
Prostate Cancer Dormancy and Reactivation in Bone Marrow


{kind=link}
Abstract
1. Introduction
2. Dormancy in Prostate Cancer
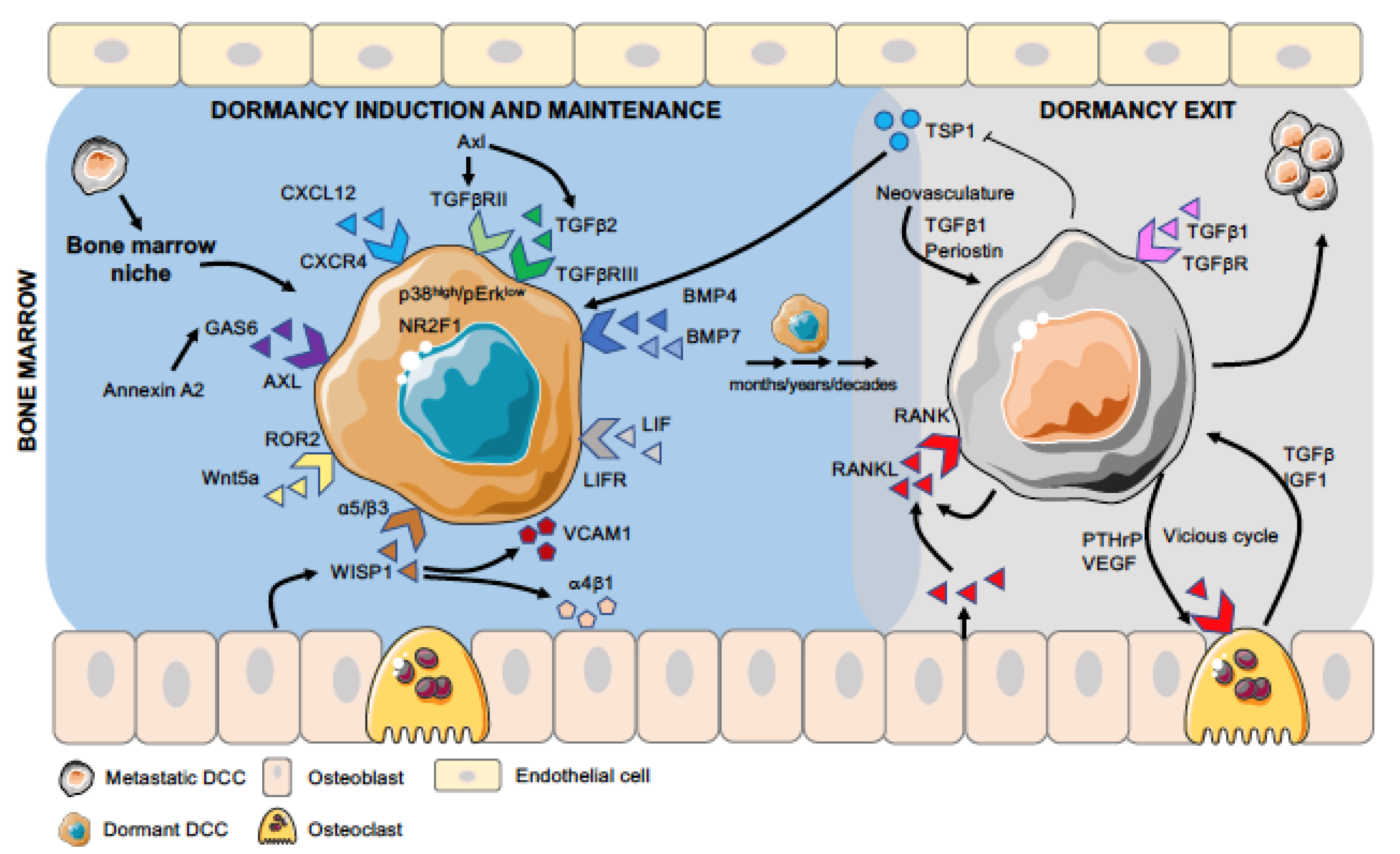
3. Niche Cell Types and Signals that Induce or Block PCa DCC Dormancy in the Bone Marrow Microenvironment
4. Therapeutic Approaches Targeting Bone–Tumor Microenvironment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dillekas, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A.; Sosa, M.S. Emerging Topics on Disseminated Cancer Cell Dormancy and the Paradigm of Metastasis. Annu. Rev. Cancer Biol. 2018, 2, 377–393. [Google Scholar] [CrossRef]
- Park, S.-Y.; Nam, J.-S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. How dormant cancer persists and reawakens. Science 2018, 361, 1314–1315. [Google Scholar] [CrossRef]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; De Stanchina, E.; Massague, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef]
- Giancotti, G. Filippo, Mechanisms Governing Metastatic Dormancy and Reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef]
- Townson, J.L.; Chambers, A.F. Dormancy of solitary metastatic cells. Cell Cycle 2006, 5, 1744–1750. [Google Scholar] [CrossRef]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar] [CrossRef]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van Der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Viale, P.H. The American Cancer Society’s Facts & Figures: 2020 Edition. J. Adv. Pract. Oncol. 2020, 11, 135–136. [Google Scholar]
- Ku, S.-Y.; Gleave, M.E.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.G.; Oh, W.K. The evolving landscape of immunotherapy in advanced prostate cancer. Immunotherapy 2019, 11, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; SOerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Roudier, M.P.; Corey, E.; True, L.D.; Hiagno, C.S.; Ott, S.M.; Vessella, R.L. Histological, Immunophenotypic and Histomorphometric Characterization of Prostate Cancer Bone Metastases. In Cancer Treatment and Research; Springer: New York, NY, USA, 2004; pp. 311–339. [Google Scholar]
- Gandaglia, G.; Abdollah, F.; Schiffmann, J.; Trudeau, V.; Shariat, S.F.; Kim, S.P.; Perrotte, P.; Montorsi, F.; Briganti, A.; Trinh, Q.; et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate 2014, 74, 210–216. [Google Scholar] [CrossRef]
- Vinjamoori, A.H.; Jagannathan, J.P.; Shinagare, A.B.; Taplin, M.-E.; Oh, W.K.; Van den Abbeele, A.D.; Ramaiya, N.H. Atypical metastases from prostate cancer: 10-year experience at a single institution. AJR Am. J. Roentgenol. 2012, 199, 367–372. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep. 2014, 3, 481. [Google Scholar] [CrossRef] [PubMed]
