
An Integrative Systems Biology Approach Identifies Molecular Signatures Associated with Gallbladder Cancer Pathogenesis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieval of GBC RNA-seq Dataset
2.2. Differential Gene Co-Expression Network Analysis
2.3. Module Preservation Analysis
2.4. Gene Ontology and Pathway Analysis of the Non-Preserved Module
2.5. Screening of Hub Genes from Non-Preserved Modules through Intramodular Connectivity and PPIs Network Analysis
2.6. Gene Regulatory Network (GRN) Analysis
2.7. EMT Scores Calculation
3. Results
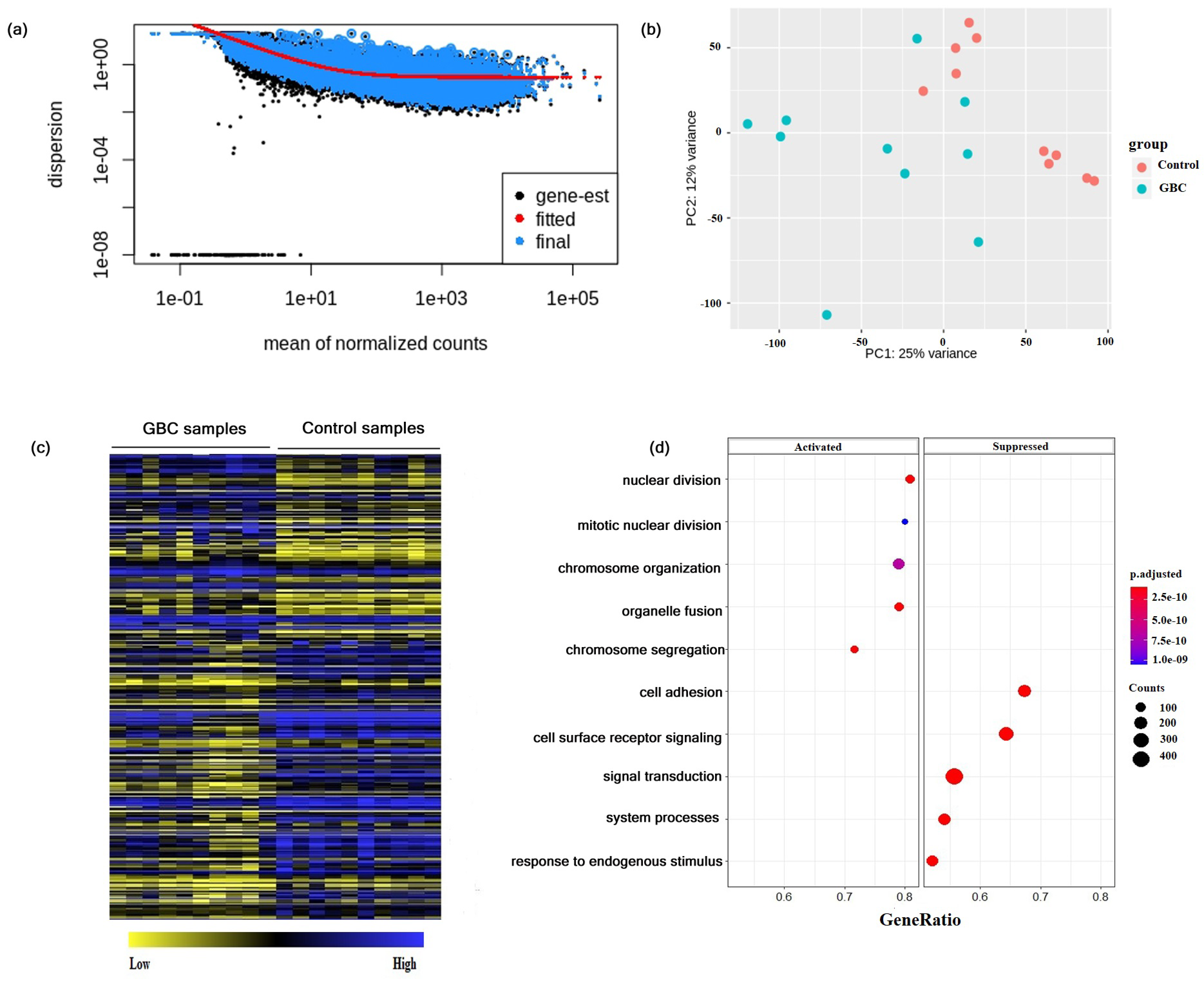
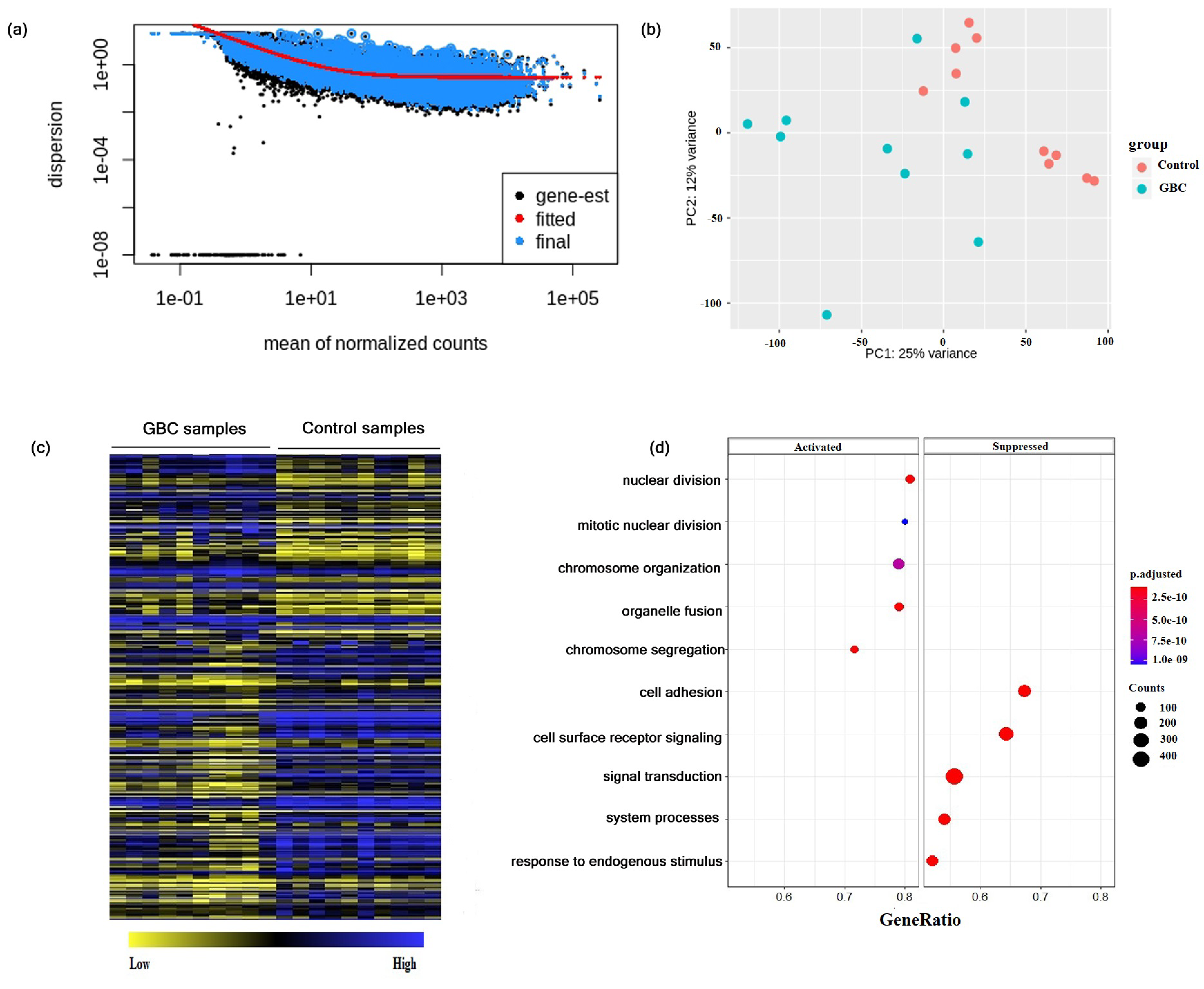
3.1. Differential Gene Expressions in Gallbladder Cancer
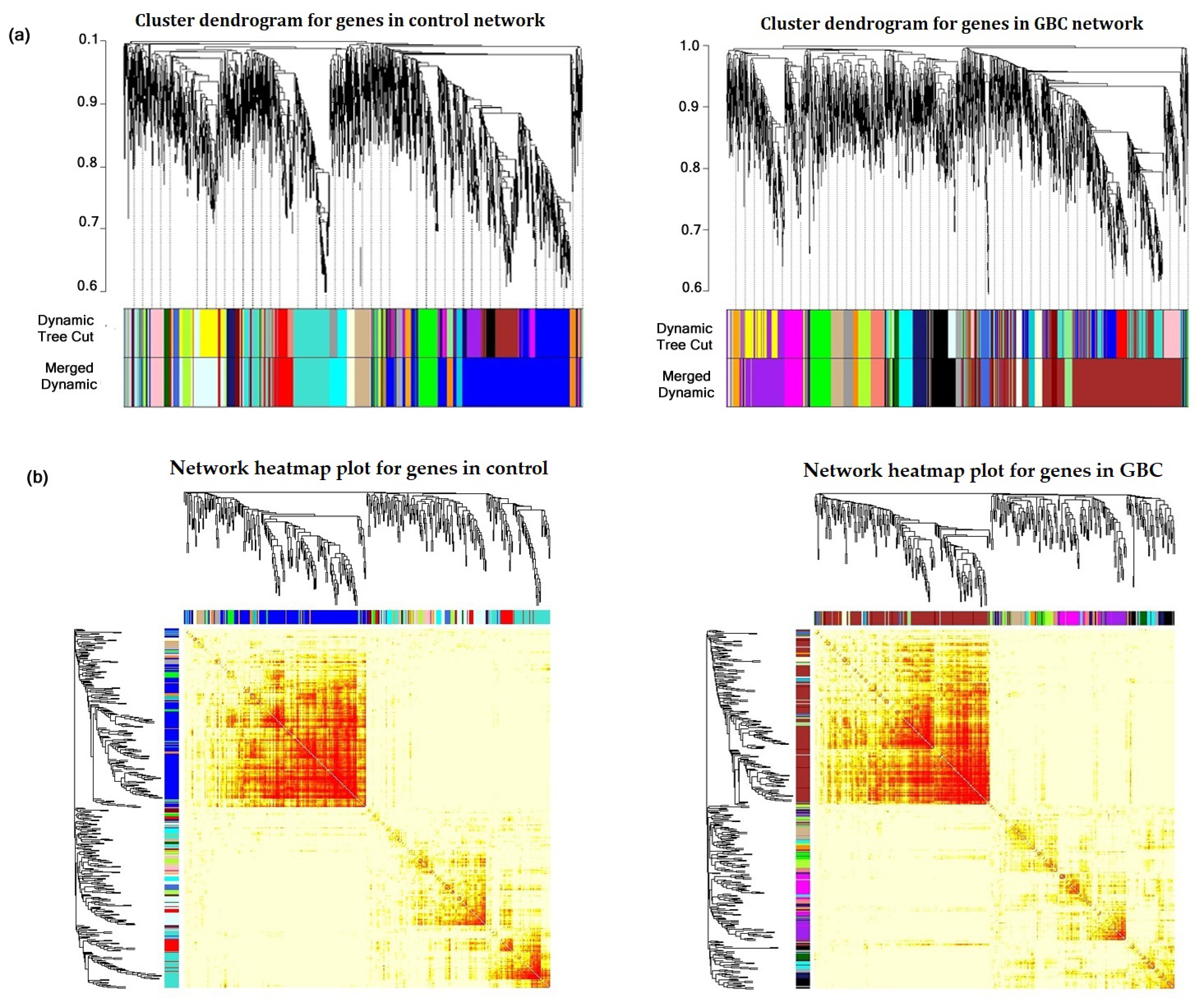
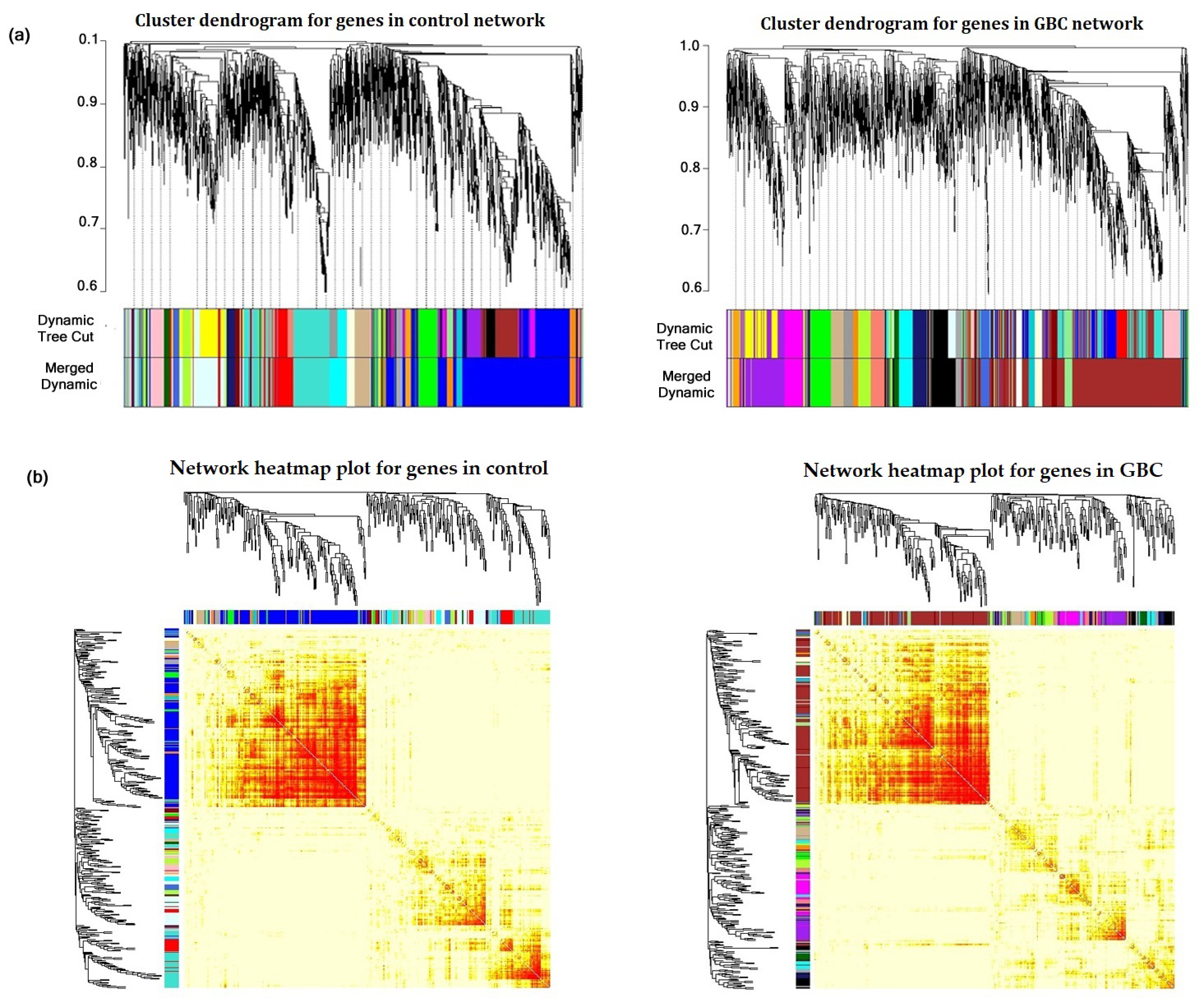
3.2. Construction of Gene Co-Expression Network and Module Detection
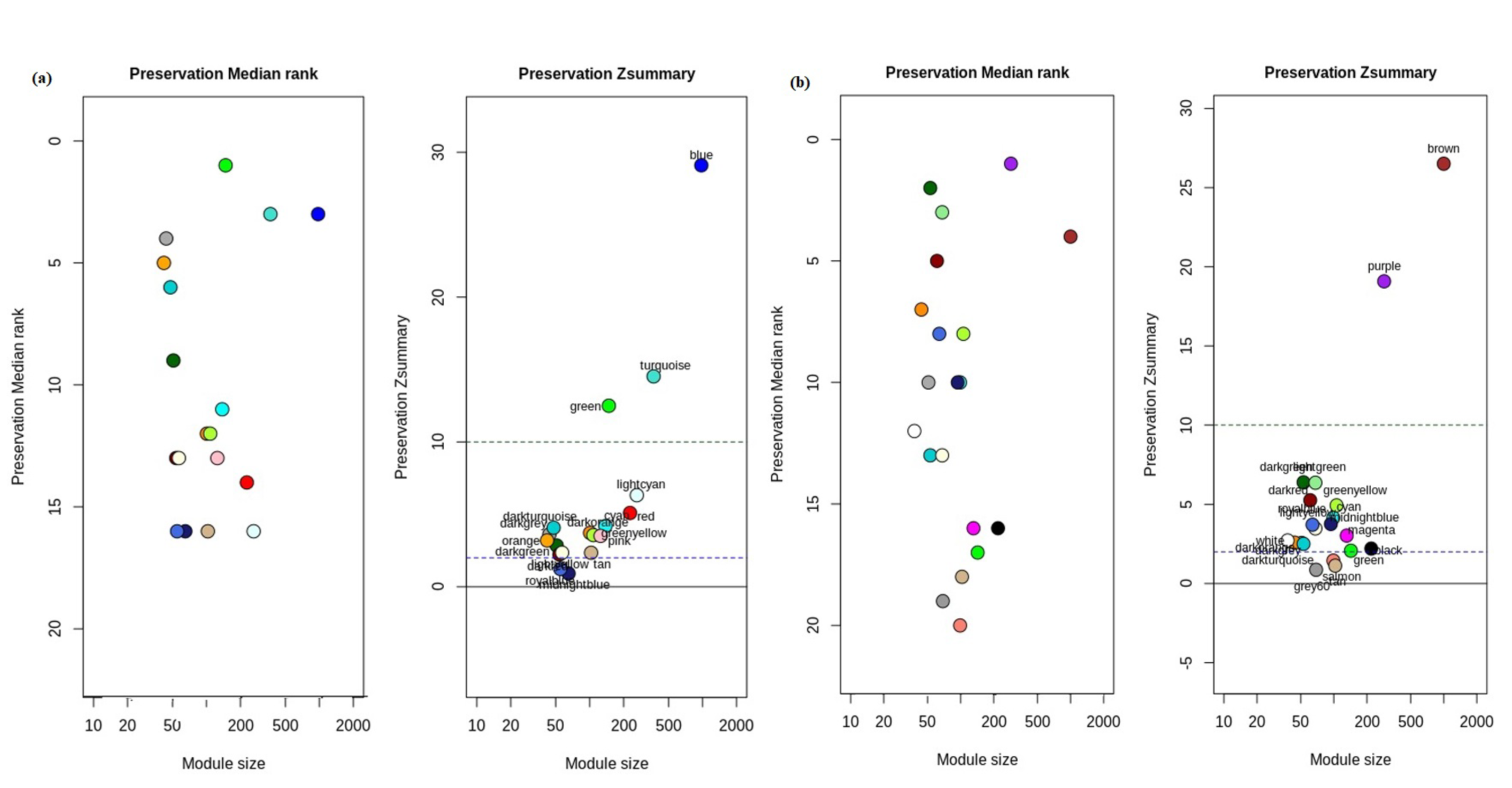
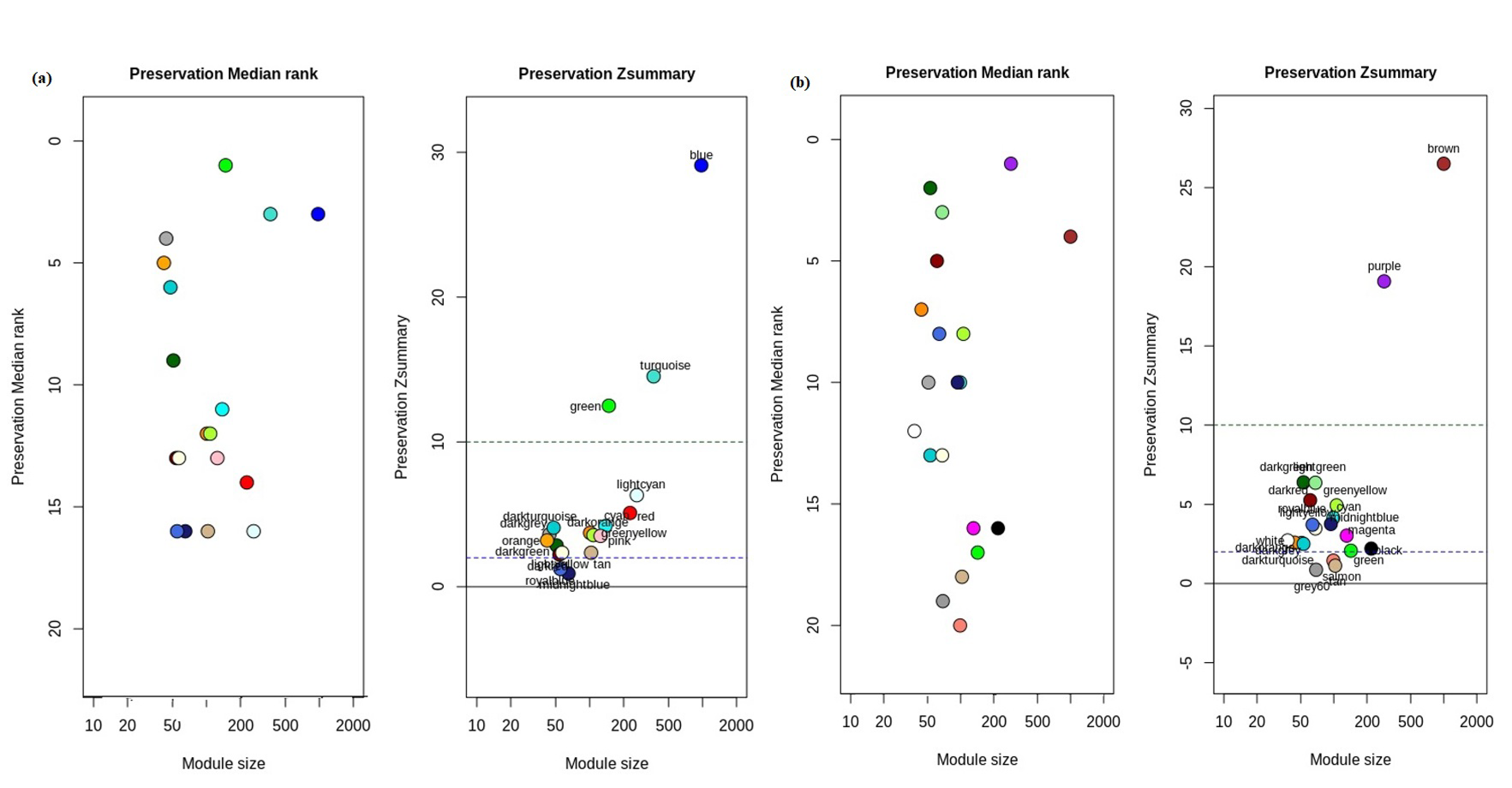
3.3. Detection of Non-Preserved Module from GBC and Control Co-Expression Network
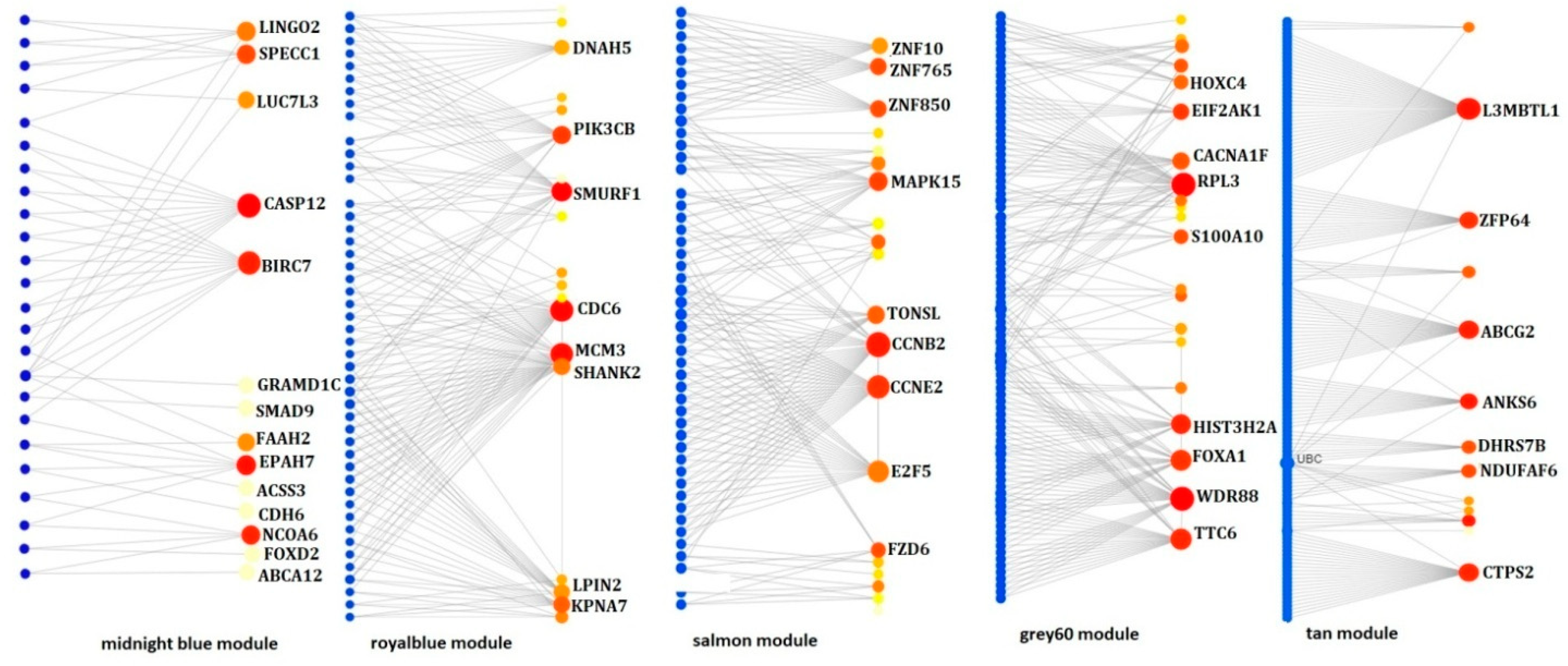
3.4. Hub Gene Identification from Non-Preserved Modules
3.5. Functional and Annotation and Pathway Associated with Genes of the Non-Preserved Modules
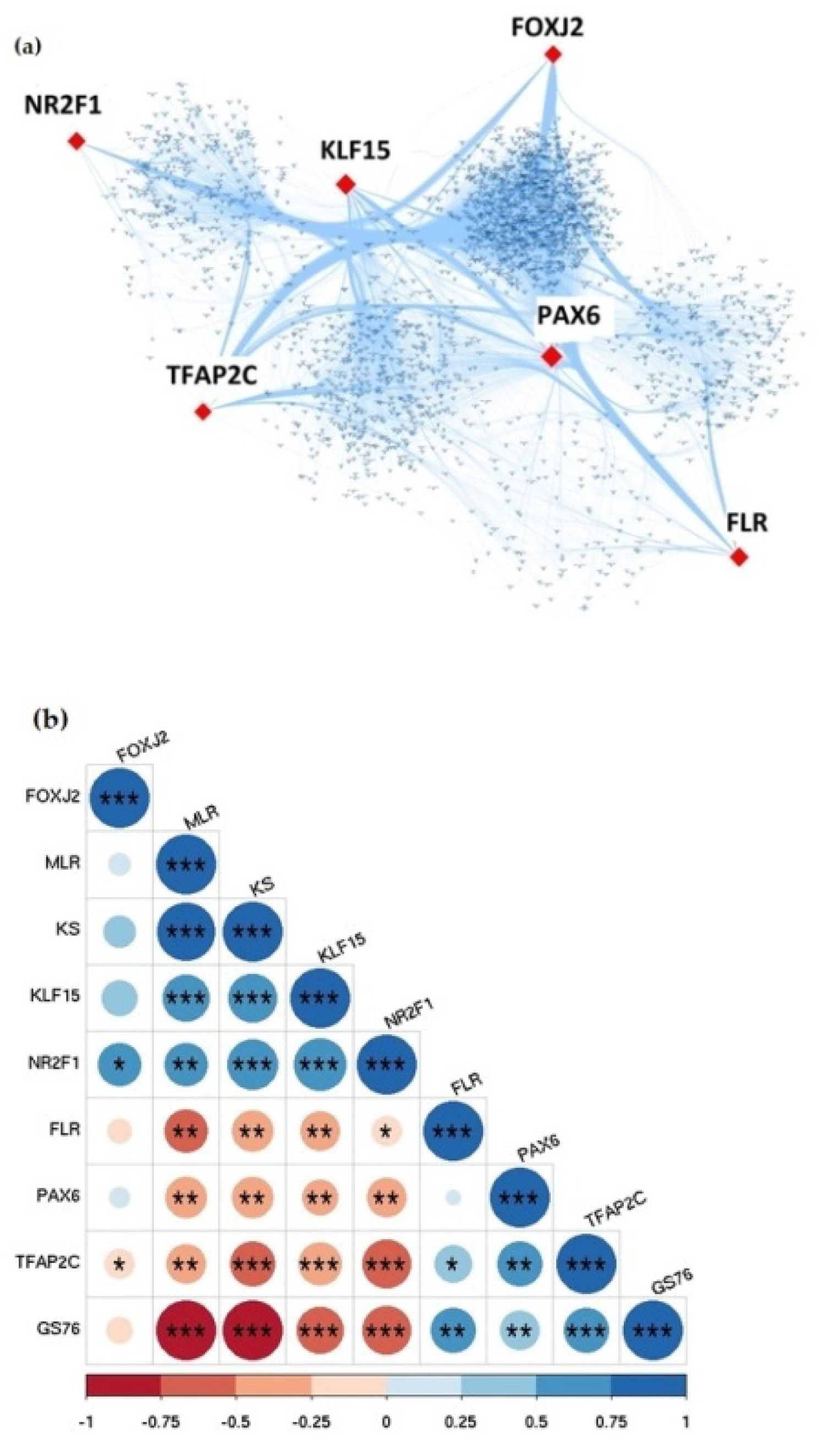
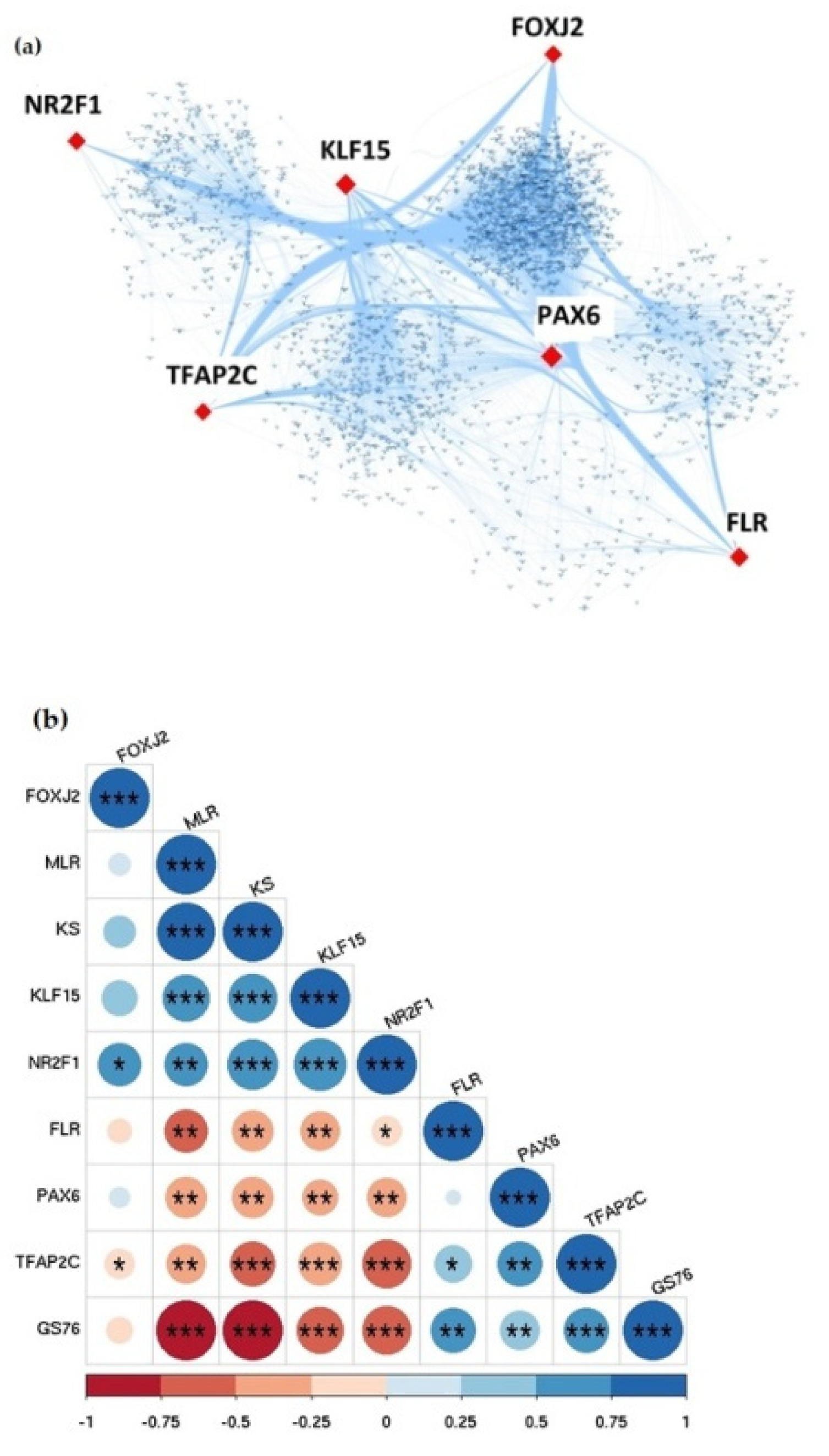
3.6. Identification of Hub Transcription Factors in GBC through TG-TF Regulatory Network Analysis
3.7. EMT Analysis Identified Differential EMT Patterns in Hub Transcription Factors
4. Discussions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wistuba, I.I.; Gazdar, A.F. Gallbladder cancer: Lessons from a rare tumour. Nat. Rev. Cancer 2004, 4, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Lazcano-Ponce, E.C.; Miquel, J.F.; Munoz, N.; Herrero, R.; Ferrecio, C.; Wistuba, I.I.; Alonso de Ruiz, P.; Aristi Urista, G.; Nervi, F. Epidemiology and Molecular Pathology of Gallbladder Cancer. CA Cancer J. Clin. 2001, 51, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Randi, G.; Franceschi, S.; La Vecchia, C. Gallbladder cancer worldwide: Geographical distribution and risk factors. Int. J. Cancer 2006, 118, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Thandra, K.C.; Barsouk, A. Epidemiology of gallbladder cancer. Clin. Exp. Hepatol. 2019, 5, 93–102. [Google Scholar] [CrossRef]
