
Adiponectin Modulation by Genotype and Maternal Choline Supplementation in a Mouse Model of Down Syndrome and Alzheimer’s Disease



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Maternal Diet Protocol
2.2. Tissue Preparation
2.3. RNA Purification
2.4. RT-qPCR
2.5. Protein Assays
3. Results
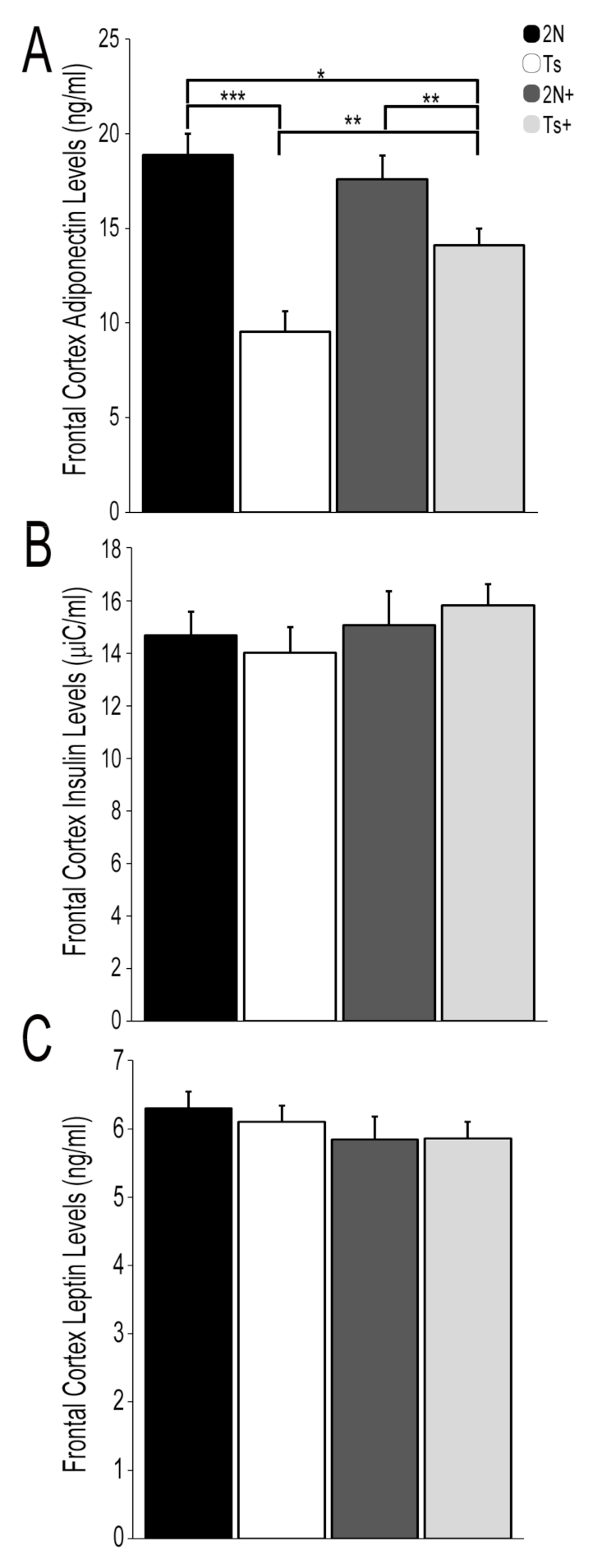
3.1. Protein Hormone Levels in Trisomic Mice and the Impact of MCS
3.2. APN Receptor mRNA and Protein Levels in Trisomic Mice and the Impact of MCS
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bittles, A.H.; Bower, C.; Hussain, R.; Glasson, E.J. The four ages of Down syndrome. Eur. J. Public Health 2006, 17, 221–225. [Google Scholar] [CrossRef]
- So, S.A.; Urbano, R.C.; Hodapp, R.M. Hospitalizations of infants and young children with Down syndrome: Evidence from inpatient person-records from a statewide administrative database. J. Intellect. Disabil. Res. 2007, 51, 1030–1038. [Google Scholar] [CrossRef]
- Lott, I.T. Neurological phenotypes for Down syndrome across the life span. Chang. Brains Appl. Brain Plast. Adv. Recover. Hum. Abil. 2012, 197, 101–121. [Google Scholar] [CrossRef]
- Presson, A.P.; Partyka, G.; Jensen, K.M.; Devine, O.J.; Rasmussen, S.A.; McCabe, L.L.; McCabe, E.R. Current estimate of Down syndrome population prevalence in the United States. J. Pediatr. 2013, 163, 1163–1168. [Google Scholar] [CrossRef]
- Mai, C.T.; Isenburg, J.L.; Canfield, M.A.; Meyer, R.E.; Correa, A.; Alverson, C.J.; Lupo, P.J.; Riehle-Colarusso, T.; Cho, S.J.; Aggarwal, D.; et al. National population-based estimates for major birth defects, 2010–2014. Birth Defects Res. 2019, 111, 1420–1435. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Gridley, G.; Cnattingius, S.; Mellemkjaer, L.; Linet, M.; Adami, H.-O.; Olsen, J.H.; Nyren, O.; Fraumeni, J.F. Mortality and Cancer Incidence Among Individuals with Down Syndrome. Arch. Intern. Med. 2003, 163, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Dick, M.B.; Doran, E.; Phelan, M.; Lott, I.T. Cognitive Profiles on the Severe Impairment Battery Are Similar in Alzheimer Disease and Down Syndrome with Dementia. Alzheimer Dis. Assoc. Disord. 2016, 30, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, R.; Guidi, S.; Ciani, E. Is it possible to improve neurodevelopmental abnormalities in Down syndrome? Rev. Neurosci. 2011, 22, 419–455. [Google Scholar] [CrossRef] [PubMed]
- Strupp, B.J.; Powers, B.E.; Velazquez, R.; Ash, J.A.; Kelley, C.M.; Alldred, M.J.; Strawderman, M.; Caudill, M.A.; Mufson, E.J.; Ginsberg, S.D. Maternal choline supplementation: A potential prenatal treatment for Down syndrome and Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Annus, T.; Wilson, L.R.; Hong, Y.T.; Acosta-Cabronero, J.; Fryer, T.D.; Cardenas-Blanco, A.; Smith, R.; Boros, I.; Coles, J.P.; Aigbirhio, F.I.; et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimer’s Dement. 2016, 12, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Beacher, F.; Daly, E.; Simmons, A.; Prasher, V.; Morris, R.; Robinson, C.; Lovestone, S.; Murphy, K.; Murphy, D.G.M. Alzheimer’s disease and Down’s syndrome: An in vivo MRI study. Psychol. Med. 2009, 39, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.C. Alzheimer disease: Treatment of Alzheimer disease in Down syndrome. Nat. Rev. Neurol. 2012, 8, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Oster-Granite, M.L.; Reeves, R.H.; Gearhart, J.D. Down syndrome, Alzheimer’s disease and the trisomy 16 mouse. Trends Neurosci. 1988, 11, 390–394. [Google Scholar] [CrossRef]