- Sowder, M.E.; Johnson, R.W. Bone as a Preferential Site for Metastasis. JBMR Plus 2019, 3, e10126. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Guise, T.; Kang, Y. The Biology of Bone Metastasis. Cold Spring Harb. Perspect. Med. 2018, 8, a031252. [Google Scholar] [CrossRef]
- Guise, T.A. The vicious cycle of bone metastases. J. Musculoskelet Neuronal Interact 2002, 2, 570–572. [Google Scholar]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef]
- Globus, R.K.; Patterson-Buckendahl, P.; Gospodarowicz, D. Regulation of bovine bone cell proliferation by fibroblast growth factor and transforming growth factor beta. Endocrinology 1988, 123, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Bienz, M.; Saad, F. Management of bone metastases in prostate cancer: A review. Curr. Opin. Support Palliat. Care 2015, 9, 261–267. [Google Scholar] [CrossRef]
- Seeman, E.; Delmas, P.D. Bone quality--the material and structural basis of bone strength and fragility. N. Engl. J. Med. 2006, 354, 2250–2261. [Google Scholar] [CrossRef] [PubMed]
- Weckermann, D.; Polzer, B.; Ragg, T.; Blana, A.; Schlimok, G.; Arnholdt, H.; Bertz, S.; Harzmann, R.; Klein, C.A. Perioperative activation of disseminated tumor cells in bone marrow of patients with prostate cancer. J. Clin. Oncol. 2009, 27, 1549–1556. [Google Scholar] [CrossRef]
- Morgan, T.M.; Lange, P.H.; Porter, M.P.; Lin, D.W.; Ellis, W.J.; Gallaher, I.S.; Vessella, R.L. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin. Cancer Res. 2009, 15, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Adam, A.P.; George, A.; Schewe, D.; Bragado, P.; Iglesias, B.V.; Ranganathan, A.C.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009, 69, 5664–5672. [Google Scholar] [CrossRef]
- Nobre, A.R.; Risson, E.; Singh, D.K.; Di Martino, J.S.; Cheung, J.F.; Wang, J.; Johnson, J.; Russnes, H.G.; Bravo-Cordero, J.J.; Birbrair, A.; et al. Bone marrow NG2+/Nestin+ mesenchymal stem cells drive DTC dormancy via TGF-β2. Nat. Cancer 2021, 2, 327–339. [Google Scholar] [CrossRef]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFbetaRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef]
- Lawson, M.A.; Mcdonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef]
- Cackowski, F.C.; Wang, Y.; Decker, J.T.; Sifuentes, C.; Weindorf, S.; Jung, Y.; Wang, Y.; Decker, A.M.; Yumoto, K.; Szerlip, N.; et al. Detection and isolation of disseminated tumor cells in bone marrow of patients with clinically localized prostate cancer. Prostate 2019, 79, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Cackowski, F.C.; Eber, M.R.; Rhee, J.; Decker, A.M.; Yumoto, K.; Berry, J.E.; Lee, E.; Shiozawa, Y.; Jung, Y.; Aguirre-Ghiso, J.A.; et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J. Cell Biochem. 2017, 118, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef]
