
Molecular Diagnosis of Pompe Disease in the Genomic Era: Correlation with Acid Alpha-Glucosidase Activity in Dried Blood Spots

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Acid Alpha-Glucosidase Enzymatic Activity Assays
2.2. Gene Panel and Next Generation Sequencing Method
2.3. Splicing Analysis
2.4. Bioinformatics
2.5. Statistical Analysis
3. Results
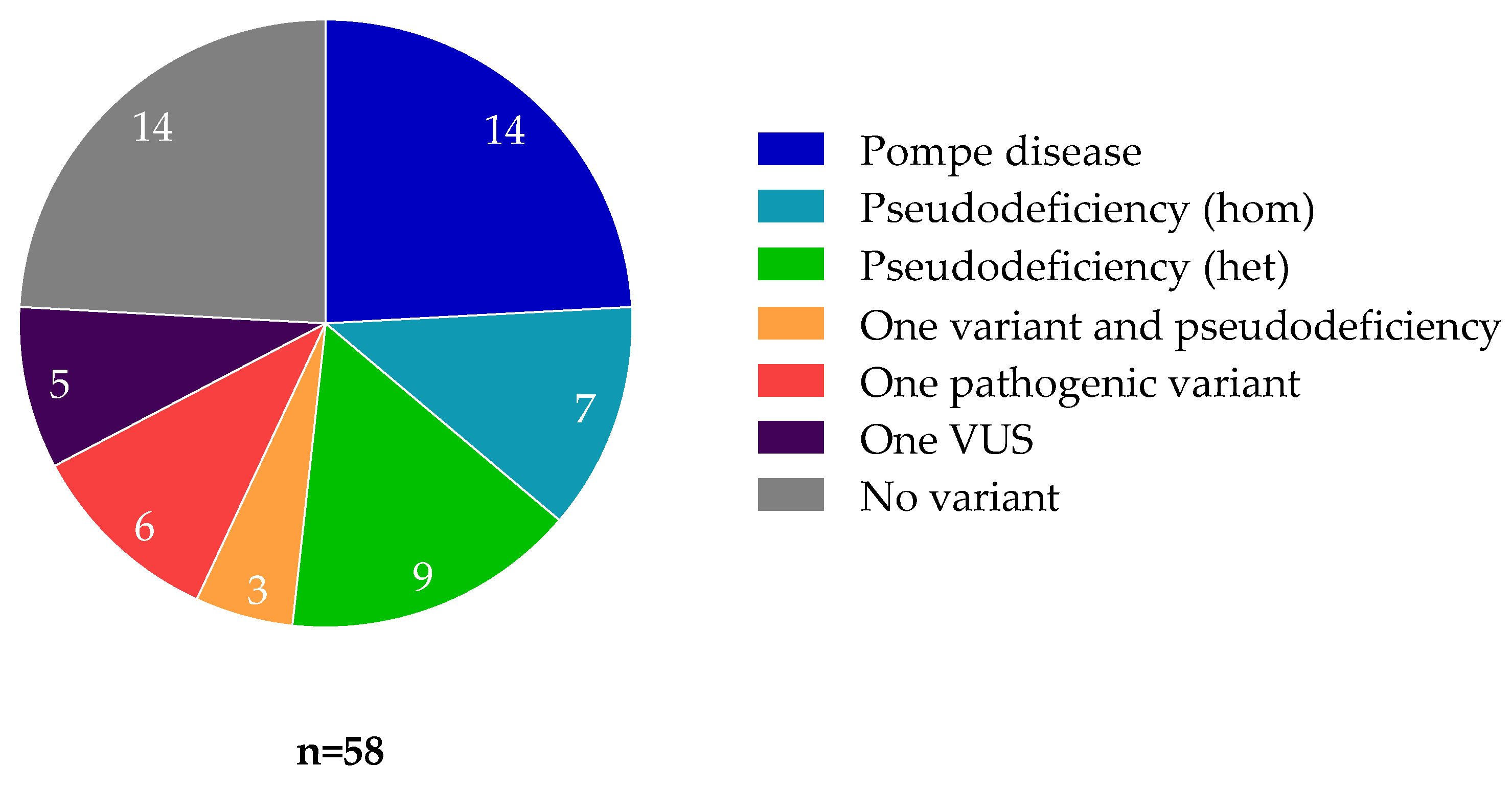
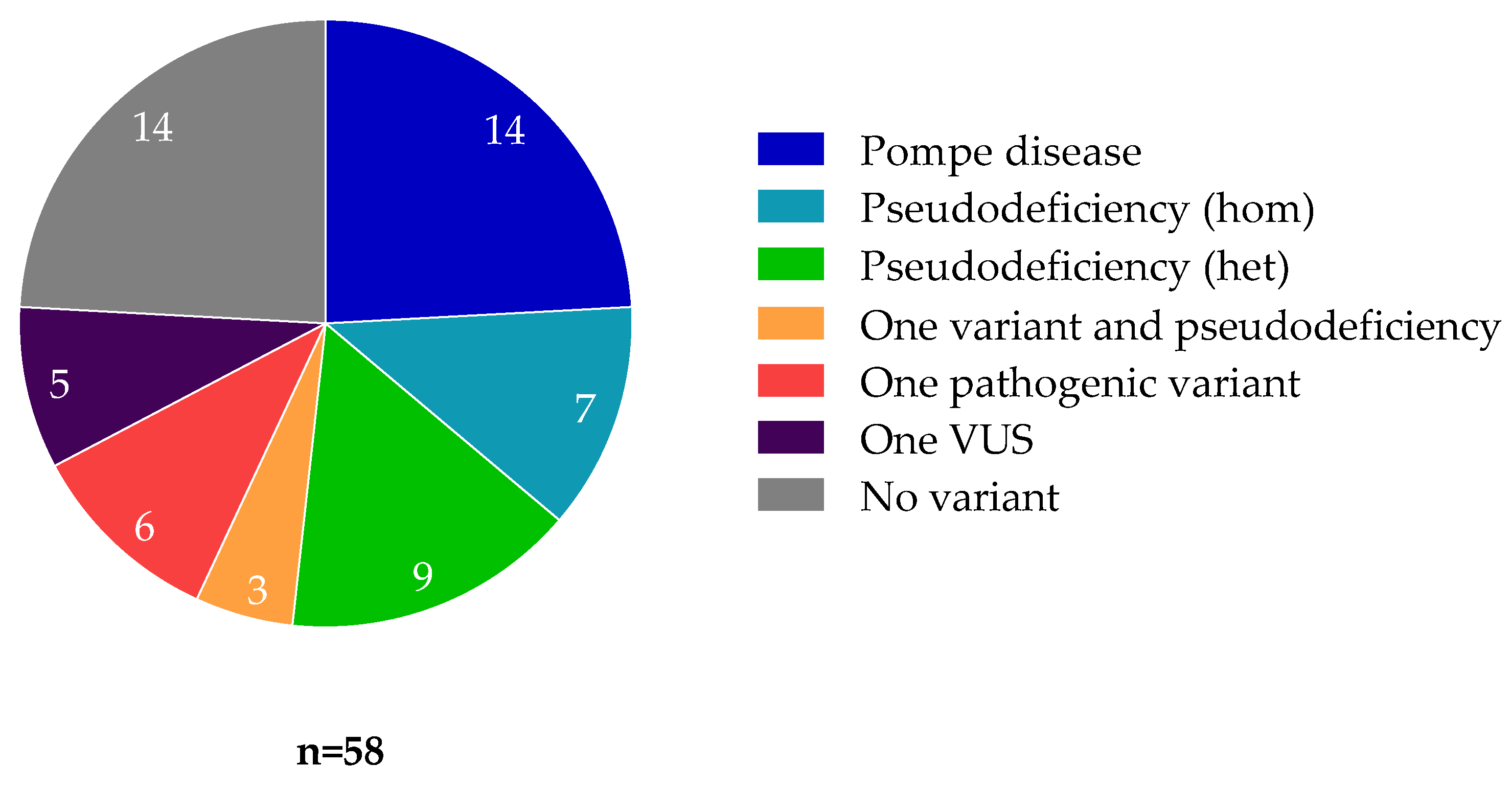
3.1. Patients with Pompe Disease
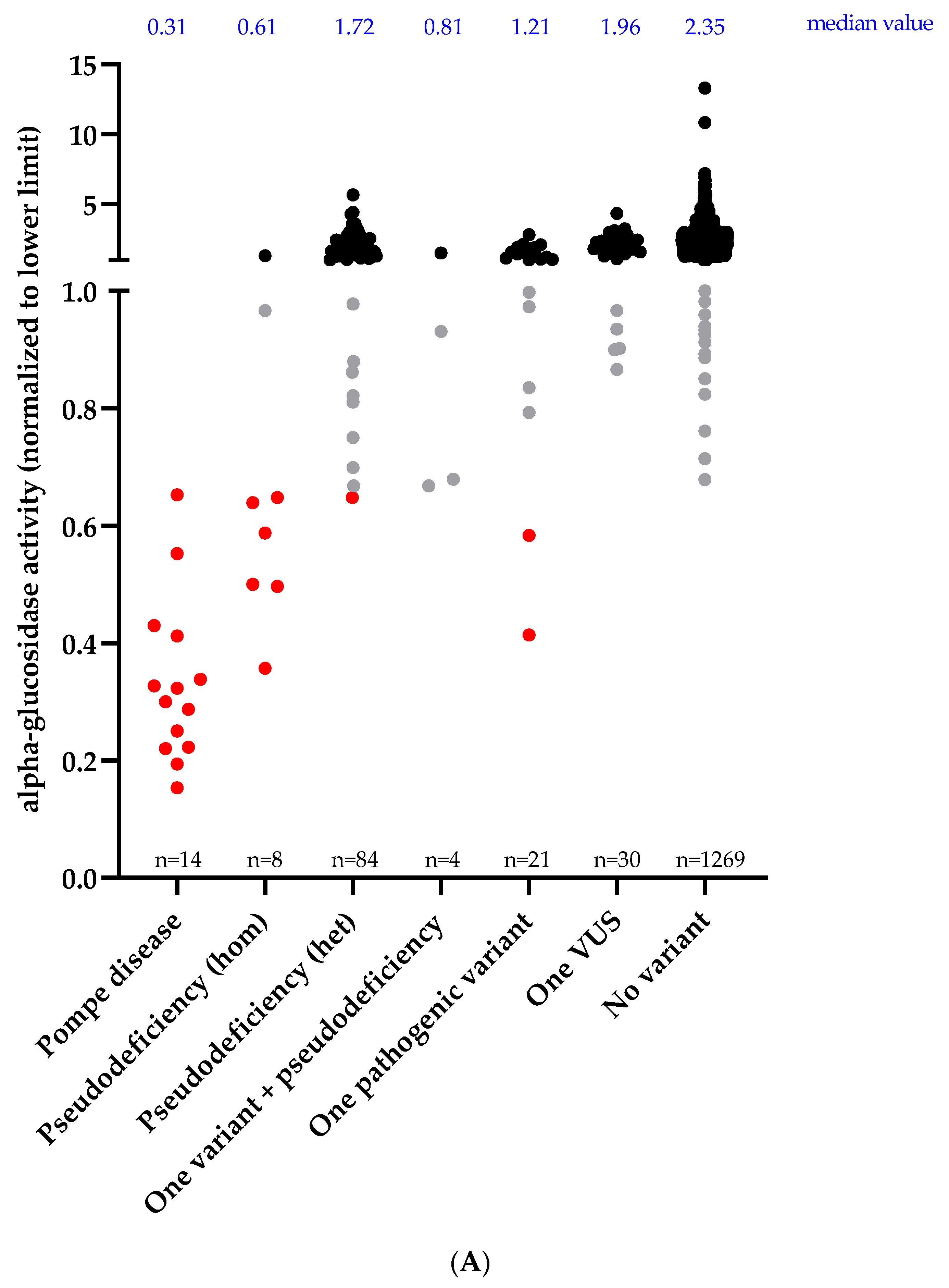
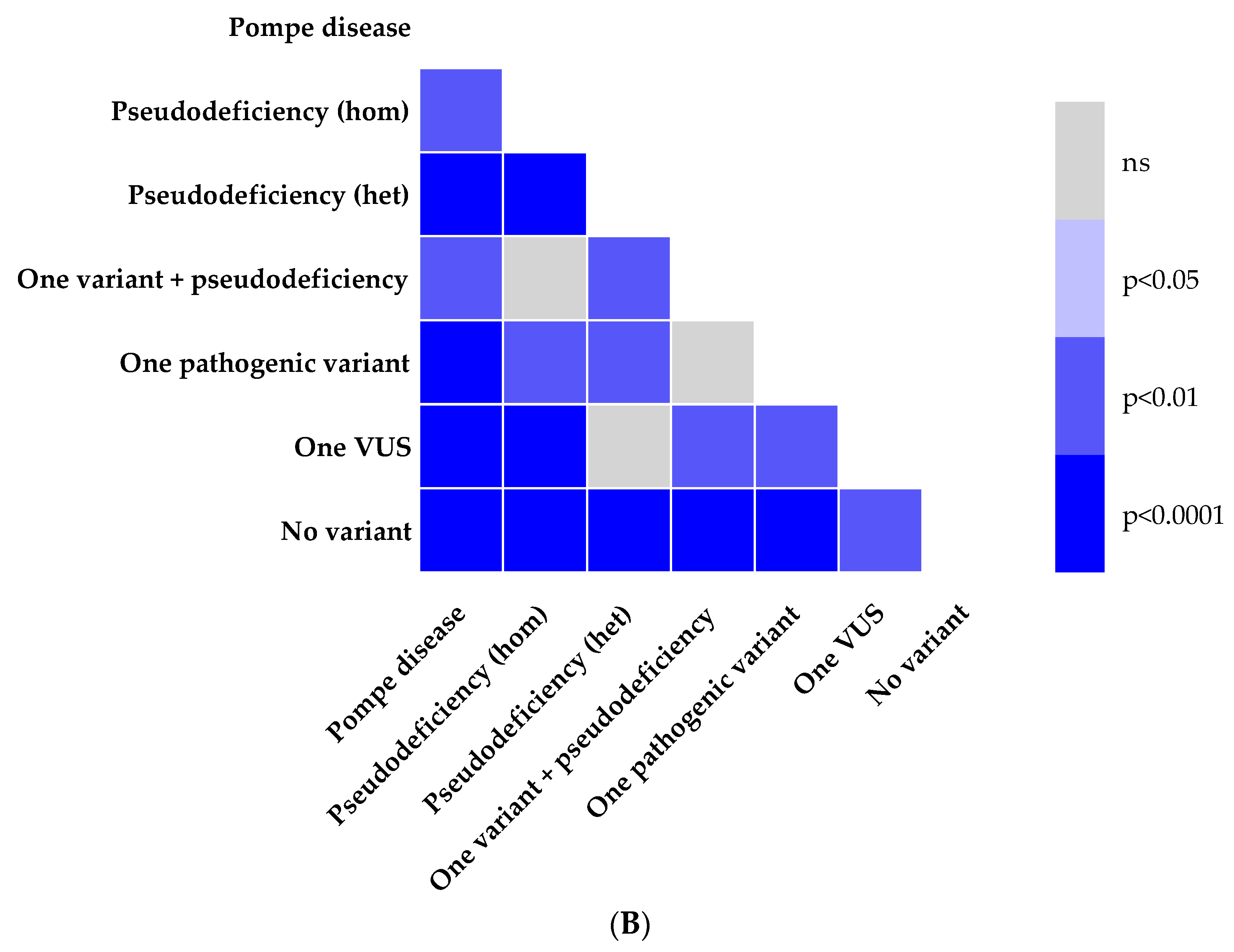
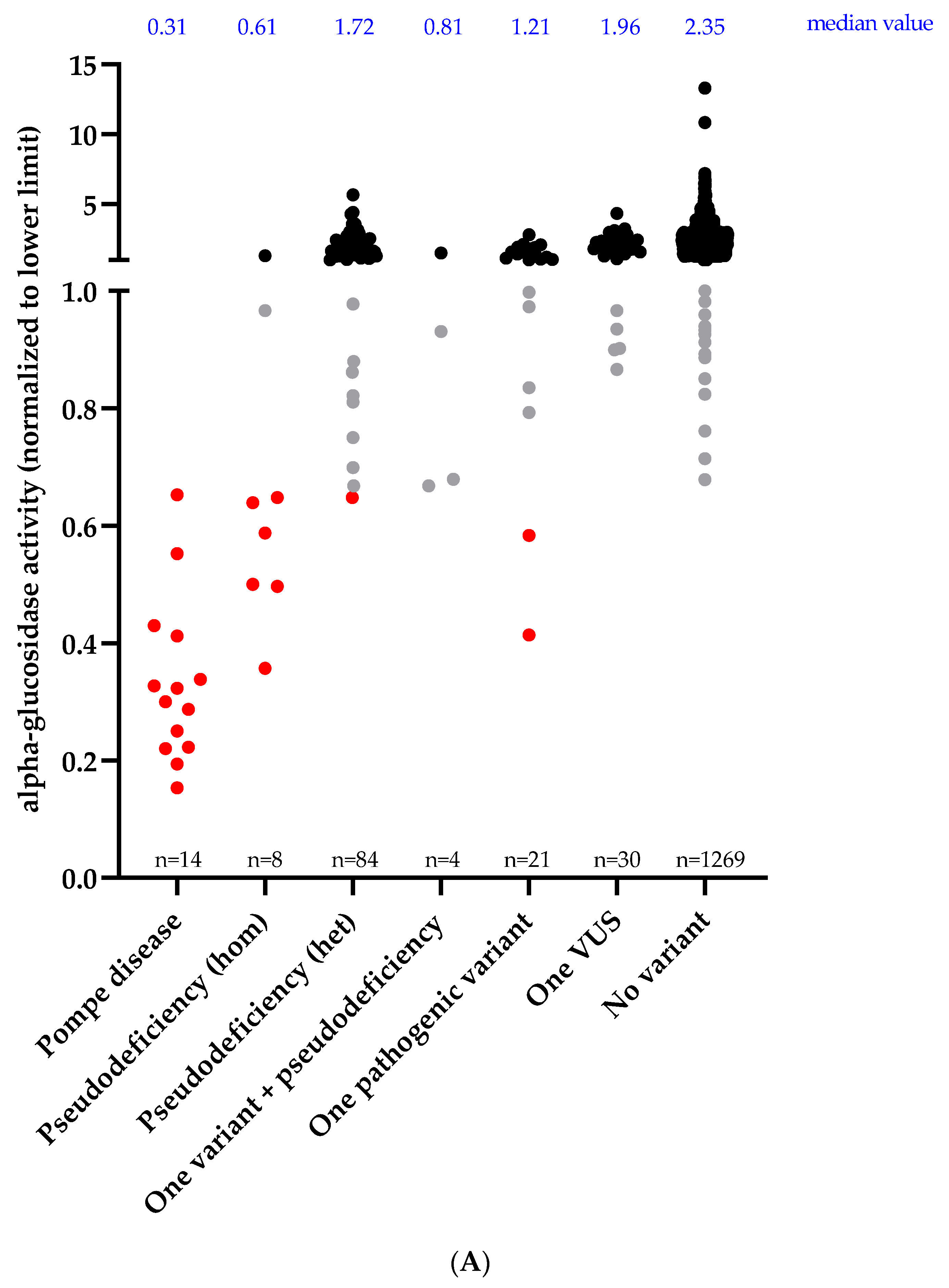
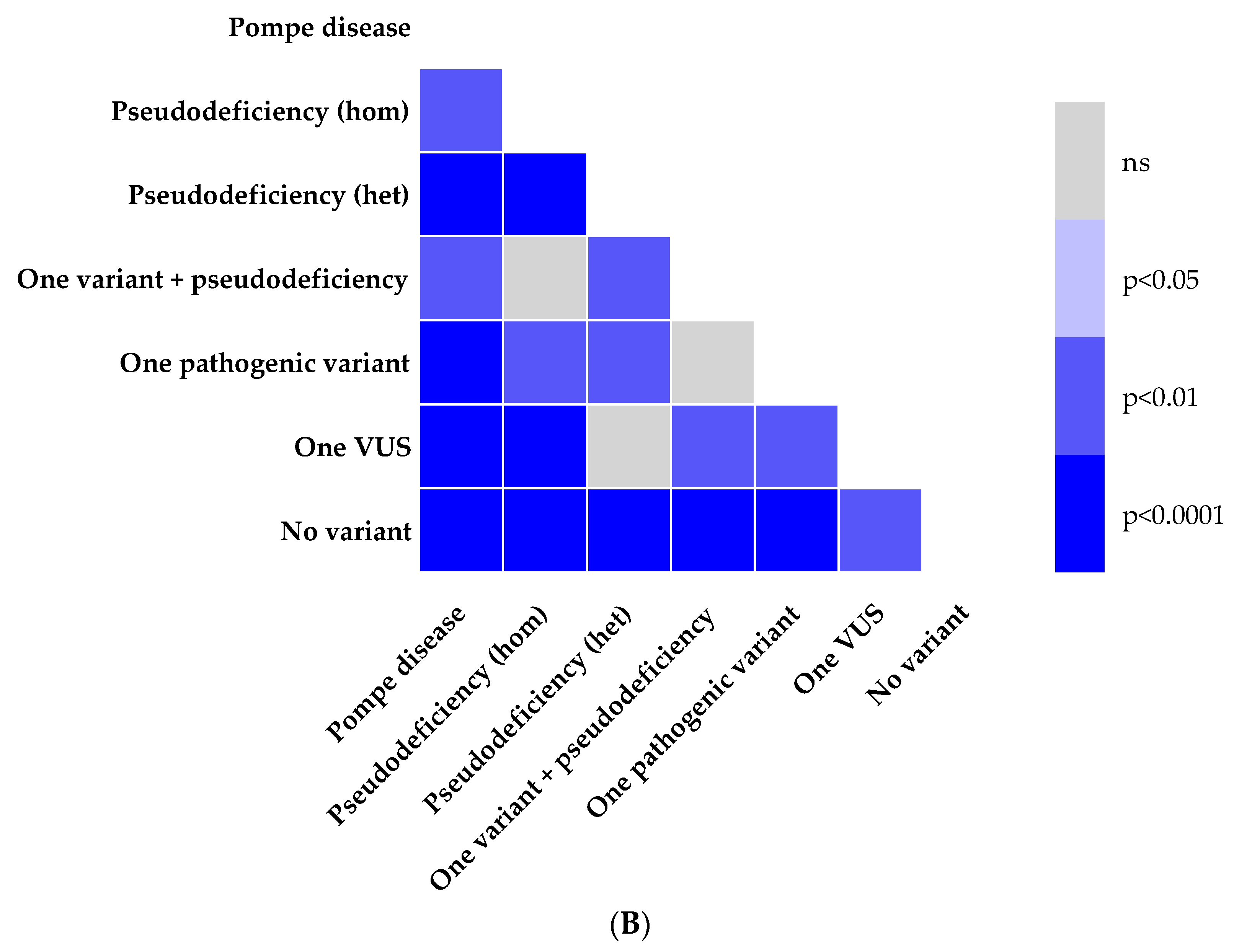
3.2. Enzymatic Activity and Genotype
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dasouki, M.; Jawdat, O.; Almadhoun, O.; Pasnoor, M.; McVey, A.L.; Abuzinadah, A.R.; Herbelin, L.; Barohn, R.J.; Dimachkie, M.M. Pompe Disease. Neurol. Clin. 2014, 32, 751–776. [Google Scholar] [CrossRef] [Green Version]
- Martiniuk, F.; Chen, A.; Mack, A.; Arvanitopoulos, E.; Chen, Y.; Rom, W.; Codd, W.J.; Hanna, B.; Alcabes, P.; Raben, N.; et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am. J. Med. Genet. 1998, 79, 69–72. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Hwu, W.-L.; Lee, N.-C. Pompe Disease: Early Diagnosis and Early Treatment Make a Difference. Pediatr. Neonatol. 2013, 54, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Ploeg, A.T.; Reuser, A.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Manganelli, F.; Ruggiero, L. Clinical features of Pompe disease. Acta Myol. 2013, 32, 82–84. [Google Scholar] [PubMed]
- Dardis, A.; Zanin, I.; Zampieri, S.; Stuani, C.; Pianta, A.; Romanello, M.; Baralle, F.E.; Bembi, B.; Buratti, E. Functional characterization of the common c.-32-13T>G mutation of GAA gene: Identification of potential therapeutic agents. Nucleic Acids Res. 2014, 42, 1291–1302. [Google Scholar] [CrossRef] [Green Version]