- Hartley, D.; Blumenthal, T.; Carrillo, M.; DiPaolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.-C.; Iqbal, K.; Krams, M.; Wisniewski, T. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimer’s Dement. 2015, 11, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T.; Dierssen, M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010, 9, 623–633. [Google Scholar] [CrossRef]
- Mann DM, A.; Yates, P.O.; Marcyniuk, B. Alzheimer’s presenile dementia, senile dementia of Alzheimer type and Down’s syndrome in middle age form an age related continuum of pathological changes. Neuropathol. Appl. Neurobiol. 1984, 10, 185–207. [Google Scholar] [CrossRef]
- Rueda, N.; Flórez, J.; Martínez-Cué, C. Mouse Models of Down Syndrome as a Tool to Unravel the Causes of Mental Disabilities. Neural Plast. 2012, 2012, 1–26. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Kleschevnikov, A.; Masliah, E.; Wu, C.; Takimoto-Kimura, R.; Salehi, A.; Mobley, W.C. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2009, 512, 453–466. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Masliah, E.; Kleschevnikov, A.; Villar, A.J.; Epstein, C.J.; Salehi, A.; Mobley, W.C. Synaptic structural abnormalities in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2004, 480, 281–298. [Google Scholar] [CrossRef]
- Granholm AC, E.; Sanders, L.A.; Crnic, L.S. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down’s syndrome. Exp. Neurol. 2000, 161, 647–663. [Google Scholar] [CrossRef]
- Kelley, C.M.; Powers, B.E.; Velázquez, R.; Ash, J.A.; Ginsberg, S.D.; Strupp, B.J.; Mufson, E.J. Sex Differences in the Cholinergic Basal Forebrain in the Ts65Dn Mouse Model of Down Syndrome and Alzheimer’s Disease. Brain Pathol. 2013, 24, 33–44. [Google Scholar] [CrossRef]
- Kelley, C.M.; Powers, B.E.; Velazquez, R.; Ash, J.A.; Ginsberg, S.; Strupp, B.J.; Mufson, E.J. Maternal choline supplementation differentially alters the basal forebrain cholinergic system of young-adult Ts65Dn and disomic mice. J. Comp. Neurol. 2014, 522, 1390–1410. [Google Scholar] [CrossRef] [PubMed]
- Tenneti, N.; Dayal, D.; Sharda, S.; Panigrahi, I.; Didi, M.; Attri, S.V.; Sachdeva, N.; Bhalla, A.K. Concentrations of leptin, adiponectin and other metabolic parameters in non-obese children with Down syndrome. J. Pediatr. Endocrinol. Metab. 2017, 30, 831–837. [Google Scholar] [CrossRef]
- Dierssen, M.; Fructuoso, M.; De Lagrán, M.M.; Perluigi, M.; Barone, E. Down Syndrome Is a Metabolic Disease: Altered Insulin Signaling Mediates Peripheral and Brain Dysfunctions. Front. Neurosci. 2020, 14, 670. [Google Scholar] [CrossRef] [PubMed]
- Milunsky, A.; Neurath, P.W. Diabetes mellitus in Down’s syndrome. Arch. Environ. Health Int. J. 1968, 17, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Taggart, L.; Coates, V.; Truesdale-Kennedy, M. Management and quality indicators of diabetes mellitus in people with intellectual disabilities. J. Intellect. Disabil. Res. 2012, 57, 1152–1163. [Google Scholar] [CrossRef]
- Capone, G.; Stephens, M.; Santoro, S.; Chicoine, B.; Bulova, P.; Peterson, M.; Jasien, J.; Smith, A.J. Down Syndrome Medical Interest Group (DSMIG-USA) Adult Health Workgroup Co-occurring medical conditions in adults with Down syndrome: A systematic review toward the development of health care guidelines. Part II. Am. J. Med. Genet. Part A 2020, 182, 1832–1845. [Google Scholar] [CrossRef]
- Leibson, C.L.; Rocca, W.A.; Hanson, V.A.; Cha, R.; Kokmen, E.; O’Brien, P.C.; Palumbo, P.J. Risk of Dementia among Persons with Diabetes Mellitus: A Population-based Cohort Study. Am. J. Epidemiol. 1997, 145, 301–308. [Google Scholar] [CrossRef]
- Reguero, M.; de Cedrón, M.G.; Wagner, S.; Reglero, G.; Quintela, J.; de Molina, A.R. Precision Nutrition to Activate Thermogenesis as a Complementary Approach to Target Obesity and Associated-Metabolic-Disorders. Cancers 2021, 13, 866. [Google Scholar] [CrossRef]