- Cackowski, F.C.; Taichman, R.S. Minimal Residual Disease in Prostate Cancer. In Advances in Experimental Medicine and Biology; Springer International Publishing: New York, NY, USA, 2018; pp. 47–53. [Google Scholar]
- Furesi, G.; Rauner, M.; Hofbauer, L.C. Emerging Players in Prostate Cancer–Bone Niche Communication. Trends Cancer 2020, 7, 112–121. [Google Scholar] [CrossRef]
- Sousa, S.; Määttä, J. The role of tumour-associated macrophages in bone metastasis. J. Bone Oncol. 2016, 5, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef]
- Nwajei, F.; Konopleva, M. The Bone Marrow Microenvironment as Niche Retreats for Hematopoietic and Leukemic Stem Cells. Adv. Hematol. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, A.; Hsu, Y.-M.S.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’Ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef]
- Tzeng, Y.-S.; Li, H.; Kang, Y.-L.; Chen, W.-C.; Cheng, W.-C.; Lai, D.-M. Loss of Cxcl12/Sdf-1 in adult mice decreases the quiescent state of hematopoietic stem/progenitor cells and alters the pattern of hematopoietic regeneration after myelosuppression. Blood 2011, 117, 429–439. [Google Scholar] [CrossRef]
- Nie, Y.; Han, Y.-C.; Zou, Y.-R. CXCR4 is required for the quiescence of primitive hematopoietic cells. J. Exp. Med. 2008, 205, 777–783. [Google Scholar] [CrossRef]
- Bonig, H.; Priestley, G.V.; Nilsson, L.M.; Jiang, Y.; Papayannopoulou, T. PTX-sensitive signals in bone marrow homing of fetal and adult hematopoietic progenitor cells. Blood 2004, 104, 2299–2306. [Google Scholar] [CrossRef]
- Decker, A.M.; Jung, Y.; Cackowski, F.C.; Yumoto, K.; Wang, J.; Taichman, R.S. Sympathetic Signaling Reactivates Quiescent Disseminated Prostate Cancer Cells in the Bone Marrow. Mol. Cancer Res. 2017, 15, 1644–1655. [Google Scholar] [CrossRef]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Levesque, J.P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef]
- Cackowski, F.C.; Taichman, R.S. Parallels between hematopoietic stem cell and prostate cancer disseminated tumor cell regulation. Bone 2019, 119, 82–86. [Google Scholar] [CrossRef]
- Sun, Y.-X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, H.; et al. Skeletal Localization and Neutralization of the SDF-1(CXCL12)/CXCR4 Axis Blocks Prostate Cancer Metastasis and Growth in Osseous Sites In Vivo. J. Bone Miner. Res. 2004, 20, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Sbrissa, D.; Semaan, L.; Govindarajan, B.; Li, Y.; Caruthers, N.J.; Stemmer, P.M.; Cher, M.L.; Sethi, S.; Vaishampayan, U.; Shisheva, A.; et al. A novel cross-talk between CXCR4 and PI4KIIIalpha in prostate cancer cells. Oncogene 2019, 38, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Chang, A.-C.; Chen, P.-C.; Lin, Y.-F.; Su, C.-M.; Liu, J.-F.; Lin, T.-H.; Chuang, S.-M.; Tang, C.-H. Osteoblast-secreted WISP-1 promotes adherence of prostate cancer cells to bone via the VCAM-1/integrin α4β1 system. Cancer Lett. 2018, 426, 47–56. [Google Scholar] [CrossRef]