- Pandey, A.; Stawiski, E.W.; Durinck, S.; Gowda, H.; Goldstein, L.D.; Barbhuiya, M.A.; Schröder, M.S.; Sreenivasamurthy, S.K.; Kim, S.-W.; Phalke, S.; et al. Integrated genomic analysis reveals mutated ELF3 as a potential gallbladder cancer vaccine candidate. Nat. Commun. 2020, 11, 4225. [Google Scholar] [CrossRef]
- Nemunaitis, J.M.; Brown-Glabeman, U.; Soares, H.; Belmonte, J.; Liem, B.; Nir, I.; Phuoc, V.; Gullapalli, R.R. Gallbladder cancer: Review of a rare orphan gastrointestinal cancer with a focus on populations of New Mexico. BMC Cancer 2018, 18, 665. [Google Scholar] [CrossRef]
- Bizama, C.; García, P.; Espinoza, J.A.; Weber, H.; Leal, P.; Nervi, B.; Roa, J.C. Targeting specific molecular pathways holds promise for advanced gallbladder cancer therapy. Cancer Treat. Rev. 2015, 41, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, R.; Tulsyan, S.; Hussain, S.; Mittal, B.; Singh Saluja, S.; Singh, S.; Tanwar, P.; Khan, A.; Javle, M.; Hassan, M.M.; et al. Genetic landscape of gallbladder cancer: Global overview. Mutat. Res. Rev. Mutat. Res. 2018, 778, 61–71. [Google Scholar] [CrossRef]
- Singh, M.K.; Chetri, K.; Pandey, U.B.; Kapoor, V.K.; Mittal, B.; Choudhuri, G. Mutational spectrum of K-ras oncogene among Indian patients with gallbladder cancer. J. Gastroenterol. Hepatol. 2004, 19, 916–921. [Google Scholar] [CrossRef]
- Leone, F.; Cavalloni, G.; Pignochino, Y.; Sarotto, I.; Ferraris, R.; Piacibello, W.; Venesio, T.; Capussotti, L.; Risio, M.; Aglietta, M. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin. Cancer Res. 2006, 12, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Ajioka, Y.; Lang, I.; Szentirmay, Z.; Kasler, M.; Nakadaira, H.; Yokoyama, N.; Watanabe, G.; Nishikura, K.; Wakai, T.; et al. Genetic changes of p53, K-ras, and microsatellite instability in gallbladder carcinoma in high-incidence areas of Japan and Hungary. World J. Gastroenterol. 2008, 14, 70–75. [Google Scholar] [CrossRef]