- Zhang, C.; Cai, W.; Zhao, H.; Zhu, M.; Cui, J.; Sun, Z. Effect of gastric bypass on BMI and lipid metabolism in type 2 diabetes mellitus. Artif. Cells Nanomed. Biotechnol. 2020, 48, 903–911. [Google Scholar] [CrossRef]
- Trombetta, B.; Carlyle, B.C.; Koenig, A.M.; Shaw, L.M.; Trojanowski, J.Q.; Wolk, D.A.; Locascio, J.J.; Arnold, S.E. The technical reliability and biotemporal stability of cerebrospinal fluid biomarkers for profiling multiple pathophysiologies in Alzheimer’s disease. PLoS ONE 2018, 13, e0193707. [Google Scholar] [CrossRef]
- Giordano, V.; Peluso, G.; Iannuccelli, M.; Benatti, P.; Nicolai, R.; Calvani, M. Systemic and Brain Metabolic Dysfunction as a New Paradigm for Approaching Alzheimer’s Dementia. Neurochem. Res. 2006, 32, 555–567. [Google Scholar] [CrossRef]
- Gutierrez-Hervas, A.; Gómez-Martínez, S.; Izquierdo-Gomez, R.; Veiga, O.L.; Perez-Bey, A.; Castro-Piñero, J.; Marcos, A. Inflammation and fatness in adolescents with and without Down syndrome: UP & DOWN study. J. Intellect. Disabil. Res. 2019, 64, 170–179. [Google Scholar] [CrossRef]
- Fructuoso, M.; Rachdi, L.; Philippe, E.; Denis, R.; Magnan, C.; Le Stunff, H.; Janel, N.; Dierssen, M. Increased levels of inflammatory plasma markers and obesity risk in a mouse model of Down syndrome. Free Radic. Biol. Med. 2018, 114, 122–130. [Google Scholar] [CrossRef]
- Peiris, H.; Duffield, M.D.; Fadista, J.; Jessup, C.F.; Kashmir, V.; Genders, A.J.; McGee, S.L.; Martin, A.M.; Saiedi, M.; Morton, N.; et al. A Syntenic Cross Species Aneuploidy Genetic Screen Links RCAN1 Expression to β-Cell Mitochondrial Dysfunction in Type 2 Diabetes. PLoS Genet. 2016, 12, e1006033. [Google Scholar] [CrossRef]
- Hölscher, C. Insulin Signaling Impairment in the Brain as a Risk Factor in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, X.; Yu, S.; Zheng, R. The Leptin Signaling. Adv. Exp. Med. Biol. 2018, 1090, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Galaz, C.; Fernández-Agulló, T.; Campoy, F.; Arribas, C.; Gallardo, N.; Andrés, A.; Ros, M.; Carrascosa, J.M. Decreased leptin uptake in hypothalamic nuclei with ageing in Wistar rats. J. Endocrinol. 2001, 171, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.; Oldreive, C.; Harvey, J. Neuroprotective actions of leptin on central and peripheral neurons in vitro. Neuroscience 2008, 154, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Signore, A.P.; Gao, Y.; Wang, S.; Zhang, F.; Hastings, T.; Yin, X.-M.; Chen, J. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J. Biol. Chem. 2007, 282, 34479–34491. [Google Scholar] [CrossRef]
- Guo, Z.; Jiang, H.; Xu, X.; Duan, W.; Mattson, M.P. Leptin-mediated Cell Survival Signaling in Hippocampal Neurons Mediated by JAK STAT3 and Mitochondrial Stabilization. J. Biol. Chem. 2008, 283, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gonzalez, R.; Antequera, D.; Vargas, T.; Spuch, C.; Bolós, M.; Carro, E. Leptin induces proliferation of neuronal progenitors and neuroprotection in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 24, 17–25. [Google Scholar] [CrossRef]
- Wang, S.; Yao, Q.; Wan, Y.; Wang, J.; Huang, C.; Li, D.; Yang, B. Adiponectin reduces brain injury after intracerebral hemorrhage by reducing NLRP3 inflammasome expression. Int. J. Neurosci. 2019, 130, 301–308. [Google Scholar] [CrossRef]
- Jeon, B.T.; Shin, H.J.; Kim, J.B.; Kim, Y.K.; Lee, D.H.; Kim, K.H.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Adiponectin protects hippocampal neurons against kainic acid-induced excitotoxicity. Brain Res. Rev. 2009, 61, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Xing, S.; Chen, C. Adiponectin gene polymorphisms contributes to ischemic stroke risk: A meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 178–184. [Google Scholar] [CrossRef]