- Engl, T.; Relja, B.; Marian, D.; Blumenberg, C.; Muller, I.; Beecken, W.-D.; Jones, J.; Ringel, E.M.; Bereiter-Hahn, J.; Jonas, D.; et al. CXCR4 Chemokine Receptor Mediates Prostate Tumor Cell Adhesion through α5 and β3 Integrins. Neoplasia 2006, 8, 290–301. [Google Scholar] [CrossRef]
- Chambard, J.C.; Lefloch, R.; Pouyssegur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- A Aguirre-Ghiso, J.; Estrada, Y.; Liu, D.; Ossowski, L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res. 2003, 63, 1684–1695. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Prunier, C.; Baker, D.; Ten Dijke, P.; Ritsma, L. TGF-beta Family Signaling Pathways in Cellular Dormancy. Trends Cancer 2019, 5, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [PubMed]
- Chery, L.; Lam, H.-M.; Coleman, I.; Lakely, B.; Coleman, R.; Larson, S.; Aguirre-Ghiso, J.A.; Xia, J.; Gulati, R.; Nelson, P.S.; et al. Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways. Oncotarget 2014, 5, 9939–9951. [Google Scholar] [CrossRef] [PubMed]
- Ruppender, N.; Larson, S.; Lakely, B.; Kollath, L.; Brown, L.; Coleman, I.; Coleman, R.; Nguyen, H.; Nelson, P.S.; Corey, E.; et al. Cellular Adhesion Promotes Prostate Cancer Cells Escape from Dormancy. PLoS ONE 2015, 10, e0130565. [Google Scholar] [CrossRef]
- Yu-Lee, L.-Y.; Lee, Y.-C.; Pan, J.; Lin, S.-C.; Pan, T.; Yu, G.; Hawke, D.H.; Pan, B.-F.; Lin, S.-H. Bone secreted factors induce cellular quiescence in prostate cancer cells. Sci. Rep. 2019, 9, 18635. [Google Scholar] [CrossRef]
- Byrne, N.M.; A Summers, M.; McDonald, M.M. Tumor Cell Dormancy and Reactivation in Bone: Skeletal Biology and Therapeutic Opportunities. JBMR Plus 2019, 3, e10125. [Google Scholar] [CrossRef]
- Taichman, R.S.; Patel, L.R.; Bedenis, R.; Wang, J.; Weidner, S.; Schumann, T.; Yumoto, K.; Berry, J.E.; Shiozawa, Y.; Pienta, K.J. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS ONE 2013, 8, e61873. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Dai, Y.; Yang, Q.; Zhang, X.; Guo, W.; Ye, L.; Huang, S.; Chen, X.; Lai, Y.; Du, H.; et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J. Exp. Med. 2018, 216, 428–449. [Google Scholar] [CrossRef] [PubMed]
- Rauner, M.; Sipos, W.; Pietschmann, P. Age-dependent Wnt gene expression in bone and during the course of osteoblast differentiation. AGE 2008, 30, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Sowder, M.E.; Giaccia, A.J. Hypoxia and Bone Metastatic Disease. Curr. Osteoporos. Rep. 2017, 15, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.S.; Rameshwar, P.; Chang, V.; Bandari, P. Oxygen saturation in the bone marrow of healthy volunteers. Blood 2002, 99, 394. [Google Scholar] [CrossRef]