- Byrne, B.J.; Kishnani, P.S.; Case, L.; Merlini, L.; Müller-Felber, W.; Prasad, S.; van der Ploeg, A. Pompe disease: Design, methodology, and early findings from the Pompe Registry. Mol. Genet. Metab. 2011, 103, 1–11. [Google Scholar] [CrossRef]
- Regnery, C.; Kornblum, C.; Hanisch, F.; Vielhaber, S.; Strigl-Pill, N.; Grunert, B.; Müller-Felber, W.; Glocker, F.X.; Spranger, M.; Deschauer, M.; et al. 36 months observational clinical study of 38 adult Pompe disease patients under alglucosidase alfa enzyme replacement therapy. J. Inherit. Metab. Dis. 2012, 35, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Van Der Ploeg, A.T.; Clemens, P.R.; Corzo, D.; Escolar, D.M.; Florence, J.; Groeneveld, G.J.; Herson, S.; Kishnani, P.S.; Laforet, P.; Lake, S.L.; et al. A Randomized Study of Alglucosidase Alfa in Late-Onset Pompe’s Disease. N. Engl. J. Med. 2010, 362, 1396–1406. [Google Scholar] [CrossRef] [Green Version]
- Prater, S.N.; Banugaria, S.G.; DeArmey, S.M.; Botha, E.G.; Stege, E.M.; Case, L.; Jones, H.N.; Phornphutkul, C.; Wang, R.; Young, S.P.; et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet. Med. 2012, 14, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Bodamer, O.A.; Scott, C.R.; Giugliani, R. Pompe Disease Newborn Screening Working Group; on behalf of the Pompe Disease Newborn Screening Working Group Newborn Screening for Pompe Disease. Pediatrics 2017, 140, S4–S13. [Google Scholar] [CrossRef] [Green Version]
- Sawada, T.; Kido, J.; Nakamura, K. Newborn Screening for Pompe Disease. Int. J. Neonatal Screen. 2020, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallwass, H.; Carr, C.; Gerrein, J.; Titlow, M.; Pomponio, R.; Bali, D.; Dai, J.; Kishnani, P.; Skrinar, A.; Corzo, D.; et al. Rapid diagnosis of late-onset Pompe disease by fluorometric assay of α-glucosidase activities in dried blood spots. Mol. Genet. Metab. 