- Maurya, S.K.; Tewari, M.; Mishra, R.R.; Shukla, H.S. Genetic abberations in gallbladder cancer. Surg. Oncol. 2012, 21, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Hu, Y.; Li, Y.; Shao, R.; Liu, F.; Liu, Y. Overview of current targeted therapy in gallbladder cancer. Signal Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Hueman, M.T.; Vollmer, C.M.; Pawlik, T.M. Evolving treatment strategies for gallbladder cancer. Ann. Surg. Oncol. 2009, 16, 2101–2115. [Google Scholar] [CrossRef] [PubMed]
- Ben-Josef, E.; Guthrie, K.A.; El-Khoueiry, A.B.; Corless, C.L.; Zalupski, M.M.; Lowy, A.M.; Thomas, C.R.; Alberts, S.R.; Dawson, L.A.; Micetich, K.C.; et al. SWOG S0809: A phase II intergroup trial of adjuvant capecitabine and gemcitabine followed by radiotherapy and concurrent capecitabine in extrahepatic cholangiocarcinoma and gallbladder carcinoma. J. Clin. Oncol. 2015, 33, 2617–2622. [Google Scholar] [CrossRef] [Green Version]
- Weigt, J.; Malfertheiner, P. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 395–397. [Google Scholar] [CrossRef]
- Lou, X.Y.; Yang, X.; Ding, Y.L.; Wang, J.J.; Yan, Q.Y.; Huang, X.G.; Guo, Y.H.; Wang, X.J.; Xiang, W.S. Synthesis of Olaparib derivatives and their antitumor activities. Chem. Res. Chinese Univ. 2013, 29, 231–235. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Paolis, D.; Urbani, A.; Salvatore, L.; Foca, L.; Tortora, G.; Minucci, A.; Concolino, P. A novel ATM pathogenic variant in an Italian women with Gallbladder cancer. Genes. 2021, 12, 313. [Google Scholar] [CrossRef]
- Ebata, T.; Ercolani, G.; Alvaro, D.; Ribero, D.; Di Tommaso, L.; Valle, J.W. Current status on cholangiocarcinoma and gallbladder cancer. Liver Cancer 2017, 6, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Werner, H.M.J.; Mills, G.B.; Ram, P.T. Cancer systems biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. Sequence analysis featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Anders, S.; Huber, W. Differential Analysis of Count Data—the DESeq2 Package. Genome Biol. 2014, 15, 10–1186. [Google Scholar]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 1–45. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Emmert-Streib, F.; Dehmer, M.; Haibe-Kains, B. Gene regulatory networks and their applications: Understanding biological and medical problems in terms of networks. Front. Cell Dev. Biol. 2014, 2, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Nada, B.S.; Ideker, T. Cytoscape: A Software Environment for Integrated Models. Genome Res. 1971, 13, 426. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.C.; Majewski, T.; Zhang, L.; Yao, H.; Bondaruk, J.; Zhang, S.; Wang, Z.; Lee, J.G.; Lee, S.; Cogdell, D.; et al. HHS Public Access. Ann. Glob. Health 2019, 27, 1781–1793. [Google Scholar] [CrossRef]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L. Scale-free networks: A decade and beyond. Science 2009, 325, 412–413. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.; George, J.T.; Tripathi, S.; Levine, H.; Jolly, M.K. Comparative Study of Transcriptomics-Based Scoring Metrics for the Epithelial-Hybrid-Mesenchymal Spectrum. Front. Bioeng. Biotechnol. 2020, 8, 220. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.K.; Shah, M.A. Targeting the cell cycle: A new approach to cancer therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Carnero, A. Targeting the cell cycle for cancer therapy. Br. J. Cancer 2002, 87, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef]
- Williams, G.H.; Stoeber, K. Cell cycle markers in clinical oncology. Curr. Opin. Cell Biol. 2007, 19, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J.; Bartkova, J. Perspective: Defects in cell cycle control and cancer. J. Pathol. 1999, 187, 95–99. [Google Scholar] [CrossRef]
- Addou-Klouche, L.; Adélaïde, J.; Finetti, P.; Cervera, N.; Ferrari, A.; Bekhouche, I.; Sircoulomb, F.; Sotiriou, C.; Viens, P.; Moulessehoul, S.; et al. Loss, mutation and deregulation of L3MBTL4 in breast cancers. Mol. Cancer 2010, 9, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvageau, M.; Sauvageau, G. Polycomb group proteins: Multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 2010, 7, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Gurvich, N.; Perna, F.; Farina, A.; Voza, F.; Menendez, S.; Hurwitz, J.; Nimer, S.D. L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability. Proc. Natl. Acad. Sci. USA 2010, 107, 22552–22557. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Jin, D.H.; Lee, J.; Kim, K.M.; Kim, S.; Kim, D.H.; Park, J. Overexpression of MAPK15 in gastric cancer is associated with copy number gain and contributes to the stability of c-Jun. Oncotarget 2015, 6, 20190–20203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colecchia, D.; Rossi, M.; Sasdelli, F.; Sanzone, S.; Strambi, A.; Chiariello, M. MAPK15 mediates BCR-ABL1-induced autophagy and regulates oncogene-dependent cell proliferation and tumor formation. Autophagy 2015, 11, 1790–1802. [Google Scholar] [CrossRef]
- Zhang, H.P.; Li, S.Y.; Wang, J.P.; Lin, J. Clinical significance and biological roles of cyclins in gastric cancer. Onco. Targets. Ther. 2018, 11, 6673–6685. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Jiang, X.; Zhang, Y.; Wang, S.; Chen, X.; Yu, X.; Ma, J.; Huang, X. Cyclin B2 Overexpression in Human Hepatocellular Carcinoma is Associated with Poor Prognosis. Arch. Med. Res. 2019, 50, 10–17. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, L.; Wang, Y.; Xi, Q.; Zhong, J.; Liu, J.; Yang, S.; Liu, R.; Wang, J.; Huang, M.; et al. High expression of CDC6 is associated with accelerated cell proliferation and poor prognosis of epithelial ovarian cancer. Pathol. Res. Pract. 2016, 212, 239–246. [Google Scholar] [CrossRef]
- Lim, N.; Townsend, P.A. Cdc6 as a novel target in cancer: Oncogenic potential, senescence and subcellular localisation. Int. J. Cancer 2020, 147, 1528–1534. [Google Scholar] [CrossRef]
- Youn, Y.; Lee, J.C.; Kim, J.; Kim, J.H.; Hwang, J.H. Cdc6 disruption leads to centrosome abnormalities and chromosome instability in pancreatic cancer cells. Sci. Rep. 2020, 10, 16518. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.N.; Vasiliou, V.; Nebert, D.W. Evolutionary divergence and functions of the ADAM and ADAMTS gene families. Hum. Genomics 2009, 4, 43–55. [Google Scholar] [CrossRef]