- Zhang, D.; Guo, M.; Zhang, W.; Lu, X.Y. Adiponectin stimulates proliferation of adult hippocampal neural stem/progenitor cells through activation of p38 mitogen-activated protein kinase (p38MAPK)/glycogen synthase kinase 3β (GSK-3β)/β-catenin signaling cascade. J. Biol. Chem. 2011, 286, 44913–44920. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, X.; Lu, X.-Y. Adiponectin Exerts Neurotrophic Effects on Dendritic Arborization, Spinogenesis, and Neurogenesis of the Dentate Gyrus of Male Mice. Endocrinology 2016, 157, 2853–2869. [Google Scholar] [CrossRef]
- Ng, R.C.-L.; Cheng, O.-Y.; Jian, M.; Kwan, J.S.-C.; Ho, P.W.-L.; Cheng, K.K.-Y.; Yeung, P.K.K.; Zhou, L.L.; Hoo, R.L.-C.; Chung, S.K.; et al. Chronic adiponectin deficiency leads to Alzheimer’s disease-like cognitive impairments and pathologies through AMPK inactivation and cerebral insulin resistance in aged mice. Mol. Neurodegener. 2016, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Valentini, D.; Alisi, A.; di Camillo, C.; Sartorelli, M.; Crudele, A.; Bartuli, A.; Nobili, V.; Villani, A. Nonalcoholic Fatty Liver Disease in Italian Children with Down Syndrome: Prevalence and Correlation with Obesity-Related Features. J. Pediatr. 2017, 189, 92–97.e1. [Google Scholar] [CrossRef]
- Corsi, M.M.; Dogliotti, G.; Pedroni, F.; Galliera, E.; Malavazos, A.E.; Villa, R.; Chiappelli, M.; Licastro, F. Adipocytokines in Down’s syndrome, an atheroma-free model: Role of adiponectin. Arch. Gerontol. Geriatr. 2009, 48, 106–109. [Google Scholar] [CrossRef]
- Waragai, M.; Adame, A.; Trinh, I.; Sekiyama, K.; Takamatsu, Y.; Une, K.; Masliah, E.; Hashimoto, M. Possible Involvement of Adiponectin, the Anti-Diabetes Molecule, in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 52, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Van Himbergen, T.M.; Beiser, A.S.; Ai, M.; Seshadri, S.; Otokozawa, S.; Au, R.; Thongtang, N.; Wolf, P.A.; Schaefer, E.J.; Schaefer, E.J. Biomarkers for insulin resistance and inflammation and the risk for all-cause dementia and Alzheimer disease: Results from the Framingham Heart Study. Arch. Neurol. 2012, 69, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.L.; Diniz, B.S.; Campos, A.C.; Miranda, A.S.; Rocha, N.P.; Talib, L.L.; Gattaz, W.F.; Forlenza, O.V. Decreased Levels of Circulating Adiponectin in Mild Cognitive Impairment and Alzheimer’s Disease. NeuroMolecular Med. 2012, 15, 115–121. [Google Scholar] [CrossRef]
- Une, K.; Takei, Y.A.; Tomita, N.; Asamura, T.; Ohrui, T.; Furukawa, K.; Arai, H. Adiponectin in plasma and cerebrospinal fluid in MCI and Alzheimer’s disease. Eur. J. Neurol. 2010, 18, 1006–1009. [Google Scholar] [CrossRef]
- Gilbert, T.; Roche, S.; Blond, E.; Bar, J.Y.; Drai, J.; Cuerq, C.; Haution-Bitker, M.; Ecochard, R.; Bonnefoy, M. Association between peripheral leptin and adiponectin levels and cognitive decline in patients with neurocognitive disorders ≥ 65 years. J. Alzheimer’s Dis. 2018, 66, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Tian, J.; Du, H. Mitochondrial Dysfunction and Synaptic Transmission Failure in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1071–1086. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Niculescu, M.D. Perinatal choline influences brain structure and function. Nutr. Rev. 2006, 64, 197–203. [Google Scholar] [CrossRef]
- Meck, W.H.; Williams, C.L. Metabolic imprinting of choline by its availability during gestation: Implications for memory and attentional processing across the lifespan. Neurosci. Biobehav. Rev. 2003, 27, 385–399. [Google Scholar] [CrossRef]
