
Insights into the Cellular and Molecular Mechanisms That Govern the Fracture-Healing Process: A Narrative Review


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Biology of Bone Formation
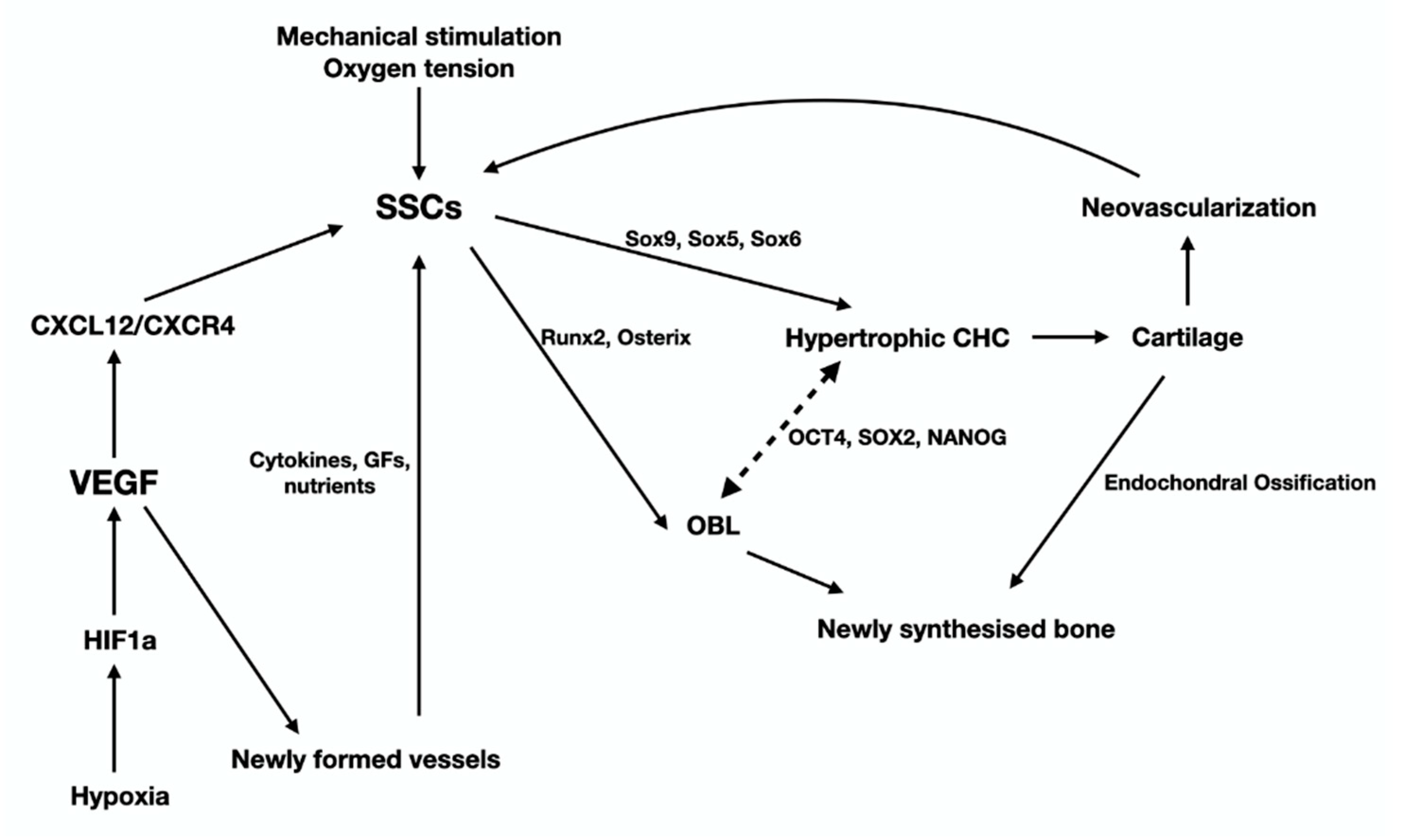
2.1. The Skeletal Stem Cells
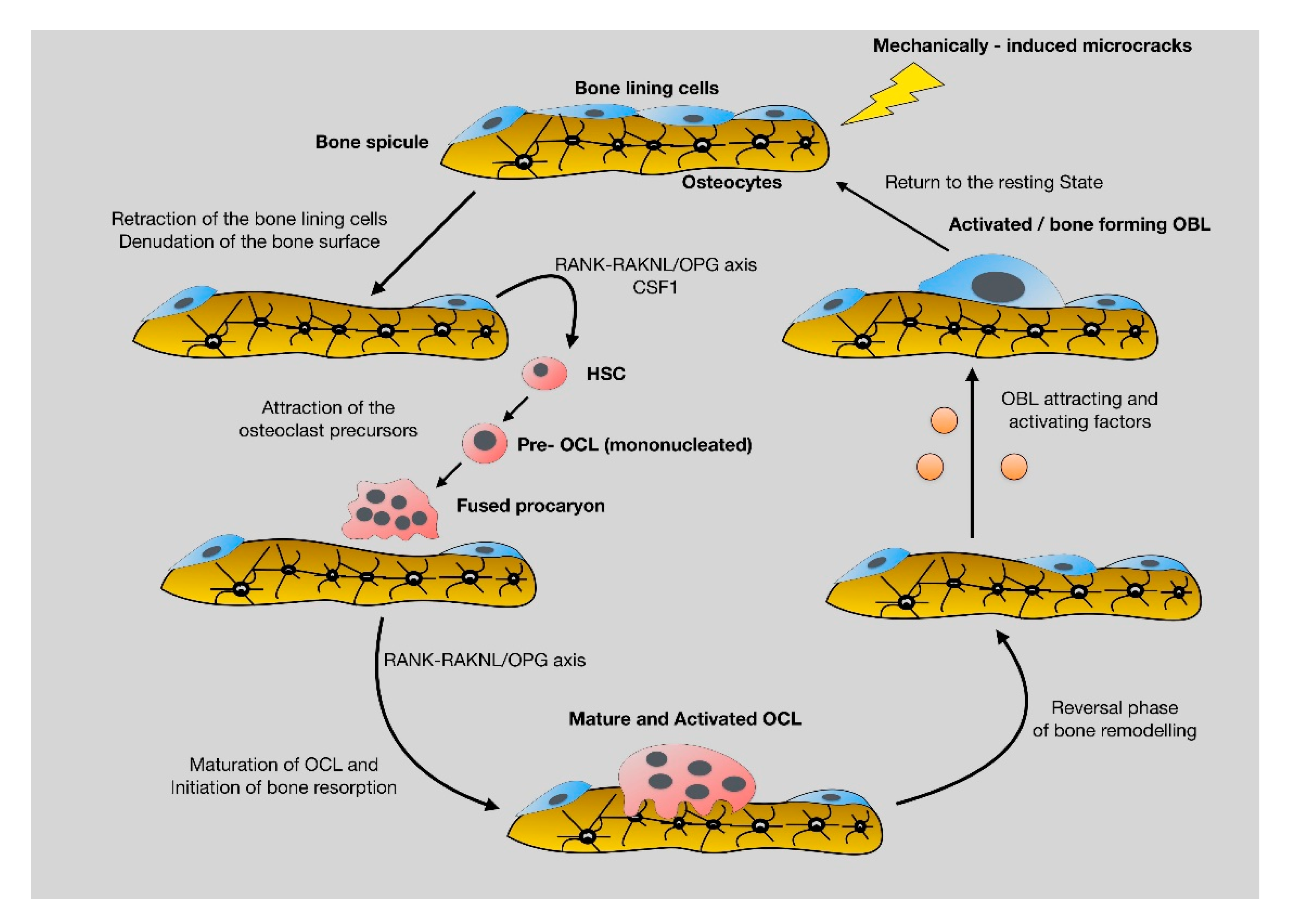
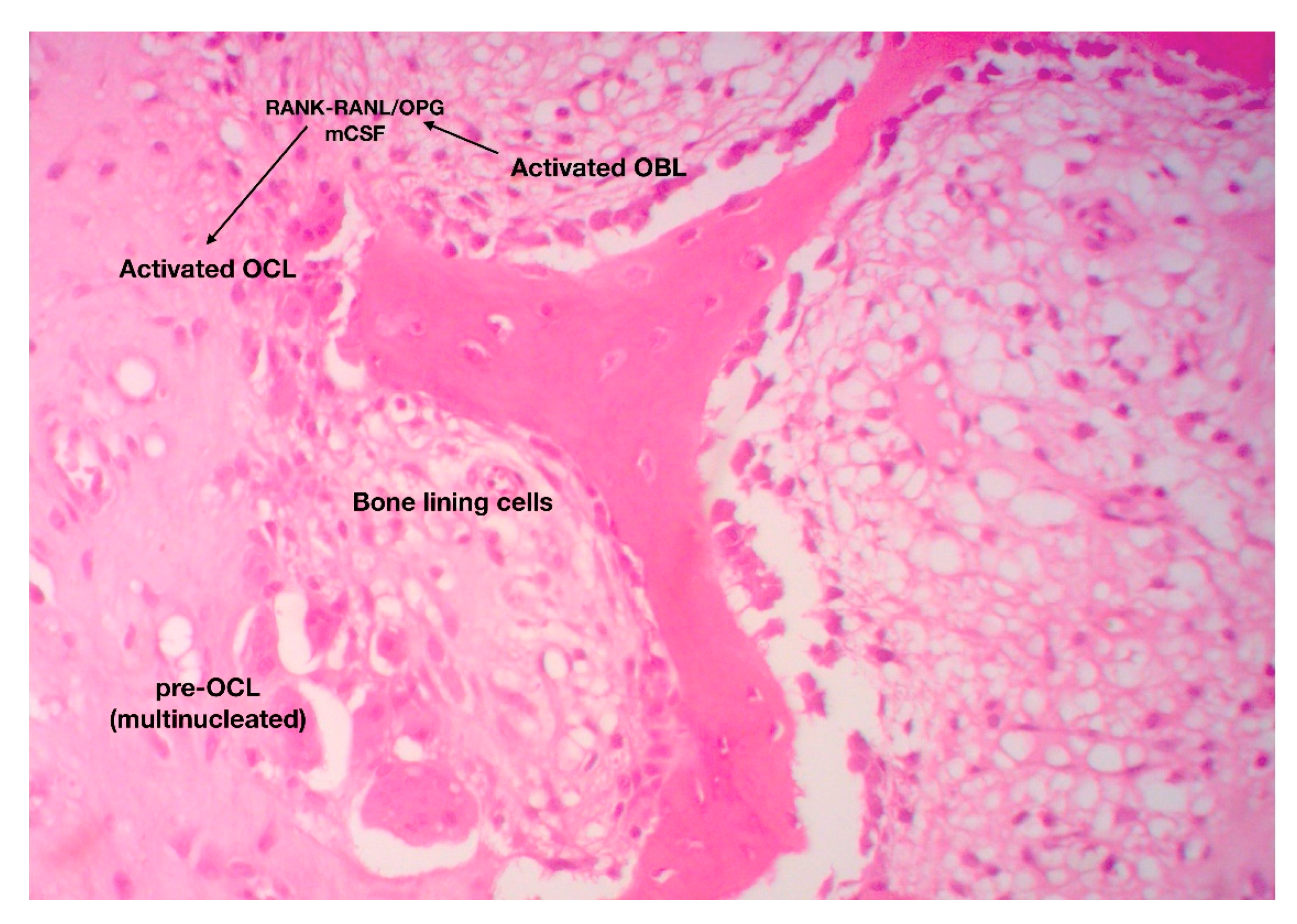
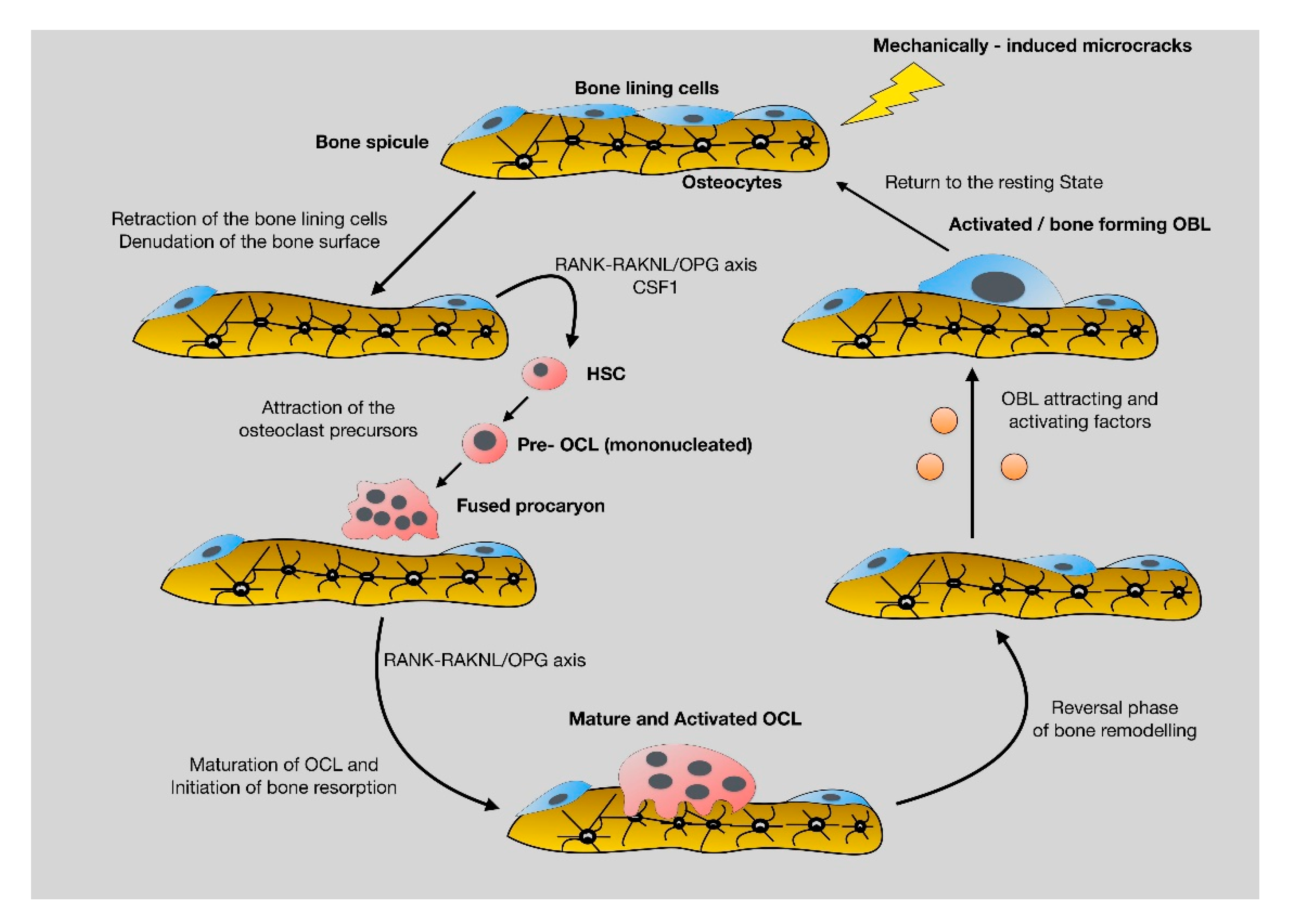
2.2. Bone Remodeling: The Continuous, Well-Balanced Interplay between Bone-Forming and Bone-Resorbing Cells
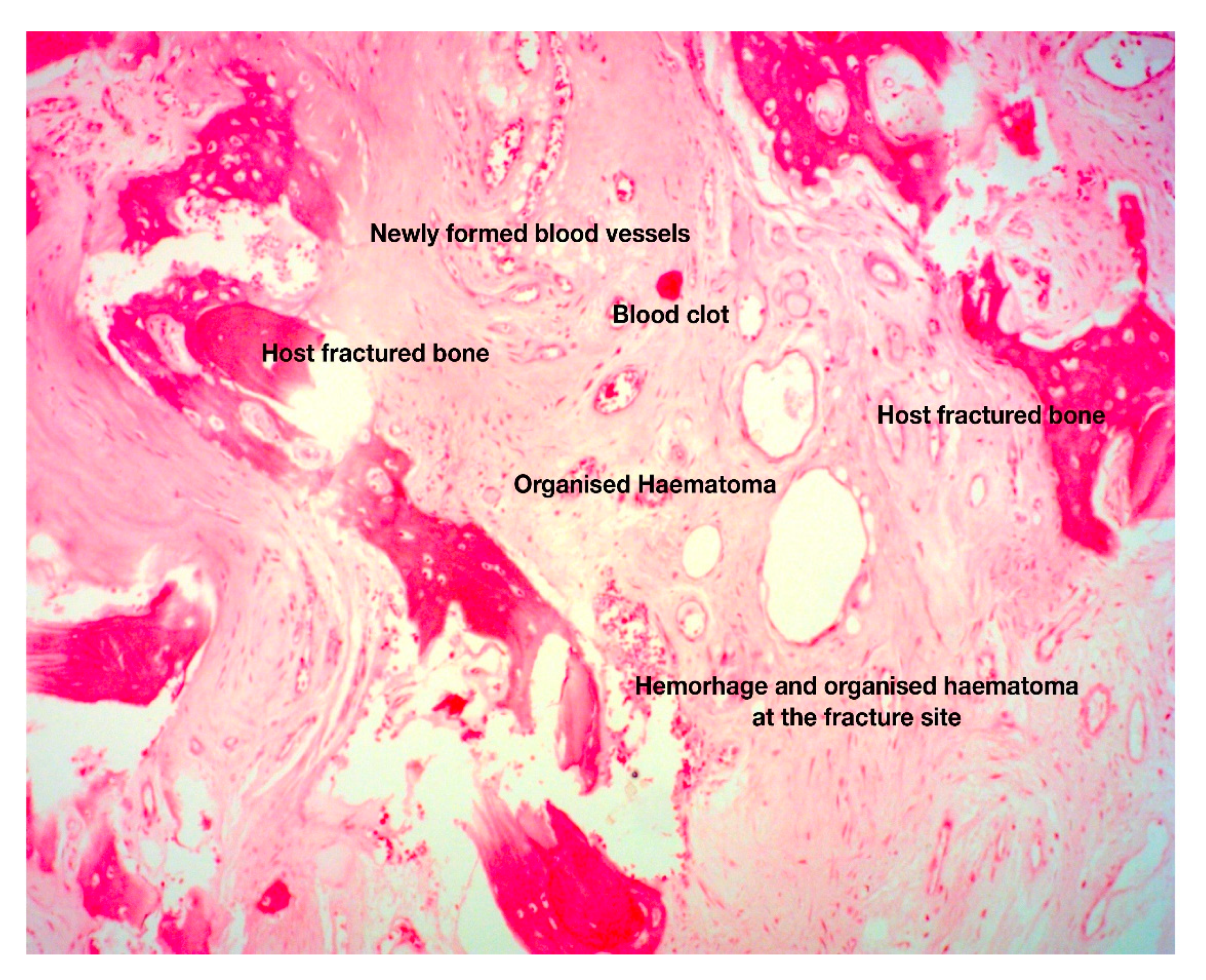
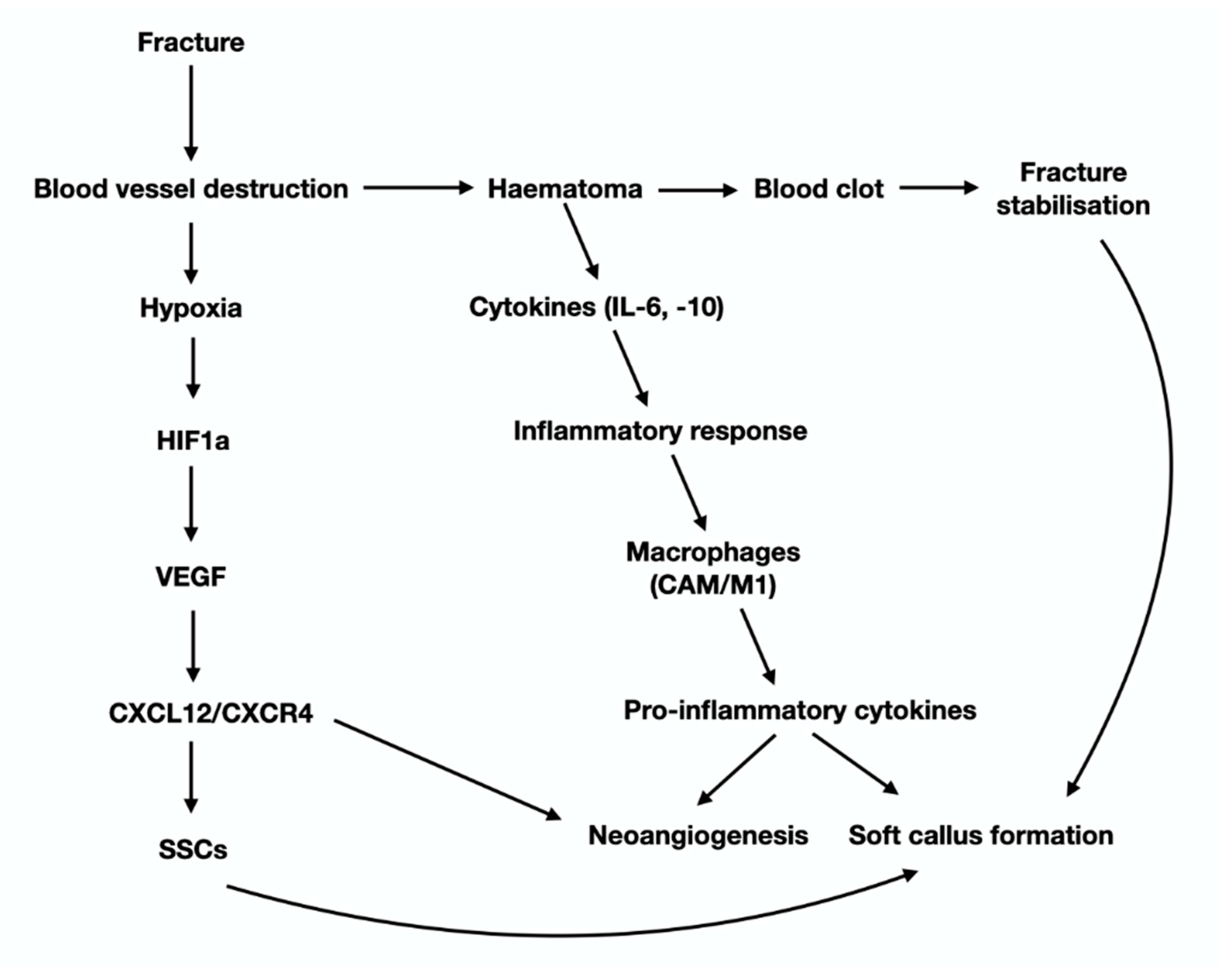
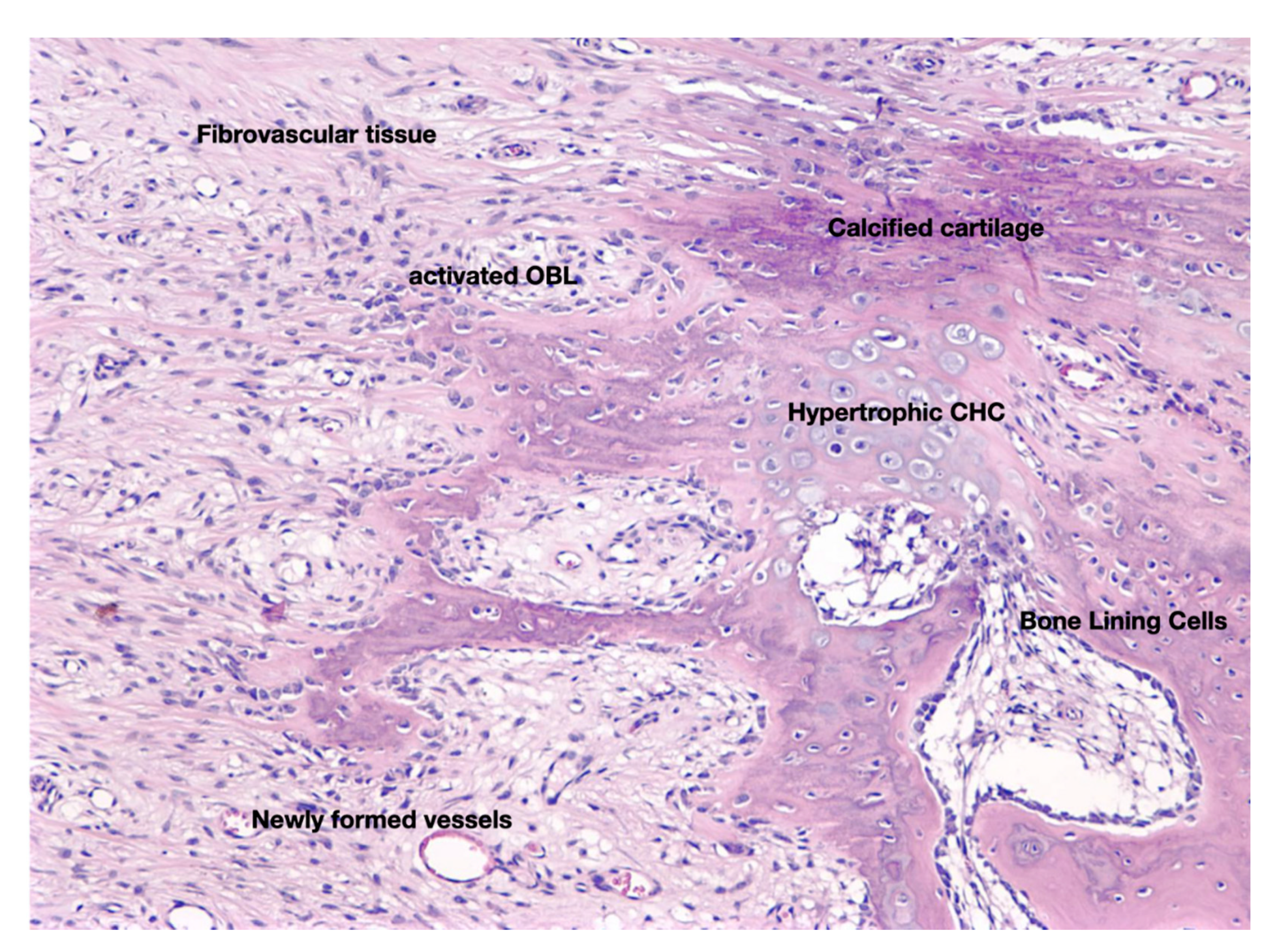
3. The Anabolic Phase of Fracture-Healing
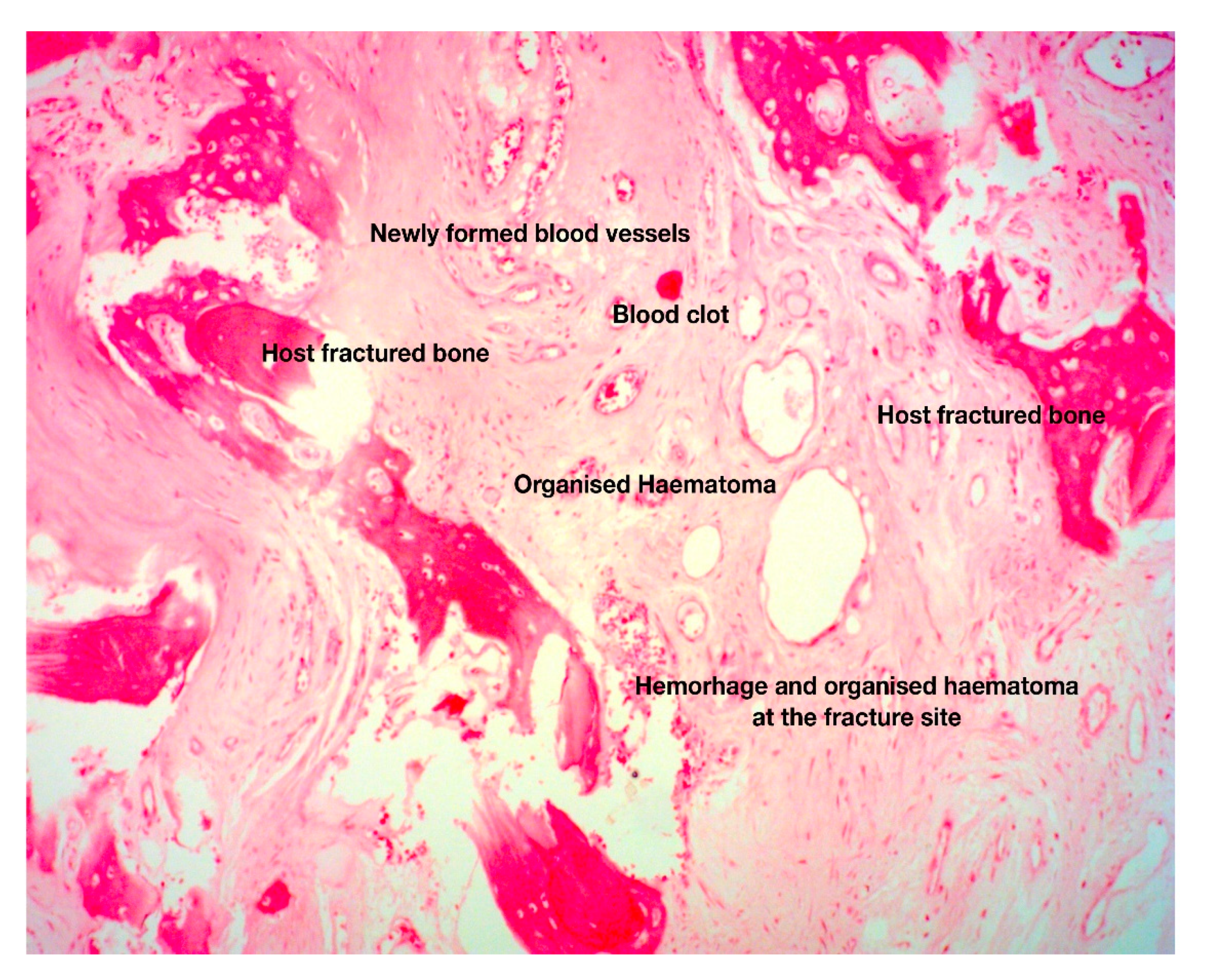
4. The Catabolic Phase of Fracture-Healing
5. The Role of Bone Marrow Fat in Fracture-Healing Process
6. Adipogenesis, Osteogenesis and Fracture-Healing
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Nandra, R.; Grover, L.; Porter, K. Fracture non-union epidemiology and treatment. Trauma 2016, 18, 3–11. [Google Scholar] [CrossRef]
- Hak, D.J.; Fitzpatrick, D.; Bishop, J.A.; Marsh, J.L.; Tilp, S.; Schnettler, R.; Simpson, H.; Alt, V. Delayed union and nonunions: Epidemiology, clinical issues, and financial aspects. Injury 2014, 45, S3–S7. [Google Scholar] [CrossRef] [PubMed]
- Melton, L.J.; Gabriel, S.E.; Crowson, C.S.; Tosteson, A.N.A.; Johnell, O.; Kanis, J.A. Cost-equivalence of different osteoporotic fractures. Osteoporos. Int. 2003, 14, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Johnell, O.; Kanis, J. Epidemiology of osteoporotic fractures. Osteoporos. Int. 2005, 16, S3–S7. [Google Scholar] [CrossRef]
