
New Insights into Gastrointestinal Involvement in Late-Onset Pompe Disease: Lessons Learned from Bench and Bedside

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.1.1. PROMIS-GI Symptom Scales
2.1.2. GI-Focused Medical History
2.1.3. Statistical Analyses
2.2. GAAKO Mouse Model (6neo/6neo)
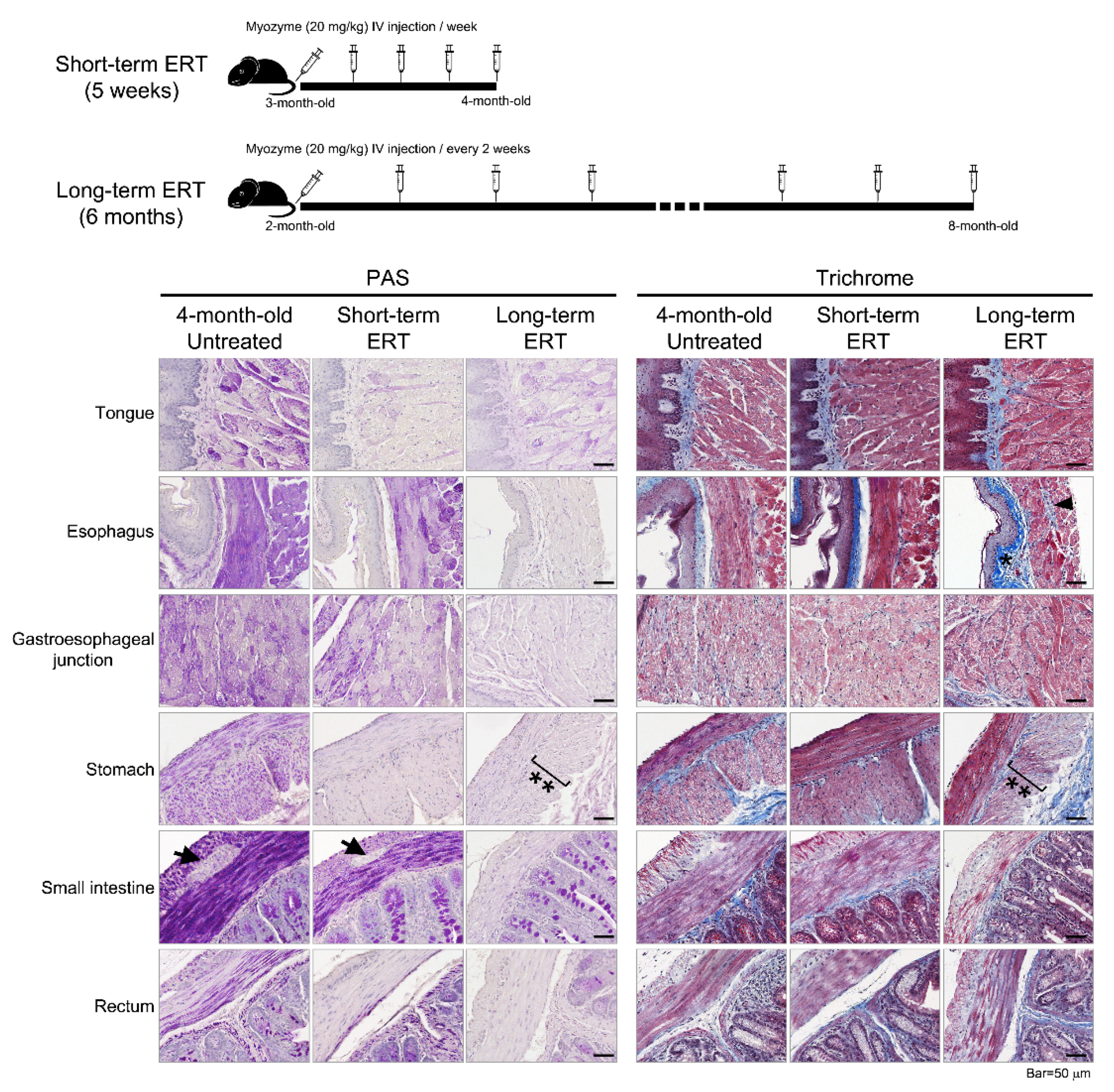
Histopathology
3. Results
3.1. Participants
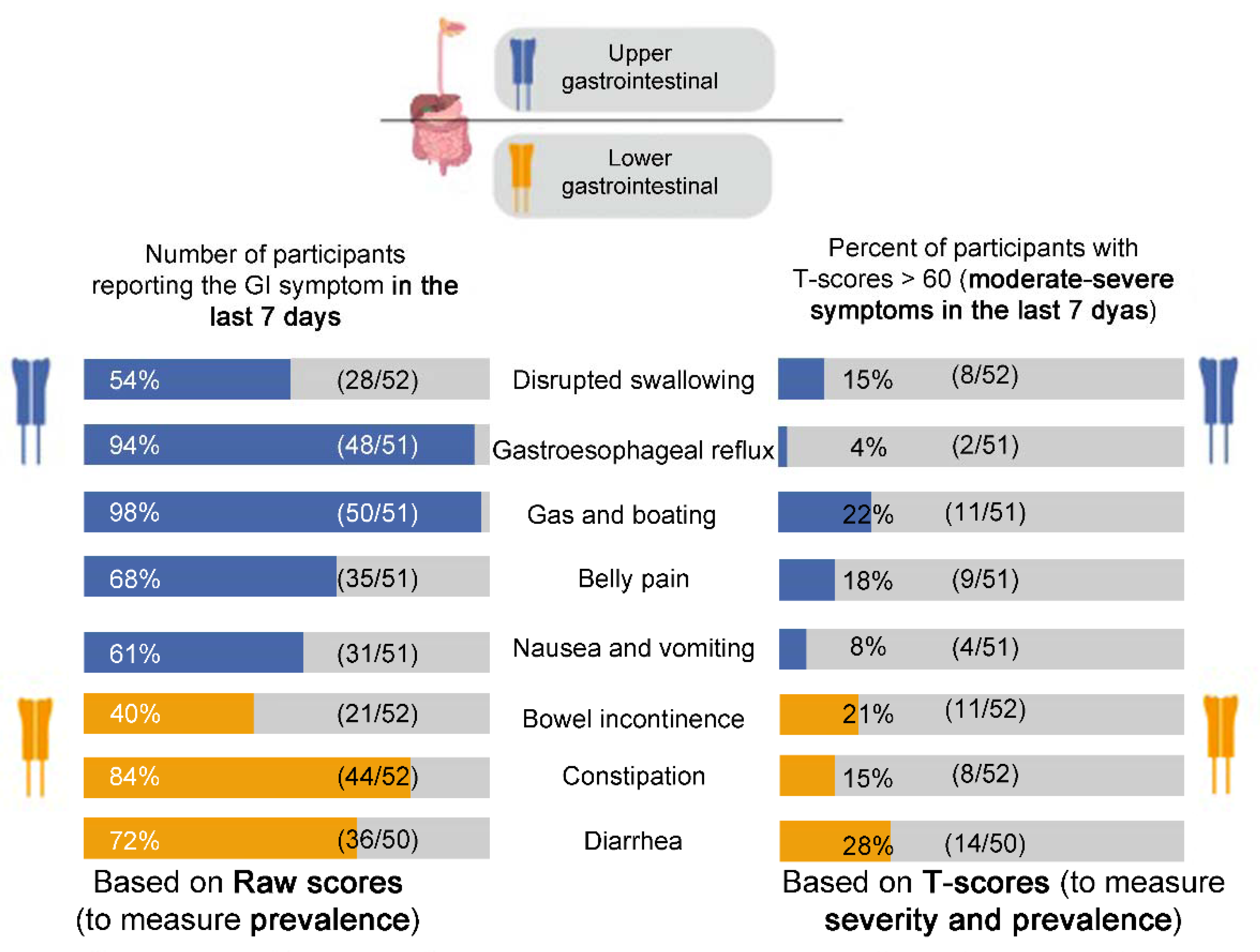
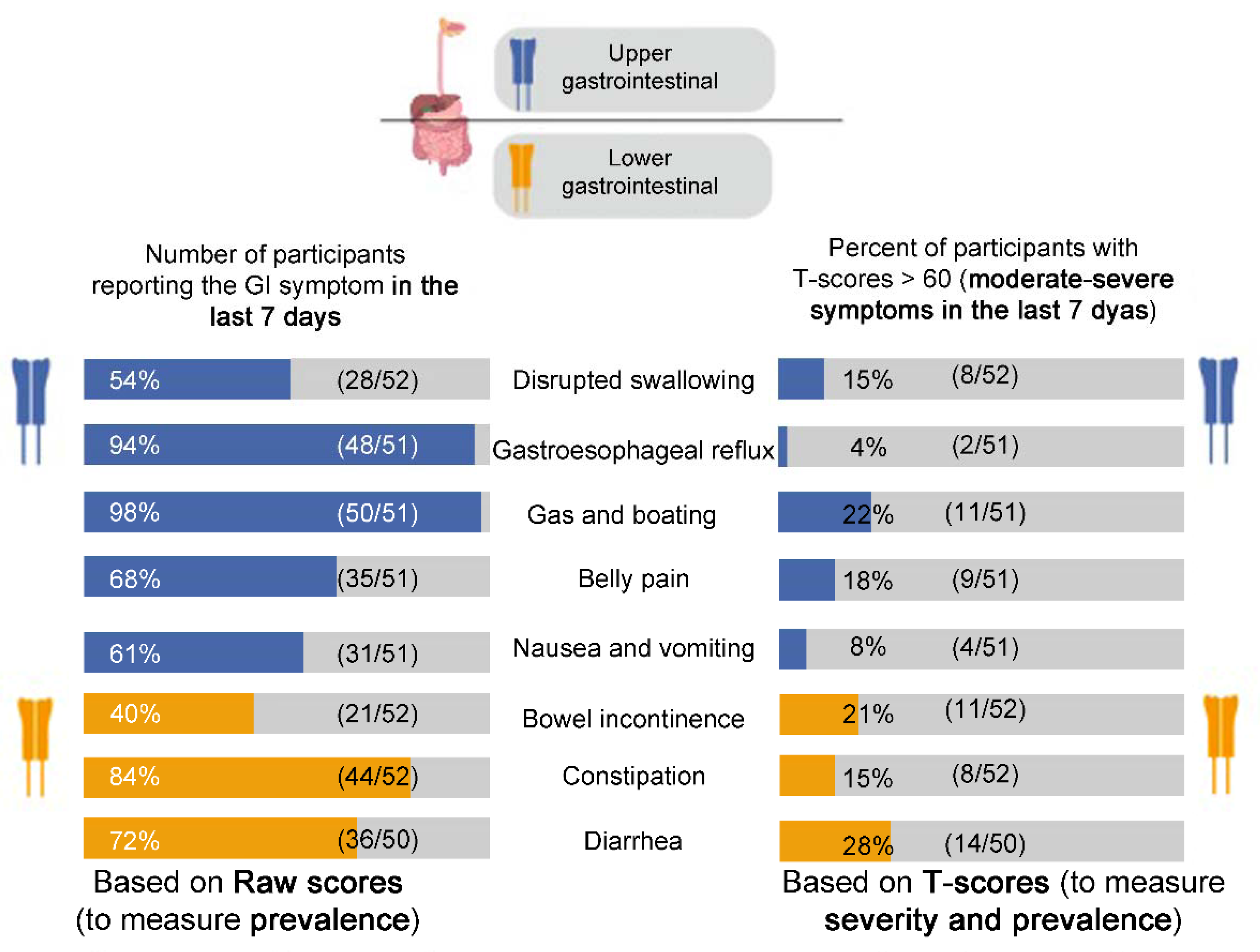
3.1.1. PROMIS-GI Symptom Scales
Raw Scores
T-Scores
3.1.2. GI-Focused Medical History
3.1.3. Statistical Analyses
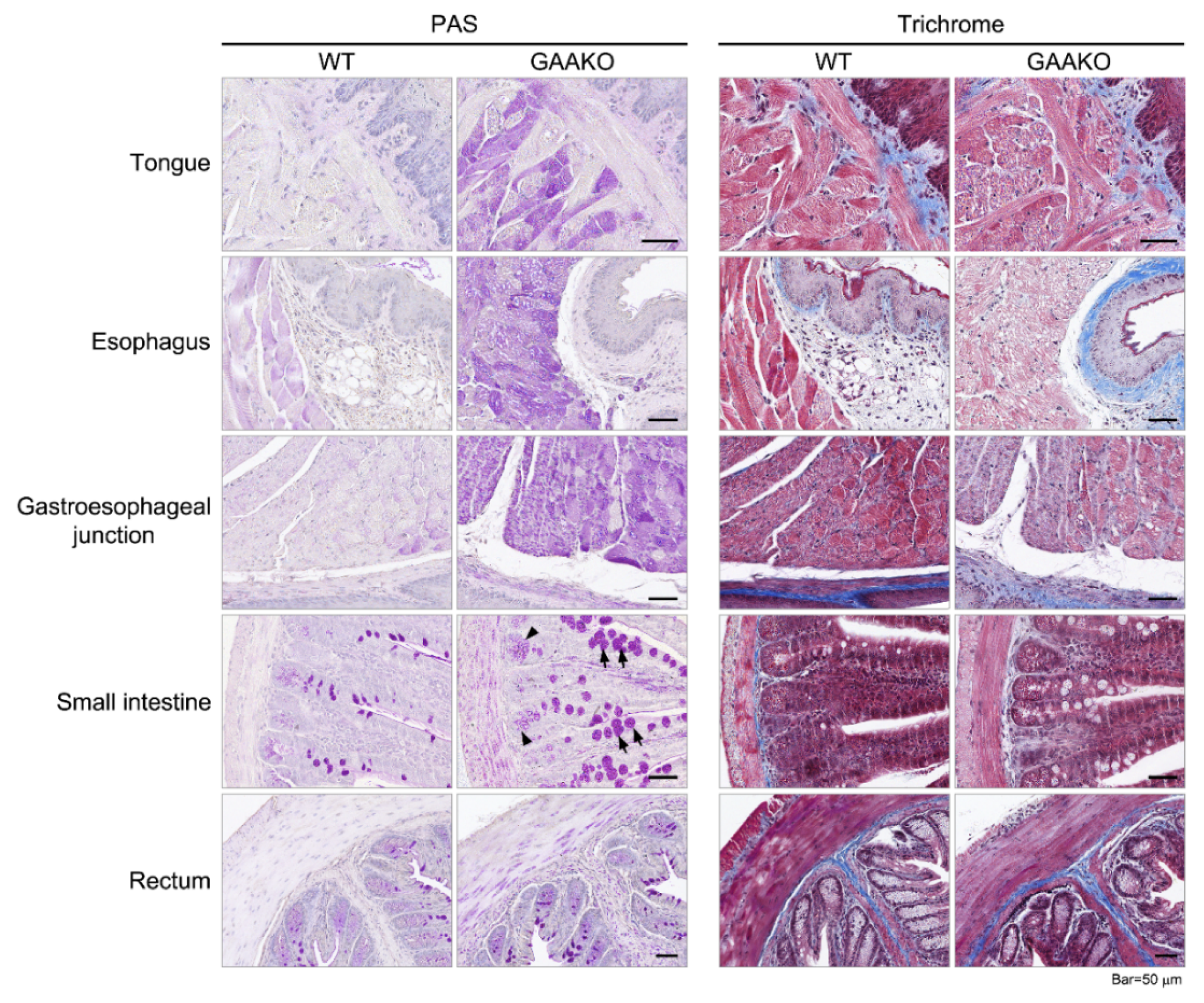
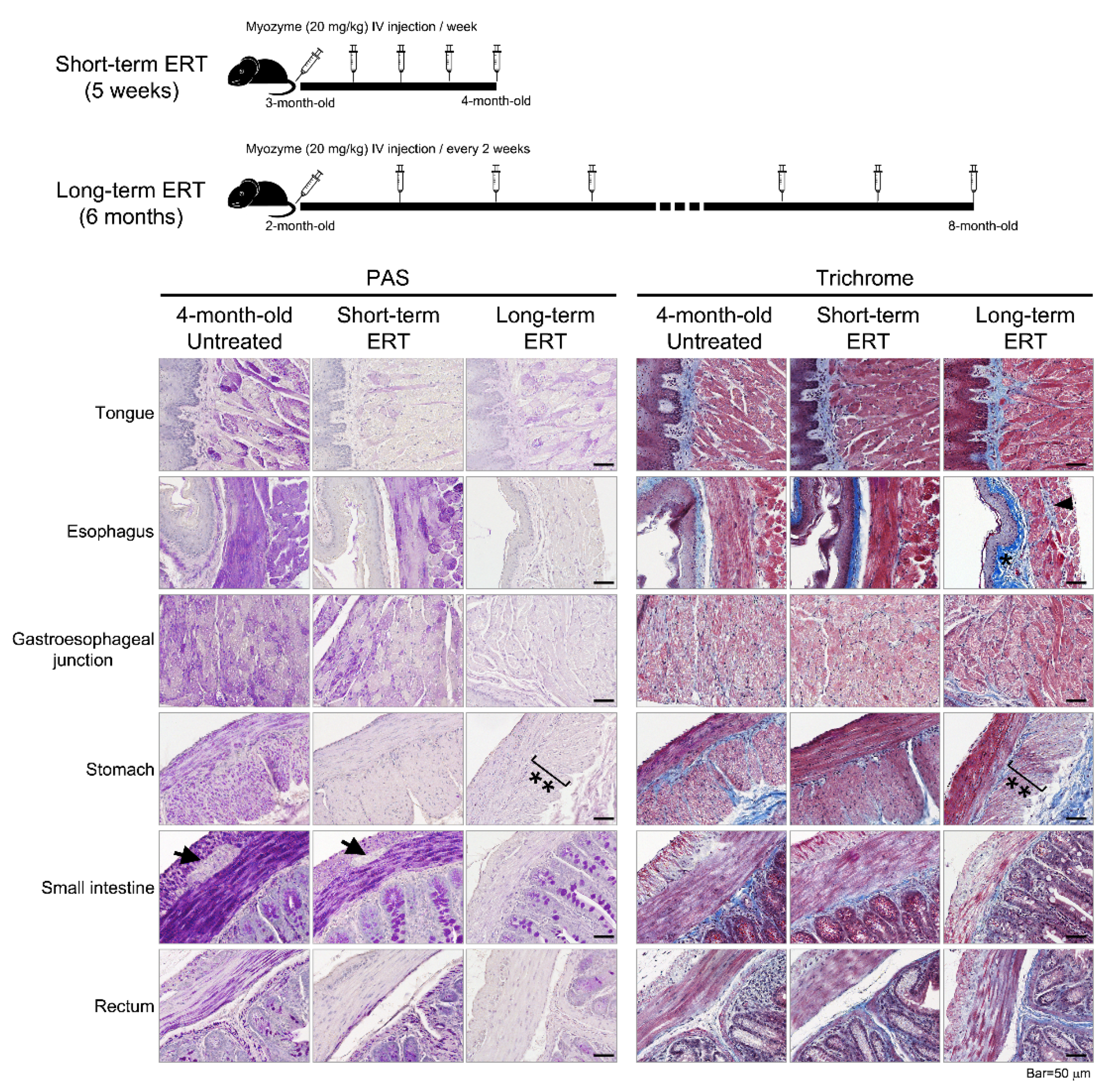
3.2. GAAKO Mouse Model (6neo/6neo)
Histopathology
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hers, H.G. Alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease). Biochem. J. 1963, 86, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Steiner, R.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.; Desai, A.K.; Kazi, Z.; Corey, K.; Austin, S.; Hobson-Webb, L.D.; Case, L.E.; Jones, H.N.; Kishnani, P.S. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017, 120, 163–172. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Corzo, D.; Nicolino, M.; Byrne, B.; Mandel, H.; Hwu, W.-L.; Leslie, N.; Levine, J.; Spencer, C.; McDonald, M.; et al. Recombinant human acid -glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology 2006, 68, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Montagnese, F.; Granata, F.; Musumeci, O.; Rodolico, C.; Mondello, S.; Barca, E.; Cucinotta, M.; Ciranni, A.; Longo, M.; Toscano, A. Intracranial arterial abnormalities in patients with late onset Pompe disease (LOPD). J. Inherit. Metab. Dis. 2016, 39, 391–398. [Google Scholar] [CrossRef]