- Ash, J.A.; Velazquez, R.; Kelley, C.M.; Powers, B.E.; Ginsberg, S.; Mufson, E.J.; Strupp, B.J. Maternal choline supplementation improves spatial mapping and increases basal forebrain cholinergic neuron number and size in aged Ts65Dn mice. Neurobiol. Dis. 2014, 70, 32–42. [Google Scholar] [CrossRef]
- Kelley, C.M.; Ash, J.A.; Powers, B.E.; Velázquez, R.; Alldred, M.J.; Ikonomovic, M.D.; Ginsberg, S.D.; Strupp, B.J.; Mufson, E.J. Effects of Maternal Choline Supplementation on the Septohippocampal Cholinergic System in the Ts65Dn Mouse Model of Down Syndrome. Curr. Alzheimer Res. 2015, 13, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Powers, B.E.; Kelley, C.M.; Velazquez, R.; Ash, J.A.; Strawderman, M.S.; Alldred, M.J.; Ginsberg, S.D.; Mufson, E.J.; Strupp, B.J. Maternal choline supplementation in a mouse model of Down syndrome: Effects on attention and nucleus basalis/substantia innominata neuron morphology in adult offspring. Neuroscience 2017, 340, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Powers, B.E.; Velázquez, R.; Kelley, C.M.; Ash, J.A.; Strawderman, M.S.; Alldred, M.J.; Ginsberg, S.D.; Mufson, E.J.; Strupp, B.J. Attentional function and basal forebrain cholinergic neuron morphology during aging in the Ts65Dn mouse model of Down syndrome. Brain Struct. Funct. 2015, 221, 4337–4352. [Google Scholar] [CrossRef]
- Velazquez, R.; Ash, J.A.; Powers, B.E.; Kelley, C.M.; Strawderman, M.; Luscher, Z.I.; Ginsberg, S.; Mufson, E.J.; Strupp, B.J. Maternal choline supplementation improves spatial learning and adult hippocampal neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2013, 58, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Chen, M.; Gandhy, S.U.; Strawderman, M.; Levitsky, D.A.; MacLean, K.N.; Strupp, B.J. Perinatal choline supplementation improves cognitive functioning and emotion regulation in the Ts65Dn mouse model of Down syndrome. Behav. Neurosci. 2010, 124, 346–361. [Google Scholar] [CrossRef]
- Mellott, T.J.; Huleatt, O.M.; Shade, B.N.; Pender, S.M.; Liu, Y.B.; Slack, B.E.; Blusztajn, J.K. Perinatal choline supplementation reduces amyloidosis and increases choline acetyltransferase expression in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS ONE 2017, 12, e0170450. [Google Scholar]
- Bearer, C.; Wellmann, K.; Tang, N.; He, M.; Mooney, S.M. Choline Ameliorates Deficits in Balance Caused by Acute Neonatal Ethanol Exposure. Cerebellum 2015, 14, 413–420. [Google Scholar] [CrossRef]
- Ross, R.G.; Hunter, S.; Hoffman, M.C.; McCarthy, L.; Chambers, B.M.; Law, A.; Leonard, S.; Zerbe, G.O.; Freedman, R. Perinatal Phosphatidylcholine Supplementation and Early Childhood Behavior Problems: Evidence forCHRNA7Moderation. Am. J. Psychiatry 2016, 173, 509–516. [Google Scholar] [CrossRef]
- Scremin, O.; Roch, M.; Norman, K.; Djazayeri, S.; Liu, Y.-Y. Brain acetylcholine and choline concentrations and dynamics in a murine model of the Fragile X syndrome: Age, sex and region-specific changes. Neuroscience 2015, 301, 520–528. [Google Scholar] [CrossRef]
- Stevens, K.E.; Adams, C.E.; Yonchek, J.; Hickel, C.; Danielson, J.; Kisley, M.A. Permanent improvement in deficient sensory inhibition in DBA/2 mice with increased perinatal choline. Psychopharmacology 2008, 198, 413–420. [Google Scholar] [CrossRef]
- Ward, B.; Agarwal, S.; Wang, K.; Berger-Sweeney, J.; Kolodny, N. Longitudinal brain MRI study in a mouse model of Rett Syndrome and the effects of choline. Neurobiol. Dis. 2008, 31, 110–119. [Google Scholar] [CrossRef]
- Ward, B.C.; Kolodny, N.H.; Nag, N.; Berger-Sweeney, J.E. Neurochemical changes in a mouse model of Rett syndrome: Changes over time and in response to perinatal choline nutritional supplementation. J. Neurochem. 2009, 108, 361–371. [Google Scholar] [CrossRef]