2007, 90, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, P.; Pavan, E.; Dardis, A. Molecular genetics of Pompe disease: A comprehensive overview. Ann. Transl. Med. 2019, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Niño, M.Y.; Wijgerde, M.; de Faria, D.O.S.; Hoogeveen-Westerveld, M.; Bergsma, A.J.; Broeders, M.; van der Beek, N.A.M.E.; Hout, H.J.M.V.D.; van der Ploeg, A.T.; Verheijen, F.W.; et al. Enzymatic diagnosis of Pompe disease: Lessons from 28 years of experience. Eur. J. Hum. Genet. 2021, 29, 434–446. [Google Scholar] [CrossRef]
- Mori, M.; Haskell, G.; Kazi, Z.; Zhu, X.; DeArmey, S.M.; Goldstein, J.L.; Bali, D.; Rehder, C.; Cirulli, E.T.; Kishnani, P.S. Sensitivity of whole exome sequencing in detecting infantile- and late-onset Pompe disease. Mol. Genet. Metab. 2017, 122, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, S.; Auray-Blais, C.; Gravel, E.; Boutin, M.; Dempsey-Nunez, L.; Jacques, P.-E.; Chenier, S.; LaRue, S.; Rioux, M.-F.; Al-Hertani, W.; et al. Diagnosis of late-onset Pompe disease and other muscle disorders by next-generation sequencing. Orphanet J. Rare Dis. 2016, 11, 8. [Google Scholar] [CrossRef]
- Thuriot, F.; Gravel, E.; Buote, C.; Doyon, M.; Lapointe, E.; Marcoux, L.; Larue, S.; Nadeau, A.; Chénier, S.; Waters, P.J.; et al. Molecular diagnosis of muscular diseases in outpatient clinics. Neurol. Genet. 2020, 6, e408. [Google Scholar] [CrossRef] [Green Version]
- Johansson, L.F.; Van Dijk, F.; De Boer, E.N.; Van Dijk-Bos, K.K.; Jongbloed, J.D.; Van Der Hout, A.H.; Westers, H.; Sinke, R.J.; Swertz, M.A.; Sijmons, R.H.; et al. CoNVaDING: Single Exon Variation Detection in Targeted NGS Data. Hum. Mutat. 2016, 37, 457–464. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Almeida, V.; Conceição, I.; Fineza, I.; Coelho, T.; Silveira, F.; Santos, M.; Valverde, A.; Geraldo, A.; Maré, R.; Aguiar, T.C.; et al. Screening for Pompe disease in a Portuguese high risk population. Neuromuscul. Disord. 2017, 27, 777–781. [Google Scholar] [CrossRef]
- Jastrzębska, A.; Potulska-Chromik, A.; Łusakowska, A.; Jastrzębski, M.; Lipowska, M.; Kierdaszuk, B.; Kamińska, A.; Kostera-Pruszczyk, A. Screening for late-onset Pompe disease in Poland. Acta Neurol. Scand. 2019, 140, 239–243. [Google Scholar] [CrossRef]
- Herbert, M.; Case, L.E.; Rairikar, M.; Cope, H.; Bailey, L.; Austin, S.L.; Kishnani, P.S. Early-onset of symptoms and clinical course of Pompe disease associated with the c.-32–13 T > G variant. Mol. Genet. Metab. 2019, 126, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Vanherpe, P.; Fieuws, S.; D’Hondt, A.; Bleyenheuft, C.; Demaerel, P.; De Bleecker, J.; Bergh, P.V.D.; Baets, J.; Remiche, G.; Verhoeven, K.; et al. Late-onset Pompe disease (LOPD) in Belgium: Clinical characteristics and outcome measures. Orphanet J. Rare Dis. 2020, 15, 83. [Google Scholar] [CrossRef]
- Johnson, K.; Töpf, A.; Bertoli, M.; Phillips, L.; Claeys, K.; Stojanovic, V.R.; Perić, S.; Hahn, A.; Maddison, P.; Akay, E.; et al. Identification of GAA variants through whole exome sequencing targeted to a cohort of 606 patients with unexplained limb-girdle muscle weakness. Orphanet J. Rare Dis. 2017, 12, 173. [Google Scholar] [CrossRef] [Green Version]
- Reddy, H.; Cho, K.-A.; Lek, M.; Estrella, E.; Valkanas, E.; Jones, M.D.; Mitsuhashi, S.; Darras, B.; Amato, A.A.; Lidov, H.G.; et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J. Hum. Genet. 2017, 62, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Nallamilli, B.R.R.; Khadilkar, S.V.; Singla, M.B.; Bhutada, A.; Dastur, R.; Gaitonde, P.S.; Rufibach, L.E.; Gloster, L.; Hegde, M. Clinical and Genomic Evaluation of 207 Genetic Myopathies in the Indian Subcontinent. Front. Neurol. 2020, 11, 559327. [Google Scholar] [CrossRef]