- McCall, A.L.; Salemi, J.; Bhanap, P.; Strickland, L.M.; Elmallah, M.K. The impact of Pompe disease on smooth muscle: A review. J. Smooth Muscle Res. 2018, 54, 100–118. [Google Scholar] [CrossRef] [Green Version]
- Remiche, G.; Herbaut, A.-G.; Ronchi, D.; Lamperti, C.; Magri, F.; Moggio, M.; Bresolin, N.; Comi, G.P. Incontinence in Late-Onset Pompe Disease: An Underdiagnosed Treatable Condition. Eur. Neurol. 2012, 68, 75–78. [Google Scholar] [CrossRef] [PubMed]
- El-Gharbawy, A.H.; Bhat, G.; Murillo, J.E.; Thurberg, B.L.; Kampmann, C.; Mengel, K.-E.; Kishnani, P.S. Expanding the clinical spectrum of late-onset Pompe disease: Dilated arteriopathy involving the thoracic aorta, a novel vascular phenotype uncovered. Mol. Genet. Metab. 2011, 103, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Hobson-Webb, L.D.; Jones, H.N.; Kishnani, P.S. Oropharyngeal dysphagia may occur in late-onset Pompe disease, implicating bulbar muscle involvement. Neuromuscul. Disord. 2013, 23, 319–323. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Amartino, H.M.; Lindberg, C.; Miller, T.M.; Wilson, A.; Keutzer, J. Methods of diagnosis of patients with Pompe disease: Data from the Pompe Registry. Mol. Genet. Metab. 2014, 113, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Pardo, J.; Garcia-Sobrino, T.; López-Ferreiro, A. Gastrointestinal symptoms in late-onset Pompe disease: Early response to enzyme replacement therapy. J. Neurol. Sci. 2015, 353, 181–182. [Google Scholar] [CrossRef]
- Gesquière-Dando, A.; Attarian, S.; De Paula, A.M.; Pouget, J.; Salort-Campana, E. Fibromyalgia-like symptoms associated with irritable bowel syndrome: A challenging diagnosis of late-onset Pompe disease. Muscle Nerve 2015, 52, 300–304. [Google Scholar] [CrossRef]
