumar, A.; Nestler, E.J. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 355–367. [Google Scholar] [CrossRef]
- Ma, D.K.; Marchetto, M.C.; Guo, J.U.; Ming, G.; Gage, F.H.; Song, H. Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat. Neurosci. 2010, 13, 1338–1344. [Google Scholar] [CrossRef]
- Florio, M.; Borrell, V.; Huttner, W.B. Human-specific genomic signatures of neocortical expansion. Curr. Opin. Neurobiol. 2017, 42, 33–44. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ziller, M.; Spengler, D. Progress in iPSC-Based Modeling of Psychiatric Disorders. Int. J. Mol. Sci 2019, 20, 4896. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Ziller, M.; Spengler, D. Focus on Causality in ESC/iPSC-Based Modeling of Psychiatric Disorders. Cells 2020, 9, 366. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, A.; Spengler, D. Chromatin Remodeler CHD8 in Autism and Brain Development. J. Clin. Med. 2021, 10, 366. https://doi.org/10.3390/jcm10020366
Hoffmann A, Spengler D. Chromatin Remodeler CHD8 in Autism and Brain Development. Journal of Clinical Medicine. 2021; 10(2):366. https://doi.org/10.3390/jcm10020366
Chicago/Turabian StyleHoffmann, Anke, and Dietmar Spengler. 2021. "Chromatin Remodeler CHD8 in Autism and Brain Development" Journal of Clinical Medicine 10, no. 2: 366. https://doi.org/10.3390/jcm10020366
APA StyleHoffmann, A., & Spengler, D. (2021). Chromatin Remodeler CHD8 in Autism and Brain Development. Journal of Clinical Medicine, 10(2), 366. https://doi.org/10.3390/jcm10020366