- Caudill, M.A.; Strupp, B.J.; Muscalu, L.; Nevins, J.E.H.; Canfield, R.L. Maternal choline supplementation during the third trimester of pregnancy improves infant information processing speed: A randomized, double-blind, controlled feeding study. FASEB J. 2018, 32, 2172–2180. [Google Scholar] [CrossRef]
- Al-Sulaiti, H.; Diboun, I.; Agha, M.V.; Mohamed, F.F.S.; Atkin, S.; Dömling, A.S.; Elrayess, M.A.; Mazloum, N.A. Metabolic signature of obesity-associated insulin resistance and type 2 diabetes. J. Transl. Med. 2019, 17, 1–11. [Google Scholar] [CrossRef]
- Alldred, M.J.; Penikalapati, S.C.; Lee, S.H.; Heguy, A.; Roussos, P.; Ginsberg, S. Profiling Basal Forebrain Cholinergic Neurons Reveals a Molecular Basis for Vulnerability within the Ts65Dn Model of Down Syndrome and Alzheimer’s Disease. 2020, in press. Available online: https://assets.researchsquare.com/files/rs-88218/v1/c0c81dd3-d34b-4ed1-8876-c5b1e8b50f8d.pdf?c=1604102440 (accessed on 3 July 2021).
- Detopoulou, P.; Panagiotakos, D.B.; Antonopoulou, S.; Pitsavos, C.; Stefanadis, C. Dietary choline and betaine intakes in relation to concentrations of inflammatory markers in healthy adults: The ATTICA study. Am. J. Clin. Nutr. 2008, 87, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Alldred, M.J.; Lee, S.H.; Petkova, E.; Ginsberg, S.D. Expression profile analysis of vulnerable CA1 pyramidal neurons in young-Middle-Aged Ts65Dn mice. J. Comp. Neurol. 2015, 523, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Alldred, M.J.; Lee, S.H.; Petkova, E.; Ginsberg, S.D. Expression profile analysis of hippocampal CA1 pyramidal neurons in aged Ts65Dn mice, a model of Down syndrome (DS) and Alzheimer’s disease (AD). Brain Struct. Funct. 2015, 220, 2983–2996. [Google Scholar] [CrossRef] [PubMed]
- Duchon, A.; Raveau, M.; Chevalier, C.; Nalesso, V.; Sharp, A.J.; Herault, Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: Relevance for modeling down syndrome. Mamm. Genome 2011, 22, 674–684. [Google Scholar] [CrossRef]
- Alldred, M.J.; Chao, H.M.; Lee, S.H.; Beilin, J.; Powers, B.E.; Petkova, E.; Ginsberg, S.D. CA1 pyramidal neuron gene expression mosaics in the Ts65Dn murine model of Down syndrome and Alzheimer’s disease following maternal choline supplementation. Hippocampus 2018, 28, 251–268. [Google Scholar] [CrossRef]
- Alldred, M.J.; Chao, H.M.; Lee, S.H.; Beilin, J.; Powers, B.E.; Petkova, E.; Ginsberg, S.D. Long-term effects of maternal choline supplementation on CA1 pyramidal neuron gene expression in the Ts65Dn mouse model of Down syndrome and Alzheimer’s disease. FASEB J. 2019, 33, 9871–9884. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Alldred, M.J.; Duff, K.E.; Ginsberg, S.D. Microarray analysis of CA1 pyramidal neurons in a mouse model of tauopathy reveals progressive synaptic dysfunction. Neurobiol. Dis. 2012, 45, 751–762. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Alldred, M.J.; Che, S.; Ginsberg, S.D. Terminal Continuation (TC) RNA Amplification Enables Expression Profiling Using Minute RNA Input Obtained from Mouse Brain. Int. J. Mol. Sci. 2008, 9, 2091–2104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Mullaney, K.A.; Peterhoff, C.M.; Che, S.; Schmidt, S.D.; Boyer-Boiteau, A.; Nixon, R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Alldred, M.J.; Counts, S.E.; Cataldo, A.M.; Neve, R.L.; Jiang, Y.; Che, S. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry 2010, 68, 885–893. [Google Scholar] [CrossRef]
- ABI. Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR. Appl. Biosyst. Prod. Guide 2004, 1–60. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/cms_042380.pdf (accessed on 3 July 2021).