- Karabul, N.; Skudlarek, A.; Berndt, J.; Kornblum, C.; Kley, R.A.; Wenninger, S.; Tiling, N.; Mengel, E.; Plöckinger, U.; Vorgerd, M.; et al. Urge Incontinence and Gastrointestinal Symptoms in Adult Patients with Pompe Disease: A Cross-Sectional Survey. JIMD Rep. 2014, 17, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, D.L.; Bialer, M.G.; Mehta, L.; Desnick, R.J. Pompe disease: Dramatic improvement in gastrointestinal function following enzyme replacement therapy. A report of three later-onset patients. Mol. Genet. Metab. 2010, 101, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S. Abnormalities of cerebral arteries are frequent in patients with late-onset Pompe disease. J. Neurol. 2010, 257, 1730–1733. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbecker, K.S.; Kirschner-Hermanns, R.; Kornblum, C.; Jaekel, A.; Anding, R.; Kohler, A. Urodynamic and clinical studies in patients with late-onset Pompe disease and lower urinary tract symptoms. Neurourol. Urodyn. 2020, 39, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- McNamara, E.R.; Austin, S.; Case, L.; Wiener, J.S.; Peterson, A.C.; Kishnani, P.S.; Zschocke, J. Expanding Our Understanding of Lower Urinary Tract Symptoms and Incontinence in Adults with Pompe Disease. JIMD Rep. 2014, 20, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Walt, J.D. The pattern of involvement of adult-onset acid maltase deficiency at autopsy. Muscle Nerve 1987, 10, 272–281. [Google Scholar] [CrossRef]