- Duffy, M.J.; McKiernan, E.; O’Donovan, N.; McGowan, P.M. Role of ADAMs in cancer formation and progression. Clin. Cancer Res. 2009, 15, 1140–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullooly, M.; McGowan, P.M.; Crown, J.; Duffy, M.J. The ADAMs family of proteases as targets for the treatment of cancer. Cancer Biol. Ther. 2016, 17, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhang, X.; Bidlingmaier, S.; Behrens, C.R.; Lee, N.-K.; Liu, B. ALPPL2 Is a Highly Specific and Targetable Tumor Cell Surface Antigen. Cancer Res. 2020, 80, 4552–4564. [Google Scholar] [CrossRef]
- Dua, P.; Kang, H.S.; Hong, S.; Tsao, M.; Kim, S.; Lee, D. Alkaline Phosphatase ALPPL-2 Is a Novel Pancreatic Carcinoma-Associated Protein. Cancer Res. 2013, 73, 1934–1946. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Yu, Q.; Li, H.; Xie, C.; Wu, Y.; Ma, D.; Sheng, P.; Dai, W. BIRC7 promotes epithelial-mesenchymal transition and metastasis in papillary thyroid carcinoma through restraining autophagy. Am. J. Cancer Res. 2020, 10, 78–94. [Google Scholar]
- Liang, J.; Zhao, W.; Tong, P.; Li, P.; Zhao, Y.; Li, H. Comprehensive molecular characterization of inhibitors of apoptosis proteins ( IAPs ) for therapeutic targeting in cancer. BMC Med. Genomics 2020, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Z.; Huang, S. BIRC7 and STC2 Expression Are Associated with Tumorigenesis and Poor Outcome in Extrahepatic Cholangiocarcinoma. Technol. Cancer Res.Treat. 2020, 19, 1533033820971676. [Google Scholar] [CrossRef]
- Jaiswal, R.; Jauhari, S.; Islamia, J.M. WDR88, CCDC11, and ARPP21 genes indulge profoundly in the desmoplastic retort to prostate and breast cancer metastasis. BioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control to GBC | GBC to Control | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Midnightblue | Royalblue | Salmon | Tan | Grey60 | |||||
| Gene | Weight | Gene | Weight | Gene | Weight | Gene | Weight | Gene | Weight |