- McCulloch, C.E.; Searle, S.R.; Neuhaus, J.M. Generalized, Linear, and Mixed Models, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2011. [Google Scholar]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Nguyen, U.; Squaglia, N.; Boge, A.; Fung, P. The Simple Western™: A gel-free, blot-free, hands-free Western blotting reinvention. Nat. Methods 2011, 8. [Google Scholar] [CrossRef]
- Rastegar, S.; Parimisetty, A.; Cassam-Sulliman, N.; Narra, S.S.; Weber, S.; Rastegar, M.; Viranaicken, W.; Couret, D.; Planesse, C.; Strähle, U.; et al. Expression of adiponectin receptors in the brain of adult zebrafish and mouse: Links with neurogenic niches and brain repair. J. Comp. Neurol. 2019, 527, 2317–2333. [Google Scholar] [CrossRef]
- Psilopanagioti, A.; Papadaki, H.; Kranioti, E.F.; Alexandrides, T.; Varakis, J.N. Expression of Adiponectin and Adiponectin Receptors in Human Pituitary Gland and Brain. Neuroendocrinology 2008, 89, 38–47. [Google Scholar] [CrossRef]
- Available online: https://www.gtexportal.org/home/gene/ADIPOR1 (accessed on 3 July 2021).
- Available online: https://www.gtexportal.org/home/gene/ADIPOR2 (accessed on 3 July 2021).
- Forny-Germano, L.; De Felice, F.G.; Vieira, M.N.D.N. The Role of Leptin and Adiponectin in Obesity-Associated Cognitive Decline and Alzheimer’s Disease. Front. Neurosci. 2019, 12, 1027. [Google Scholar] [CrossRef]
- Gorska-Ciebiada, M.; Saryusz-Wolska, M.; Borkowska, A.; Ciebiada, M.; Loba, J. Adiponectin, leptin and IL-1 β in elderly diabetic patients with mild cognitive impairment. Metab. Brain Dis. 2016, 31, 257–266. [Google Scholar] [CrossRef]
- Uddin, S.; Rahman, M.; Abu Sufian, M.; Jeandet, P.; Ashraf, G.M.; Bin-Jumah, M.N.; Mousa, S.A.; Abdel-Daim, M.M.; Akhtar, M.F.; Saleem, A.; et al. Exploring the New Horizon of AdipoQ in Obesity-Related Alzheimer’s Dementia. Front. Physiol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.W.; Bin Abid, N.; Jo, M.H.; Jo, M.G.; Yoon, G.H.; Kim, M.O. Suppression of adiponectin receptor 1 promotes memory dysfunction and Alzheimer’s disease-like pathologies. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Várhelyi, Z.P.; Kálmán, J.; Oláh, Z.; Ivitz, E.V.; Fodor, E.K.; Sántha, M.; Datki, Z.L.; Pákáski, M. Adiponectin receptors are less sensitive to stress in a transgenic mouse model of Alzheimer’s disease. Front. Neurosci. 2017, 11, 199. [Google Scholar] [CrossRef]
- Ali, T.; Rehman, S.U.; Khan, A.; Badshah, H.; Bin Abid, N.; Kim, M.W.; Jo, M.H.; Chung, S.S.; Lee, H.-G.; Rutten, B.P.F.; et al. Adiponectin-mimetic novel nonapeptide rescues aberrant neuronal metabolic-associated memory deficits in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 1–22. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Di Domenico, F.; Head, E.; Butterfield, D.A.; Perluigi, M.; Barone, E. Brain insulin resistance triggers early onset Alzheimer disease in Down syndrome. Neurobiol. Dis. 2020, 137, 104772. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; West, A.A.; Caudill, M.A. Maternal choline supplementation: A nutritional approach for improving offspring health? Trends Endocrinol. Metab. 2014, 25, 263–273. [Google Scholar] [CrossRef]
- Korsmo, H.W.; Jiang, X.; Caudill, M.A. Choline: Exploring the Growing Science on Its Benefits for Moms and Babies. Nutrients 2019, 11, 1823. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Bothwell, M.; Kordower, J.H. Loss of nerve growth factor receptor-containing neurons in Alzheimer’s disease: A quantitative analysis across subregions of the basal forebrain. Exp. Neurol. 1989, 105, 221–232. [Google Scholar] [CrossRef]
- Mufson, E.J.; Ginsberg, S.D.; Ikonomovic, M.D.; DeKosky, S.T. Human cholinergic basal forebrain: Chemoanatomy and neurologic dysfunction. J. Chem. Neuroanat. 2003, 26, 233–242. [Google Scholar] [CrossRef]