- Pena, L.D.M.; Proia, A.D.; Kishnani, P.S. Postmortem Findings and Clinical Correlates in Individuals with Infantile-Onset Pompe Disease. JIMD Rep. 2015, 23, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Hobson-Webb, L.D.; Proia, A.D.; Thurberg, B.L.; Banugaria, S.; Prater, S.N.; Kishnani, P.S. Autopsy findings in late-onset Pompe disease: A case report and systematic review of the literature. Mol. Genet. Metab. 2012, 106, 462–469. [Google Scholar] [CrossRef]
- Kobayashi, H.; Shimada, Y.; Ikegami, M.; Kawai, T.; Sakurai, K.; Urashima, T.; Ijima, M.; Fujiwara, M.; Kaneshiro, E.; Ohashi, T.; et al. Prognostic factors for the late onset Pompe disease with enzyme replacement therapy: From our experience of 4 cases including an autopsy case. Mol. Genet. Metab. 2010, 100, 14–19. [Google Scholar] [CrossRef]
- Bijvoet, A.G.; Van De Kamp, E.H.; Kroos, M.A.; Ding, J.-H.; Yang, B.Z.; Visser, P.; Bakker, C.E.; Verbeet, M.P.; Oostra, B.A.; Reuser, A.J.; et al. Generalized glycogen storage and cardiomegaly in a knockout mouse model of Pompe disease. Hum. Mol. Genet. 1998, 7, 53–62. [Google Scholar] [CrossRef]
- Raben, N.; Nagaraju, K.; Lee, E.; Kessler, P.; Byrne, B.; Lee, L.; LaMarca, M.; King, C.; Ward, J.; Sauer, B.; et al. Targeted Disruption of the Acid α-Glucosidase Gene in Mice Causes an Illness with Critical Features of Both Infantile and Adult Human Glycogen Storage Disease Type II. J. Biol. Chem. 1998, 273, 19086–19092. [Google Scholar] [CrossRef] [Green Version]
- Bijvoet, A.G.A. Recombinant Human Acid α-Glucosidase: High Level Production in Mouse Milk, Biochemical Characteristics, Correction of Enzyme Deficiency in GSDII KO Mice. Hum. Mol. Genet. 