- Nallamilli, B.R.R.; Chakravorty, S.; Kesari, A.; Tanner, A.; Ankala, A.; Schneider, T.; Da Silva, C.; Beadling, R.; Alexander, J.J.; Askree, S.H.; et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann. Clin. Transl. Neurol. 2018, 5, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, J.A.; Ehuletche, M.D.R.G.; Perna, A.; Dubrovsky, A.; Franca, M.C.; Vargas, S.; Hegde, M.; Claeys, K.G.; Straub, V.; Daba, N.; et al. The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease. Orphanet J. Rare Dis. 2020, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaoui, R.; Cooper, S.; Lek, M.; Jones, K.; Corbett, A.; Reddel, S.W.; Needham, M.; Liang, C.; Waddell, L.B.; Nicholson, G.; et al. Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy. JAMA Neurol. 2015, 72, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Töpf, A.; Johnson, K.; Bates, A.; Phillips, L.; Chao, K.R.; England, E.M.; Laricchia, K.M.; Mullen, T.; Valkanas, E.; Xu, L.; et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet. Med. 2020, 22, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Savarese, M.; Torella, A.; Musumeci, O.; Angelini, C.; Astrea, G.; Bello, L.; Bruno, C.; Comi, G.P.; Di Fruscio, G.; Piluso, G.; et al. Targeted gene panel screening is an effective tool to identify undiagnosed late onset Pompe disease. Neuromuscul. Disord. 2018, 28, 586–591. [Google Scholar] [CrossRef] [Green Version]