- Collignon, A.-M.; Lesieur, J.; Vacher, C.; Chaussain, C.; Rochefort, G.Y. Strategies Developed to Induce, Direct, and Potentiate Bone Healing. Front. Physiol. 2017, 8, 927. [Google Scholar] [CrossRef] [Green Version]
- Robey, P. “Mesenchymal stem cells”: Fact or fiction, and implications in their therapeutic use. F1000Research 2017, 6, F1000. [Google Scholar] [CrossRef] [Green Version]
- Serowoky, M.A.; Arata, C.E.; Crump, J.G.; Mariani, F.V. Skeletal stem cells: Insights into maintaining and regenerating the skeleton. Development 2020, 147, dev179325. [Google Scholar] [CrossRef]
- Chan, C.K.; Seo, E.Y.; Chen, J.Y.; Lo, D.; McArdle, A.; Sinha, R.; Tevlin, R.; Seita, J.; Vincent-Tompkins, J.; Wearda, T.; et al. Identification and specification of the mouse skeletal stem cell. Cell 2015, 160, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Zhao, H.; Urata, M.; Chai, Y. Sutures Possess Strong Regenerative Capacity for Calvarial Bone Injury. Stem Cells Dev. 2016, 25, 1801–1807. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Spencer, J.A.; Koh, B.I.; Kobayashi, T.; Fujisaki, J.; Clemens, T.L.; Lin, C.P.; Kronenberg, H.M.; Scadden, D.T. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 2012, 10, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhang, X.; Bikle, D.D. Osteogenic Differentiation of Periosteal Cells during Fracture Healing. J. Cell. Physiol. 2017, 232, 913–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papachristou, D.J.; Papachroni, K.K.; Basdra, E.K.; Papavassiliou, A.G. Signaling networks and transcription factors regulating mechanotransduction in bone. BioEssays 2009, 31, 794–804. [Google Scholar] [CrossRef]