1998, 7, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Cella, D.; Riley, W.; Stone, A.; Rothrock, N.; Reeve, B.; Yount, S.; Amtmann, D.; Bode, R.; Buysse, D.; Choi, S.; et al. The Patient-Reported Outcomes Measurement Information System (PROMIS) developed and tested its first wave of adult self-reported health outcome item banks: 2005–2008. J. Clin. Epidemiol. 2010, 63, 1179–1194. [Google Scholar] [CrossRef] [Green Version]
- Rothrock, N.E.; Hays, R.D.; Spritzer, K.; Yount, S.E.; Riley, W.; Cella, D. Relative to the general US population, chronic diseases are associated with poorer health-related quality of life as measured by the Patient-Reported Outcomes Measurement Information System (PROMIS). J. Clin. Epidemiol. 2010, 63, 1195–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, D.; Hays, R.D.; Shreiner, A.B.; Melmed, G.Y.; Chang, L.; Khanna, P.P.; Bolus, R.; Whitman, C.; Paz, S.H.; Hays, T.; et al. Responsiveness to Change and Minimally Important Differences of the Patient-Reported Outcomes Measurement Information System Gastrointestinal Symptoms Scales. Dig. Dis. Sci. 2017, 62, 1186–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, B.M.R.; Hays, R.D.; Bolus, R.; Melmed, G.Y.; Chang, L.; Whitman, C.; Khanna, P.P.; Paz, S.H.; Hays, T.; Reise, S.; et al. Development of the NIH Patient-Reported Outcomes Measurement Information System (PROMIS) Gastrointestinal Symptom Scales. Am. J. Gastroenterol. 2014, 109, 1804–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almario, C.V.; Ballal, M.L.; Chey, W.D.; Nordstrom, C.; Khanna, D.; Spiegel, B.M.R. Burden of Gastrointestinal Symptoms in the United States: Results of a Nationally Representative Survey of Over 71,000 Americans. Am. J. Gastroenterol. 2018, 113, 1701–1710. [Google Scholar] [CrossRef]