- Cox, T.R.; Rumney, R.M.H.; Schoof, E.M.; Perryman, L.; Høye, A.; Agrawal, A.; Bird, D.; Ab Latif, N.B.; Forrest, H.; Evans, H.R.; et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 2015, 522, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yan, C.H.; Yuan, M.; Wei, Y.; Hu, G.; Kang, Y. In vivo Dynamics and Distinct Functions of Hypoxia in Primary Tumor Growth and Organotropic Metastasis of Breast Cancer. Cancer Res. 2010, 70, 3905–3914. [Google Scholar] [CrossRef]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef]
- Summers, M.A.; McDonald, M.M.; Croucher, P. Cancer Cell Dormancy in Metastasis. Cold Spring Harb. Perspect. Med. 2020, 10, a037556. [Google Scholar] [CrossRef]
- Ghiso, J.A.A.; Kovalski, K.; Ossowski, L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L.J. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef]
- Chu, G.C.-Y.; E Zhau, H.; Wang, R.; Rogatko, A.; Feng, X.; Zayzafoon, M.; Liu, Y.; Farach-Carson, M.C.; You, S.; Kim, J.; et al. RANK- and c-Met.-mediated signal network promotes prostate cancer metastatic colonization. Endocr. Relat. Cancer 2014, 21, 311–326. [Google Scholar] [CrossRef]
- Farrar, J.D.; Katz, K.H.; Windsor, J.; Thrush, G.; Scheuermann, R.H.; Uhr, J.W.; E Street, N. Cancer dormancy. VII. A regulatory role for CD8+ T cells and IFN-gamma in establishing and maintaining the tumor-dormant state. J. Immunol. 1999, 162, 2842–2849. [Google Scholar] [PubMed]
- Pantel, K.; Schlimok, G.; Kutter, D.; Schaller, G.; Genz, T.; Wiebecke, B.; Backmann, R.; Funke, I.; Riethmüller, G. Frequent down-regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res. 1991, 51, 4712–4715. [Google Scholar]
- Owen, K.L.; Gearing, L.J.; Zanker, D.J.; Brockwell, N.K.; Khoo, W.H.; Roden, D.L.; Cmero, M.; Mangiola, S.; Hong, M.K.; Spurling, A.J.; et al. Prostate cancer cell-intrinsic interferon signaling regulates dormancy and metastatic outgrowth in bone. EMBO Rep. 2020, 21, e50162. [Google Scholar] [CrossRef]
- Wirth, M.; Tammela, T.; Cicalese, V.; Veiga, F.G.; Delaere, K.; Miller, K.; Tubaro, A.; Schulze, M.; Debruyne, F.; Huland, H.; et al. Prevention of Bone Metastases in Patients with High.-risk Nonmetastatic Prostate Cancer Treated with Zoledronic Acid: Efficacy and Safety Results of the Zometa European Study (ZEUS). Eur. Urol. 2015, 67, 482–491. [Google Scholar] [CrossRef]
- Mason, M.D.; Sydes, M.; Glaholm, J.; Langley, R.E.; Huddart, R.A.; Sokal, M.; Stott, M.; Robinson, A.C.; James, N.D.; Parmar, M.K.B.; et al. Oral Sodium Clodronate for Nonmetastatic Prostate Cancer—Results of a Randomized Double-Blind. Placebo-Controlled Trial: Medical Research Council PR04 (ISRCTN61384873). JNCI J. Natl. Cancer Inst. 2007, 99, 765–776. [Google Scholar] [CrossRef]