- Blair, H.C.; Zaidi, M.; Huang, C.L.-H.; Sun, L. The Developmental Basis of Skeletal Cell Differentiation and the Molecular Basis of Major Skeletal Defects. Biol. Rev. 2008, 83, 401–415. [Google Scholar] [CrossRef]
- de Gorter, D.J.J.; ten Dijke, P. Signal Transduction Cascades Controlling Osteoblast Differentiation. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism; Wiley Online Books: London, UK, 2013; pp. 15–24. [Google Scholar]
- Crockett, J.C.; Mellis, D.J.; Scott, D.I.; Helfrich, M.H. New knowledge on critical osteoclast formation and activation pathways from study of rare genetic diseases of osteoclasts: Focus on the RANK/RANKL axis. Osteoporos. Int. 2011, 22, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Jacobs, C.R.; Temiyasathit, S.; Castillo, A.B. Osteocyte mechanobiology and pericellular mechanics. Annu. Rev. Biomed. Eng. 2010, 12, 369–400. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Rochefort, G.Y.; Pallu, S.; Benhamou, C.L. Osteocyte: The unrecognized side of bone tissue. Osteoporos. Int. 2010, 21, 1457–1469. [Google Scholar] [CrossRef]
- Martin, T.J.; Sims, N.A. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139. [Google Scholar] [CrossRef]
- Marsell, R.; Einhorn, T.A. The biology of fracture healing. Injury 2011, 42, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, K.A.; Al-Aql, Z.S.; Wan, C.; Fitch, J.L.; Stapleton, S.N.; Mason, Z.D.; Cole, R.M.; Gilbert, S.R.; Clemens, T.L.; Morgan, E.F.; et al. Bone Formation During Distraction Osteogenesis Is Dependent on Both VEGFR1 and VEGFR2 Signaling. J. Bone Miner. Res. 2008, 23, 