- Mufson, E.J.; Ma, S.Y.; Dills, J.; Cochran, E.J.; Leurgans, S.; Wuu, J.; Kordower, J.H. Loss of basal forebrain P75NTR immunoreactivity in subjects with mild cognitive impairment and Alzheimer’s disease. J. Comp. Neurol. 2002, 443, 136–153. [Google Scholar] [CrossRef]
- Kusminski, C.M.; McTernan, P.G.; Schraw, T.; Kos, K.; O’Hare, J.P.; Ahima, R.; Kumar, S.; Scherer, P.E. Adiponectin complexes in human cerebrospinal fluid: Distinct complex distribution from serum. Diabetologia 2007, 50, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Blusztajn, J.K.; Slack, B.E.; Mellott, T.J. Neuroprotective Actions of Dietary Choline. Nutrients 2017, 9, 815. [Google Scholar] [CrossRef] [PubMed]
- Nag, N.; Berger-Sweeney, J.E. Postnatal dietary choline supplementation alters behavior in a mouse model of Rett syndrome. Neurobiol. Dis. 2007, 26, 473–480. [Google Scholar] [CrossRef]
- Zeisel, S.H. Choline: Clinical Nutrigenetic/Nutrigenomic Approaches for Identification of Functions and Dietary Requirements. J. Nutr. Nutr. 2010, 3, 209–219. [Google Scholar] [CrossRef]
- Jacobson, S.W.; Molteno, C.D.; Meintjes, E.M.; Senekal, M.S.; Lindinger, N.M.; Dodge, N.C.; Zeisel, S.H.; Duggan, C.P.; Jacobson, J.L.; Carter, R. Feasibility and Acceptability of Maternal Choline Supplementation in Heavy Drinking Pregnant Women: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Alcohol. Clin. Exp. Res. 2018, 42, 1315–1326. [Google Scholar] [CrossRef]
- Jacobson, S.W.; Carter, R.C.; Molteno, C.D.; Stanton, M.E.; Herbert, J.S.; Lindinger, N.M.; Jacobson, J.L. Efficacy of maternal choline supplementation during pregnancy in mitigating adverse effects of prenatal alcohol exposure on growth and cognitive function: A randomized, double-blind, placebo-controlled clinical trial. Alcohol. Clin. Exp. Res. 2018, 42, 1327–1341. [Google Scholar] [CrossRef]
- Cheatham, C.L.; Goldman, B.D.; Fischer, L.M.; Da Costa, K.-A.; Reznick, J.S.; Zeisel, S.H. Phosphatidylcholine supplementation in pregnant women consuming moderate-choline diets does not enhance infant cognitive function: A randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2012, 96, 1465–1472. [Google Scholar] [CrossRef]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; More, K.J.; Breitbart, R.E.; et al. Evidence That the Diabetes Gene Encodes the Leptin Receptor: Identification of a Mutation in the Leptin Receptor Gene in db/db Mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Hashimoto, M.; Ho, G.; Sugama, S.; Takenouchi, T.; Waragai, M.; Sugino, H.; Masliah, E. Possible Role of Activin in the Adiponectin Par-adox-Induced Progress of Alzheimer’s Disease. J. Alzheimers Dis. 2021, 81, 451–458. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alldred, M.J.; Lee, S.H.; Ginsberg, S.D. Adiponectin Modulation by Genotype and Maternal Choline Supplementation in a Mouse Model of Down Syndrome and Alzheimer’s Disease. J. Clin. Med. 2021, 10, 2994. https://doi.org/10.3390/jcm10132994
Alldred MJ, Lee SH, Ginsberg SD. Adiponectin Modulation by Genotype and Maternal Choline Supplementation in a Mouse Model of Down Syndrome and Alzheimer’s Disease. Journal of Clinical Medicine. 2021; 10(13):2994. https://doi.org/10.3390/jcm10132994
Chicago/Turabian StyleAlldred, Melissa J., Sang Han Lee, and Stephen D. Ginsberg. 2021. "Adiponectin Modulation by Genotype and Maternal Choline Supplementation in a Mouse Model of Down Syndrome and Alzheimer’s Disease" Journal of Clinical Medicine 10, no. 13: 2994. https://doi.org/10.3390/jcm10132994
APA StyleAlldred, M. J., Lee, S. H., & Ginsberg, S. D. (2021). Adiponectin Modulation by Genotype and Maternal Choline Supplementation in a Mouse Model of Down Syndrome and Alzheimer’s Disease. Journal of Clinical Medicine, 10(13), 2994. https://doi.org/10.3390/jcm10132994