- Lim, J.-A.; Yi, H.; Gao, F.; Raben, N.; Kishnani, P.S.; Sun, B. Intravenous Injection of an AAV-PHP.B Vector Encoding Human Acid α-Glucosidase Rescues Both Muscle and CNS Defects in Murine Pompe Disease. Mol. Ther. Methods Clin. Dev. 2019, 12, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Schoser, B.; Bilder, D.A.; Dimmock, D.; Gupta, D.; James, E.S.; Prasad, S. The humanistic burden of Pompe disease: Are there still unmet needs? A systematic review. BMC Neurol. 2017, 17, 202. [Google Scholar] [CrossRef] [Green Version]
- Ajay, D.; McNamara, E.R.; Austin, S.; Wiener, J.S.; Kishnani, P. Lower Urinary Tract Symptoms and Incontinence in Children with Pompe Disease. JIMD Rep. 2015, 28, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Post, M. Definitions of Quality of Life: What Has Happened and How to Move On. Top. Spinal Cord Inj. Rehabil. 2014, 20, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Bodamer, O.A.; Scott, C.R.; Giugliani, R. Pompe Disease Newborn Screening Working Group; on behalf of the Pompe Disease Newborn Screening Working Group Newborn Screening for Pompe Disease. Pediatrics 2017, 140, S4–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harfouche, M.; Kishnani, P.S.; Krusinska, E.; Gault, J.; Sitaraman, S.; Sowinski, A.; Katz, I.; Austin, S.; Goldstein, M.; Mulberg, A.E. Use of the patient-reported outcomes measurement information system (PROMIS®) to assess late-onset Pompe disease severity. J. Patient-Reported Outcomes 2020, 4, 1–11. [Google Scholar] [CrossRef]
- Genzyme Corporation. Cambridge, MA. Lumizyme® (Alglucosidase Alfa) [Packet Insert] [Most Recent Major Changes in 02/2020]. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/125291lbl.pdf (accessed on 15 March 2021).