596–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddle, R.C.; Khatri, R.; Schipani, E.; Clemens, T.L. Role of hypoxia-inducible factor-1α in angiogenic–osteogenic coupling. J. Mol. Med. 2009, 87, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Kolar, P.; Schmidt-Bleek, K.; Schell, H.; Gaber, T.; Toben, D.; Schmidmaier, G.; Perka, C.; Buttgereit, F.; Duda, G.N. The early fracture hematoma and its potential role in fracture healing. Tissue Eng. Part B Rev. 2010, 16, 427–434. [Google Scholar] [CrossRef]
- Yuasa, M.; Mignemi, N.A.; Nyman, J.S.; Duvall, C.L.; Schwartz, H.S.; Okawa, A.; Yoshii, T.; Bhattacharjee, G.; Zhao, C.; Bible, J.E.; et al. Fibrinolysis is essential for fracture repair and prevention of heterotopic ossification. J. Clin. Investig. 2015, 125, 3117–3131. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, A.; Bergdolt, S.; Wiegner, R.; Radermacher, P.; Huber-Lang, M.; Ignatius, A. The crucial role of neutrophil granulocytes in bone fracture healing. Eur. Cell Mater. 2016, 32, 152–162. [Google Scholar] [CrossRef]
- Kon, T.; Cho, T.-J.; Aizawa, T.; Yamazaki, M.; Nooh, N.; Graves, D.; Gerstenfeld, L.C.; Einhorn, T.A. Expression of Osteoprotegerin, Receptor Activator of NF-κB Ligand (Osteoprotegerin Ligand) and Related Proinflammatory Cytokines During Fracture Healing. J. Bone Miner. Res. 2001, 16, 1004–1014. [Google Scholar] [CrossRef]
- Pettit, A.R.; Chang, M.K.; Hume, D.A.; Raggatt, L.-J. Osteal macrophages: A new twist on coupling during bone dynamics. Bone 2008, 43, 976–982. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Sakamoto, K.; Minamizato, T.; Katsube, K.; Nakanishi, S. Regulation of osteoblast differentiation mediated by BMP, Notch, and CCN3/NOV. Jpn. Dent. Sci. Rev. 2008, 44, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.P.H.; Xu, J.; Xue, M.; Jackson, C. Matrix metalloproteinases in bone development and pathology: Current knowledge and potential clinical utility. Met. Med. 2016, 3, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Sinder, B.P.; Pettit, A.R.; McCauley, L.K. Macrophages: Their Emerging Roles in Bone. J. Bone Miner. Res. 2015, 30, 2140–2149. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Mailhot, G.; Yang, M.; Mason-Savas, A.; MacKay, C.A.; Leav, I.; Odgren, P.R. BMP-5 expression increases during chondrocyte differentiation in vivo and in vitro and promotes proliferation and cartilage matrix synthesis in primary chondrocyte cultures. J. Cell. Physiol. 2008, 214, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Xu, H.; Yin, H.; Zhao, X.; Li, D.; Zhu, Q.; Wang, Y. The role of BMP6 in the proliferation and differentiation of chicken cartilage cells. PLoS ONE 2019, 14, e0204384. [Google Scholar] [CrossRef] [Green Version]
- Rosati, R.; Horan, G.S.B.; Pinero, G.J.; Garofalo, S.; Keene, D.R.; Horton, W.A.; Vuorio, E.; de Crombrugghe, B.; Behringer, R.R. Normal long bone growth and development in type X collagen-null mice. Nat. Genet. 1994, 8, 129–135. [Google Scholar] [CrossRef]
- Day, T.F.; Guo, X.; Garrett-Beal, L.; Yang, Y. Wnt/β-Catenin Signaling in Mesenchymal Progenitors Controls Osteoblast and Chondrocyte Differentiation during Vertebrate Skeletogenesis. Dev. Cell 2005, 8, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Hill, T.P.; Später, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/β-Catenin