- Winder, T.L.; Tan, C.A.; Klemm, S.; White, H.; Westbrook, J.M.; Wang, J.Z.; Entezam, A.; Truty, R.; Nussbaum, R.L.; McNally, E.M.; et al. Clinical utility of multigene analysis in over 25,000 patients with neuromuscular disorders. Neurol. Genet. 2020, 6, e412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.; Desai, A.K.; Kazi, Z.; Corey, K.; Austin, S.; Hobson-Webb, L.D.; Case, L.E.; Jones, H.N.; Kishnani, P.S. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017, 120, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Preisler, N.; Lukacs, Z.; Vinge, L.; Madsen, K.L.; Husu, E.; Hansen, R.S.; Duno, M.; Andersen, H.; Laub, M.; Vissing, J. Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophies. Mol. Genet. Metab. 2013, 110, 287–289. [Google Scholar] [CrossRef]
- Gutiérrez-Rivas, E.; Bautista, J.; Vílchez, J.; Muelas, N.; Diaz-Manera, J.; Illa, I.; Martínez-Arroyo, A.; Olive, M.; Sanz, I.; Arpa, J.; et al. Targeted screening for the detection of Pompe disease in patients with unclassified limb-girdle muscular dystrophy or asymptomatic hyperCKemia using dried blood: A Spanish cohort. Neuromuscul. Disord. 2015, 25, 548–553. [Google Scholar] [CrossRef]
- Bergsma, A.J.; Groen, S.L.I.T.; Verheijen, F.W.; Van Der Ploeg, A.T.; Pijnappel, W.W.M.P. From Cryptic Toward Canonical Pre-mRNA Splicing in Pompe Disease: A Pipeline for the Development of Antisense Oligonucleotides. Mol. Ther. Nucleic Acids 2016, 5, e361. [Google Scholar] [CrossRef]
- Reuser, A.J.J.; Van Der Ploeg, A.T.; Chien, Y.; Llerena, J.; Abbott, M.; Clemens, P.R.; Kimonis, V.E.; Leslie, N.; Maruti, S.S.; Sanson, B.; et al. GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry. Hum. Mutat. 2019, 40, 2146–2164. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Muñoz, E.A.; Milko, L.V.; Harrison, S.M.; Azzariti, D.R.; Kurtz, C.L.; Lee, K.; Mester, J.L.; Weaver, M.A.; Currey, E.; Craigen, W.; et al. ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum. Mutat. 2018, 39, 1614–1622. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Pathogenic/Likely Pathogenic Variants | Occurrence |