- Banikazemi, M.; Ullman, T.; Desnick, R.J. Gastrointestinal manifestations of Fabry disease: Clinical response to enzyme replacement therapy. Mol. Genet. Metab. 2005, 85, 255–259. [Google Scholar] [CrossRef]
- Verderese, C.L. Gaucher’s disease: A pilot study of the symptomatic responses to enzyme replacement therapy. J. Neurosci. Nurs. 1993, 25, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Funk, B. Expression of the insulin-like growth factor-II/mannose-6-phosphate receptor in multiple human tissues during fetal life and early infancy. J. Clin. Endocrinol. Metab. 1992, 75, 424–431. [Google Scholar] [PubMed] [Green Version]
- Cardone, M.; Porto, C.; Tarallo, A.; Vicinanza, M.; Rossi, B.; Polishchuk, E.; Donaudy, F.; Andria, G.; De Matteis, M.A.; Parenti, G. Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics 2008, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Shea, L.; Raben, N. Autophagy in skeletal muscle: Implications for Pompe disease. Int. J. Clin. Pharmacol. Ther. 2009, 47, S42–S47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margeta, M. Autophagy Defects in Skeletal Myopathies. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 261–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoser, B. Pompe disease: What are we missing? Ann. Transl. Med. 2019, 7, 292. [Google Scholar] [CrossRef] [PubMed]
- Meinke, P.; Limmer, S.; Hintze, S.; Schoser, B. Assessing metabolic profiles in human myoblasts from patients with late-onset Pompe disease. Ann. Transl. Med. 2019, 7, 277. [Google Scholar] [CrossRef] [PubMed]
- PROMIS. Available online: http://www.healthmeasures.net/explore-measurement-systems/promis (accessed on 15 February 2021).