- Smith, M.R.; Kabbinavar, F.; Saad, F.; Hussain, A.A.; Gittelman, M.M.; Bilhartz, D.D.; Wynne, C.C.; Murray, R.R.; Zinner, N.N.; Schulman, C.; et al. Natural History of Rising Serum Prostate-Specific Antigen in Men With Castrate Nonmetastatic Prostate Cancer. J. Clin. Oncol. 2005, 23, 2918–2925. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damião, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef]
- Buschhaus, J.M.; Humphries, B.A.; Eckley, S.S.; Robison, T.H.; Cutter, A.C.; Rajendran, S.; Haley, H.R.; Bevoor, A.S.; Luker, K.E.; Luker, G.D. Targeting disseminated estrogen-receptor-positive breast cancer cells in bone marrow. Oncogene 2020, 39, 5649–5662. [Google Scholar] [CrossRef] [PubMed]
- Grabinski, N.; Bartkowiak, K.; Grupp, K.; Brandt, B.; Pantel, K.; Jücker, M. Distinct functional roles of Akt isoforms for proliferation, survival, migration and EGF-mediated signalling in lung cancer derived disseminated tumor cells. Cell. Signal. 2011, 23, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bernstein, E.; Aguirre-Ghiso, J.A. Epigenetic Regulation of Cancer Dormancy as a Plasticity Mechanism for Metastasis Initiation. In Cancer Drug Discovery and Development; Springer International Publishing: New York, NY, USA, 2017; pp. 1–16. [Google Scholar]
- Wang, W.; Goswami, S.; Sahai, E.; Wyckoff, J.B.; Segall, J.E.; Condeelis, J.S. Tumor cells caught in the act of invading: Their strategy for enhanced cell motility. Trends Cell Biol. 2005, 15, 138–145. [Google Scholar] [CrossRef]
- Condeelis, J.; Singer, R.H.; Segall, J.E. The Great Escape: When Cancer Cells Hijack the Genes for Chemotaxis and Motility. Annu. Rev. Cell Dev. Biol. 2005, 21, 695–718. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, D.K.; Patel, V.G.; Oh, W.K.; Aguirre-Ghiso, J.A. Prostate Cancer Dormancy and Reactivation in Bone Marrow. J. Clin. Med. 2021, 10, 2648. https://doi.org/10.3390/jcm10122648
Singh DK, Patel VG, Oh WK, Aguirre-Ghiso JA. Prostate Cancer Dormancy and Reactivation in Bone Marrow. Journal of Clinical Medicine. 2021; 10(12):2648. https://doi.org/10.3390/jcm10122648
Chicago/Turabian StyleSingh, Deepak K., Vaibhav G. Patel, William K. Oh, and Julio A. Aguirre-Ghiso. 2021. "Prostate Cancer Dormancy and Reactivation in Bone Marrow" Journal of Clinical Medicine 10, no. 12: 2648. https://doi.org/10.3390/jcm10122648
APA StyleSingh, D. K., Patel, V. G., Oh, W. K., & Aguirre-Ghiso, J. A. (2021). Prostate Cancer Dormancy and Reactivation in Bone Marrow. Journal of Clinical Medicine, 10(12), 2648. https://doi.org/10.3390/jcm10122648