Signaling Prevents Osteoblasts from Differentiating into Chondrocytes. Dev. Cell 2005, 8, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.P.; Ferro, F.; Yang, F.; Taylor, A.J.; Chang, W.; Miclau, T.; Marcucio, R.S.; Bahney, C.S. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development 2017, 144, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, H.; Teitelbaum, S.; Ghiselli, R.; Gluck, S. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science 1989, 245, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Palagano, E.; Blair, H.C.; Pangrazio, A.; Tourkova, I.; Strina, D.; Angius, A.; Cuccuru, G.; Oppo, M.; Uva, P.; Van Hul, W.; et al. Buried in the Middle but Guilty: Intronic Mutations in the TCIRG1 Gene Cause Human Autosomal Recessive Osteopetrosis. J. Bone Miner. Res. 2015, 30, 1814–1821. [Google Scholar] [CrossRef]
- Schlesinger, P.H.; Blair, H.C.; Teitelbaum, S.L.; Edwards, J.C. Characterization of the Osteoclast Ruffled Border Chloride Channel and Its Role in Bone Resorption. J. Biol. Chem. 1997, 272, 18636–18643. [Google Scholar] [CrossRef] [Green Version]
- Lelliott, C.; Vidal-Puig, A.J. Lipotoxicity, an imbalance between lipogenesis de novo and fatty acid oxidation. Int. J. Obes. 2004, 28, S22–S28. [Google Scholar] [CrossRef] [Green Version]
- Einhorn, T.A.; Gerstenfeld, L.C. Fracture healing: Mechanisms and interventions. Nat. Rev. Rheumatol. 2015, 11, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Einhorn, T.A. Can an anti-fracture agent heal fractures? Clin. Cases Miner. Bone Metab. 2010, 7, 11–14. [Google Scholar] [PubMed]