- Korlimarla, A.; Lim, J.-A.; Kishnani, P.S.; Sun, B. An emerging phenotype of central nervous system involvement in Pompe disease: From bench to bedside and beyond. Ann. Transl. Med. 2019, 7, 289. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Study duration Total number of patients (n)
| 1 year, 3 months 58 n = 52 n = 38 (32/52 who also completed the PROMIS-GI + additional 6 patients who only had GI-focused medical history in their medical records) | |
| Demographics | Median age = 51.5 ± 15.5 years (age range: 18–79 years) 35 females, 23 males | |
| Patients on ERT (treated group) | Patients not on ERT (untreated group) | |
|
| |
| For longitudinal analysis of PROMIS-GI scales | n = 18 (1 baseline and 1 follow-up) n = 1 (1 baseline and 2 follow-ups) | |
| PROMIS-GI Symptom Domain | Prevalence (Using Raw Scores) | Prevalence/Severity [Using Mean T-Scores (SD)] | Minimally Important Differences (MIDs) in T-Scores (n = 19) *** | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Study Population | Reference Population [29] | Study Population | Reference Population [28] | ||||||||
| Patients with Pompe Disease n = 52 | GP n = 1177 | GI Clinical Sample n = 865 | Patients with Pompe Disease | GP | GI Clinical Sample | Estimated Reference Values for MIDs [29] | n with Improvement | n with Worsening | n with No MID in T-Scores | ||
| Upper GI | disrupted swallowing | 54% | 5.8% | u | 49.15 (9.60) | 50 (10) | 51 (10) | u | N/A | ||
| gastroesophageal reflux * | 94% | 16–30.9% | 33% | 46.76 (8.06) | 50 (10) | 51 (10) | +5 points for improvement, −1 point for worsening | 4 | 6 | 9 | |
| gas/bloating | 98% | 20.6% | u | 54.57 (7.68) | 50 (10) | 57 (10) | ±6 points | 4 | 4 | 10 | |
| belly pain | 68% | 24.8% | u | 46.34 (12.06) | 50 (10) | 57 (11) | ±6 points | 4 | 5 | 9 | |
| nausea/vomiting | 61% | 9.5–19% | 24% | 47.07 (7.35) | 50 (10) | 53 (10) | u | N/A | |||
| Lower GI | constipation * | 84% | 19.7–47% | 39% total | 50.05 (8.54) | 50 (10) | 54 (10) | ±5–6 points | 2 | 4 | 12 |
| diarrhea | 72% | 6.6–20% | u | 52.18 (10.38) | 50 (10) | 56 (11) | ±5–6 points | 3 | 2 | 13 | |
| incontinence | 40% | 8.3% | u | 48.67 (9.25) | 50 (10) | 53 (11) | u | N/A | |||
| Other GI condition [28] | IBS * IBD * systemic sclerosis * others | N/A | 11% 4% 1% 47% | 40% 28% 18% 39% ** | N/A | N/A | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korlimarla, A.; Lim, J.-A.; McIntosh, P.; Zimmerman, K.; Sun, B.D.; Kishnani, P.S. New Insights into Gastrointestinal Involvement in Late-Onset Pompe Disease: Lessons Learned from Bench and Bedside. J. Clin. Med. 2021, 10, 3395. https://doi.org/10.3390/jcm10153395
Korlimarla A, Lim J-A, McIntosh P, Zimmerman K, Sun BD, Kishnani PS. New Insights into Gastrointestinal Involvement in Late-Onset Pompe Disease: Lessons Learned from Bench and Bedside. Journal of Clinical Medicine. 2021; 10(15):3395. https://doi.org/10.3390/jcm10153395
Chicago/Turabian StyleKorlimarla, Aditi, Jeong-A Lim, Paul McIntosh, Kanecia Zimmerman, Baodong D. Sun, and Priya S. Kishnani. 2021. "New Insights into Gastrointestinal Involvement in Late-Onset Pompe Disease: Lessons Learned from Bench and Bedside" Journal of Clinical Medicine 10, no. 15: 3395. https://doi.org/10.3390/jcm10153395
APA StyleKorlimarla, A., Lim, J.-A., McIntosh, P., Zimmerman, K., Sun, B. D., & Kishnani, P. S. (2021). New Insights into Gastrointestinal Involvement in Late-Onset Pompe Disease: Lessons Learned from Bench and Bedside. Journal of Clinical Medicine, 10(15), 3395. https://doi.org/10.3390/jcm10153395