| AL009178.3 | 2.87 | ADAM18 | 2.87 | MAPK15 | 12.51 | L3MBTL1 | 11.38 | ALPPL2 | 8.87 |
| SPATC1 | 2.71 | CNTN4 | 2.71 | TRAPPC9 | 12.08 | ZNF337-AS1 | 10.47 | PATE4 | 8.01 |
| CTSV | 2.70 | NUP62CL | 2.70 | OPLAH | 11.72 | AC099661.1 | 9.64 | AP00842.3 | 7.85 |
| AL353746.1 | 2.64 | QTRT2 | 2.64 | OTUD6B | 10.97 | AC240565 | 8.68 | GPATCH1 | 7.80 |
| AL360270.1 | 2.61 | LINCO1517 | 2.61 | TAF2 | 10.72 | C1QTNF | 8.14 | AP000977.1 | 7.06 |
| Control to GBC | GBC to Control | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Midnightblue | Royalblue | Salmon | Tan | Grey60 | |||||
| Gene | Degree | Gene | Degree | Gene | Degree | Gene | Degree | Gene | Degree |
| CDC6 | 30 | BIRC7 | 12 | CCNB2 | 30 | L3MBTL1 | 34 | WDR88 | 31 |
| MCM3 | 28 | CASP12 | 11 | CCNE2 | 25 | ABCG2 | 23 | RPL3 | 31 |
| SMURF1 | 22 | UBC | 6 | E2F5 | 22 | CTPS2 | 12 | TIC6 | 23 |
| PIK3AB | 17 | EPH7 | 6 | MAPK15 | 15 | SIM2 | 7 | FOXA1 | 22 |
| SHANK2 | 14 | LINGO2 | 5 | TONSL | 12 | PTP4A1 | 7 | HIST3H2A | 18 |
| Modules | Biological Processes (GO Terms) | Counts | Genes | p-Value |
|---|---|---|---|---|
| midnight blue | intracellular signal transduction | 4 | NEK11, DGKB, GUCY1B2, NUDT4 | 0.0390 |
| royalblue | negative regulation of DNA replication | 2 | S100A11, CDC6 | 0.0390 |
| salmon | dorsal/ventral axis specification | 2 | PAX6, RGS20 | 0.0370 |
| neuron migration | 3 | CELSR3, PAX6, PTK2 | 0.0420 | |
| negative regulation of keratinocytes proliferation | 2 | CTSV, EPPK1 | 0.0430 | |
| interphase of mitotic cell cycle | 4 | CCNB2, CCNE2, E2F5, TAF2 | 0.0382 | |
| grey60 | Negative regulation of translation | 2 | FXF1, EIFAK1 | 0.00824 |
| Cell fate commitment | 3 | FOXA1, HOXA11, TFAP2C | 0.0131 | |
| Developmental growth | HOXA11, TFAP2C, PLAC1 | 0.0186 | ||
| tan | planar cell polarity pathway involved in axon guidance | 2 | VANGL2, RYK | 0.0120 |
| Epidermal cell differentiation | 2 | OVOL2, SPINK5 | 0.0160 | |
| Negative regulation of serine type endopeptidase activity | 2 | SPINK1, SPINK5 | 0.0280 |
| Module | KEGG Pathways | Counts | Genes | p-Value |
|---|---|---|---|---|