|---|---|
| c.32-13T>G | 12 |
| c.655G>A (p.Gly219Arg) | 2 |
| c.1115A>T (p.His372Leu) | 2 |
| c.1-?_2859+?del (p.Met1_Cys952del) | 1 |
| c.258dupC (p.Asn87fs) | 1 |
| c.525delT (p.Glu176fs) | 1 |
| c.706delG (p.Val236fs) | 1 |
| c.896T>C (p.Leu299Pro) | 1 |
| c.1396dupG (p.Val466fs) | 1 |
| c.1551+1G>C | 1 |
| c.1805C>T (p.Thr602Ile) | 1 |
| c.1912G>T (p.Gly638Trp) | 1 |
| c.1927G>A (p.Gly643Arg) | 1 |
| c.2242dupG (p.Glu748fs) | 1 |
| c.2577G>A (p.Trp859*) | 1 |
| Reference | Genetic Approach | Cohort | Number of Patients | Proportion of Pompe Patients | Recruitment |
|---|---|---|---|---|---|
| Ghaoui et al., 2015 PMID: 26436962 [30] | WES | LGMD | 100 | 1.00% | Australia, retrospective research, muscle biopsies |
| Reddy et al., 2017 PMID: 27708273 [26] | WES | LGMD | 55 | 1.82% | United States, research protocol |
| Chakravorty et al., 2020 PMID: 33250842 [27] | WES | Myopathy | 201 | 1.00% | India, tertiary care hospital |
| Mean proportion of Pompe disease patients (whole-exome sequencing) | 1.27% | ||||
| Töpf et al., 2020 PMID: 32528171 [31] | WES (429 genes analyzed) | LGMD and/or elevated CK | 1001 | 1.00% | Europe and Middle East, 43 neuromuscular referral centers |
| Johnson et al., 2017 PMID: 29149851 [25] | WES (169 genes analyzed) | LGMD and/or elevated CK | 606 | 1.98% | England, referred by clinicians |
| Nallamilli et al., 2018 PMID: 30564623 [28] | Targeted sequencing (35 genes) | LGMD | 4656 | 0.82% | United States, Emory Genetics Laboratory |
| Savarese et al., 2018 PMID: 29880332 [32] | Targeted sequencing (93 genes) | LGMD, myopathies and/or isolated hyperCKemia | 504 | 3.17% | Italy, tertiary centers for neuromuscular disorders |
| Bevilacqua et al., 2020 PMID: 31931849 [29] | Targeted sequencing (10 genes) | LGMD | 2103 | 0.43% | Latin America, 20 institutions |