- Scherer, P.E. Adipose Tissue. Lipid Storage Compart. Endocr. Organ 2006, 55, 1537–1545. [Google Scholar] [CrossRef] [Green Version]
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Gesta, S.; Tseng, Y.-H.; Kahn, C.R. Developmental Origin of Fat: Tracking Obesity to Its Source. Cell 2007, 131, 242–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jesus, L.A.; Carvalho, S.D.; Ribeiro, M.O.; Schneider, M.; Kim, S.-W.; Harney, J.W.; Larsen, P.R.; Bianco, A.C. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J. Clin. Investig. 2001, 108, 1379–1385. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneki, H.; Guo, R.; Chen, D.; Yao, Z.; Schwarz, E.M.; Zhang, Y.E.; Boyce, B.F.; Xing, L. Tumor Necrosis Factor Promotes Runx2 Degradation through Up-regulation of Smurf1 and Smurf2 in Osteoblasts. J. Biol. Chem. 2006, 281, 4326–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Miyanishi, K.; Suen, A.; Epstein, N.J.; Tomita, T.; Smith, R.L.; Goodman, S.B. Human interleukin-1-induced murine osteoclastogenesis is dependent on RANKL, but independent of TNF-α. Cytokine 2004, 26, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef]
- Motyl, K.J.; Bishop, K.A.; DeMambro, V.E.; Bornstein, S.A.; Le, P.; Kawai, M.; Lotinun, S.; Horowitz, M.C.; Baron, R.; Bouxsein, M.L.; et al. Altered thermogenesis and impaired bone remodeling in Misty mice. J. Bone Miner. Res. 2013, 28, 1885–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, H.C.; Kalyvioti, E.; Papachristou, N.I.; Tourkova, I.L.; Syggelos, S.A.; Deligianni, D.; Orkoula, M.G.; Kontoyannis, C.G.; Karavia, E.A.; Kypreos, K.E.; et al. Apolipoprotein A-1 regulates osteoblast and lipoblast precursor cells in mice. Lab. Investig. 2016, 96, 763–772. [Google Scholar] [CrossRef]