| midnightblue | Glycerolipid metabolism | 2 | HLA-DMA | 0.00683 |
| Toxoplasmosis | 2 | DGKB, LIPC | 0.0222 | |
| Apoptosis | 2 | CTSV, CASP12 | 0.0313 | |
| Glycosaminoglycan degradation | 1 | HYAL4 | 0.0382 | |
| royalblue | Phosphotidyl inositol signaling | 2 | PIK3CB, ITPKA | 0.00762 |
| Cell cycle | 2 | MCM3, CDC6 | 0.0204 | |
| salmon | Cell cycle | 3 | E2F5, CCNB2, CCNE2 | 0.0450 |
| Small cell lungs cancer | 2 | CCNE2, PTK2 | 0.0385 | |
| P53 signaling | 2 | CCNE2, CCEB2 | 0.0240 | |
| Ubiquine biosynthesis | 1 | COQ2T | 0.0364 | |
| grey60 | Thiamine metaboilism | 1 | ALPPL2 | 0.0266 |
| Necroptosis | 2 | H2AW, RNF103-CHMP3 | 0.0292 | |
| Alcoholism | 2 | H2AW, H2BO1 | 0.0355 | |
| Histidine metabolism | 1 | ALDH3B2 | 0.0380 | |
| tan | Wnt signaling pathway | 2 | VANGL2, RYK | 0.0321 |
| Steroid biosynthesis | 1 | CYP24A1 | 0.0339 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, N.; Kshattry, M.; Mandal, S.; Jolly, M.K.; Bhattacharyya, D.K.; Barah, P. An Integrative Systems Biology Approach Identifies Molecular Signatures Associated with Gallbladder Cancer Pathogenesis. J. Clin. Med. 2021, 10, 3520. https://doi.org/10.3390/jcm10163520
Roy N, Kshattry M, Mandal S, Jolly MK, Bhattacharyya DK, Barah P. An Integrative Systems Biology Approach Identifies Molecular Signatures Associated with Gallbladder Cancer Pathogenesis. Journal of Clinical Medicine. 2021; 10(16):3520. https://doi.org/10.3390/jcm10163520
Chicago/Turabian StyleRoy, Nabanita, Mrinmoy Kshattry, Susmita Mandal, Mohit Kumar Jolly, Dhruba Kumar Bhattacharyya, and Pankaj Barah. 2021. "An Integrative Systems Biology Approach Identifies Molecular Signatures Associated with Gallbladder Cancer Pathogenesis" Journal of Clinical Medicine 10, no. 16: 3520. https://doi.org/10.3390/jcm10163520
APA StyleRoy, N., Kshattry, M., Mandal, S., Jolly, M. K., Bhattacharyya, D. K., & Barah, P. (2021). An Integrative Systems Biology Approach Identifies Molecular Signatures Associated with Gallbladder Cancer Pathogenesis. Journal of Clinical Medicine, 10(16), 3520. https://doi.org/10.3390/jcm10163520