| Winder et al., 2020 PMID: 32337338 [33] | Targeted sequencing (266 genes) | Cardiomyopathy/skeletal muscle, muscular dystrophy, neuromuscular disorders, LGMD | 6493 | 0.25% | United States, Invitae Laboratory |
| Mean proportion of Pompe disease patients (targeted sequencing) | 1.28% | ||||
| This study | Targeted sequencing (89 genes) | Suspected muscle disorders | 2030 | 0.69% (entire cohort) 1.15% (LGMD) | Canada, outpatient clinics |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thuriot, F.; Gravel, E.; Hodson, K.; Ganopolsky, J.; Rakic, B.; Waters, P.J.; Gravel, S.; Lévesque, S. Molecular Diagnosis of Pompe Disease in the Genomic Era: Correlation with Acid Alpha-Glucosidase Activity in Dried Blood Spots. J. Clin. Med. 2021, 10, 3868. https://doi.org/10.3390/jcm10173868
Thuriot F, Gravel E, Hodson K, Ganopolsky J, Rakic B, Waters PJ, Gravel S, Lévesque S. Molecular Diagnosis of Pompe Disease in the Genomic Era: Correlation with Acid Alpha-Glucosidase Activity in Dried Blood Spots. Journal of Clinical Medicine. 2021; 10(17):3868. https://doi.org/10.3390/jcm10173868
Chicago/Turabian StyleThuriot, Fanny, Elaine Gravel, Katherine Hodson, Jorge Ganopolsky, Bojana Rakic, Paula J. Waters, Serge Gravel, and Sébastien Lévesque. 2021. "Molecular Diagnosis of Pompe Disease in the Genomic Era: Correlation with Acid Alpha-Glucosidase Activity in Dried Blood Spots" Journal of Clinical Medicine 10, no. 17: 3868. https://doi.org/10.3390/jcm10173868
APA StyleThuriot, F., Gravel, E., Hodson, K., Ganopolsky, J., Rakic, B., Waters, P. J., Gravel, S., & Lévesque, S. (2021). Molecular Diagnosis of Pompe Disease in the Genomic Era: Correlation with Acid Alpha-Glucosidase Activity in Dried Blood Spots. Journal of Clinical Medicine, 10(17), 3868. https://doi.org/10.3390/jcm10173868