- Papachristou, N.I.; Blair, H.C.; Kypreos, K.E.; Papachristou, D.J. High-density lipoprotein (HDL) metabolism and bone mass. J. Endocrinol. 2017, 233, R95–R107. [Google Scholar] [CrossRef]
- Muruganandan, S.; Sinal, C.J. The impact of bone marrow adipocytes on osteoblast and osteoclast differentiation. IUBMB Life 2014, 66, 147–155. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papachristou, D.J.; Georgopoulos, S.; Giannoudis, P.V.; Panagiotopoulos, E. Insights into the Cellular and Molecular Mechanisms That Govern the Fracture-Healing Process: A Narrative Review. J. Clin. Med. 2021, 10, 3554. https://doi.org/10.3390/jcm10163554
Papachristou DJ, Georgopoulos S, Giannoudis PV, Panagiotopoulos E. Insights into the Cellular and Molecular Mechanisms That Govern the Fracture-Healing Process: A Narrative Review. Journal of Clinical Medicine. 2021; 10(16):3554. https://doi.org/10.3390/jcm10163554
Chicago/Turabian StylePapachristou, Dionysios J., Stavros Georgopoulos, Peter V. Giannoudis, and Elias Panagiotopoulos. 2021. "Insights into the Cellular and Molecular Mechanisms That Govern the Fracture-Healing Process: A Narrative Review" Journal of Clinical Medicine 10, no. 16: 3554. https://doi.org/10.3390/jcm10163554
APA StylePapachristou, D. J., Georgopoulos, S., Giannoudis, P. V., & Panagiotopoulos, E. (2021). Insights into the Cellular and Molecular Mechanisms That Govern the Fracture-Healing Process: A Narrative Review. Journal of Clinical Medicine, 10(16), 3554. https://doi.org/10.3390/jcm10163554