Clinical Utility of Plasma KRAS, NRAS and BRAF Mutational Analysis with Real Time PCR in Metastatic Colorectal Cancer Patients—The Importance of Tissue/Plasma Discordant Cases

 , , ,
, , ,  ,
,  , and
, and 
Abstract
1. Introduction
2. Experimental Section
2.1. Materials and Methods
2.1.1. Patients
2.1.2. Molecular Analysis
Cell-Free DNA Extraction and Amplification
Tissue DNA Extraction and RAS and BRAF Mutational Status
2.2. Statistical Analysis
3. Results
3.1. Patients’ Characteristics
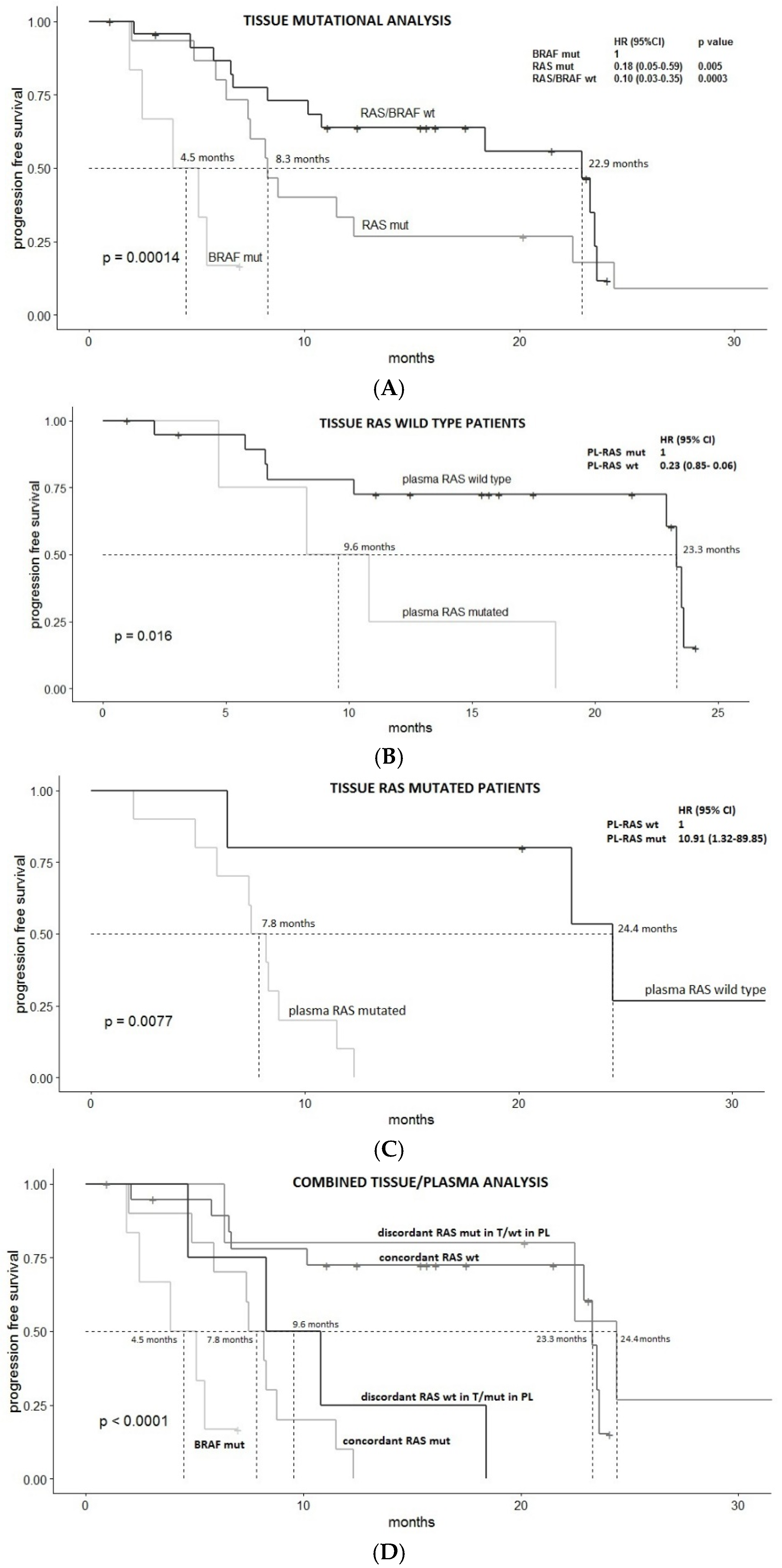
3.2. Survival Analysis
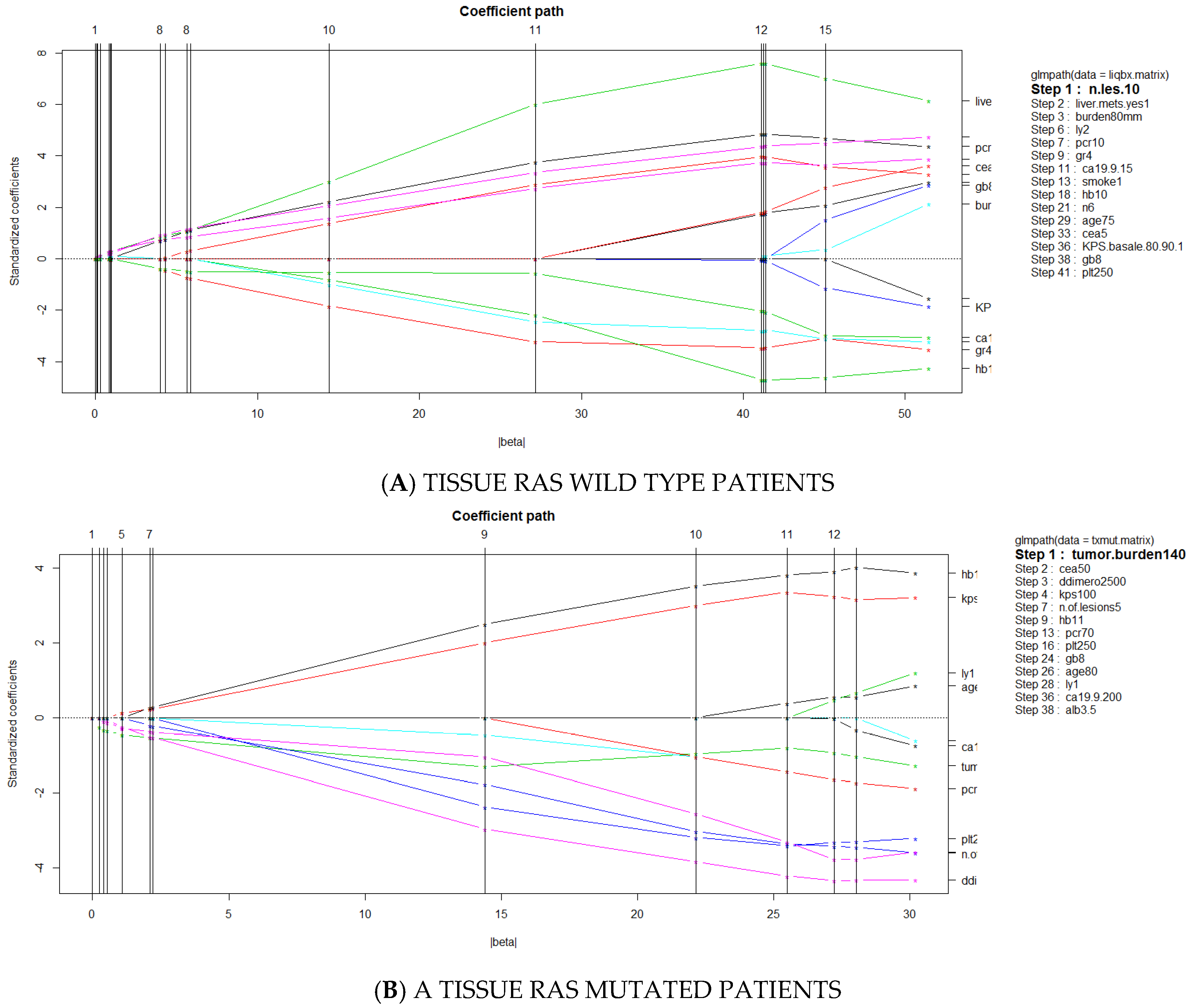
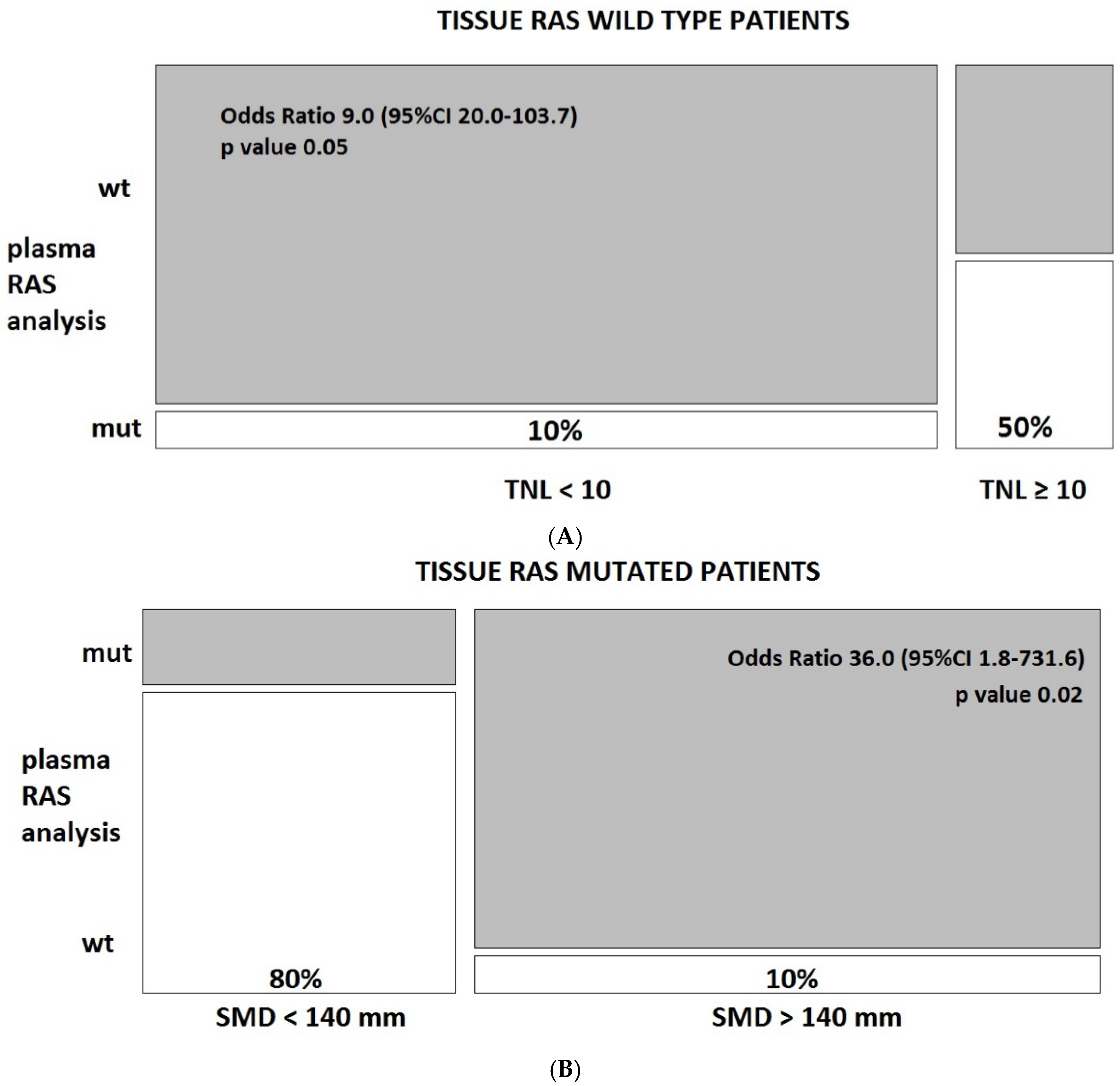
3.3. Predictors of Discordant Cases
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mandel, P.; Metais, P. Les acides nucléiques du plasma sanguin chez l’homme. CR Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S. R Analysis of the size distributions of fetal and maternal cell-free DNA by paired-end sequencing. Clin. Chem. 2010, 56, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Su, L.; Qian, C. Circulating tumor DNA: A promising biomarker in the liquid biopsy of cancer. Oncotarget 2016, 7, 48832–48841. [Google Scholar] [CrossRef] [PubMed]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- van der Vaart, M.; Pretorius, P.J. The origin of circulating free DNA. Clin. Chem. 2007, 53, 2215. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Yoshino, T. Clinical Utility of Analyzing Circulating Tumor DNA in Patients with Metastatic Colorectal Cancer. Oncologist 2018, 23, 1310–1318. [Google Scholar] [CrossRef]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Chabner, B.A. Application of cell-free DNA analysis to cancer treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti- EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, G.; Southward, K.; Handley, K.; Magill, L.; Beaumont, C.; Stahlschmidt, J. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J. Clin. Oncol. 2011, 29, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Elez, E.; Chianese, C.; Sanz-García, E.; Martinelli, E.; Noguerido, A.; Mancuso, F.M.; Caratù, G.; Matito, J.; Grasselli, J.; Cardone, C.; et al. Impact of circulating tumor DNA mutant allele fraction on prognosis in RAS-mutant metastatic colorectal cancer. Mol. Oncol. 2019, 13, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- García-Foncillas, J.; Tabernero, J.; Élez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; López, R.; Muinelo-Romay, L.; Montagut, C. Prospective multicentre real-world RAS mutation comparison between OncoBEAM-based liquid biopsy and tissue analysis in metastatic colorectal cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Kagawa, Y.; Kato, T.; Akagi, K.; Denda, T.; Nishina, T.; Komatsu, Y.; Oki, E.; Kudo, T.; Kumamoto, H.; et al. A multicentre, prospective study of plasma circulating tumour DNA test for detecting RAS mutation in patients with metastatic colorectal cancer. Br. J. Cancer 2019, 120, 982–986. [Google Scholar] [CrossRef]
- Yao, J.; Zang, W.; Ge, Y.; Weygant, N.; Yu, P.; Li, L.; Rao, G.; Jiang, Z.; Yan, R.; He, L.; et al. RAS/BRAF Circulating Tumor DNA Mutations as a Predictor of Response to First-Line Chemotherapy in Metastatic Colorectal Cancer Patients. Can. J. Gastroenterol. Hepatol. 2018, 2018, 4248971. [Google Scholar] [CrossRef]
- Garcia-Carbonero, N.; Martinez-Useros, J.; Li, W.; Orta, A.; Perez, N.; Carames, C.; Hernandez, T.; Moreno, I.; Serrano, G.; Garcia-Foncillas, J. KRAS and BRAF Mutations as Prognostic and Predictive Biomarkers for Standard Chemotherapy Response in Metastatic Colorectal Cancer: A Single Institutional Study. Cells 2020, 9, 219. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Djanani, A.; Eller, S.; Öfner, D.; Troppmair, J.; Maglione, M. The Role of BRAF in Metastatic Colorectal Carcinoma-Past, Present, and Future. Int. J. Mol. Sci. 2020, 21, 9001. [Google Scholar] [CrossRef]
- Godsey, J.H.; Silvestro, A.; Barrett, J.C.; Bramlett, K.; Chudova, D.; Deras, I.; Dickey, J.; Hicks, J.; Johann, D.J.; Leary, R.; et al. Generic Protocols for the Analytical Validation of Next-Generation Sequencing-Based ctDNA Assays: A Joint Consensus Recommendation of the BloodPAC’s Analytical Variables Working Group. Clin. Chem. 2020, 66, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Hosen, M.I.; Forey, N.; Durand, G.; Voegele, C.; Bilici, S.; Avogbe, P.H.; Delhomme, T.M.; Foll, M.; Manel, A.; Vian, E.; et al. Development of Sensitive Droplet Digital PCR Assays for Detecting Urinary TERT Promoter Mutations as Non-Invasive Biomarkers for Detection of Urothelial Cancer. Cancers 2020, 12, 3541. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chang, L.; Guan, Y.; Yang, L.; Xia, X.; Cui, L.; Yi, X.; Lin, G. Application of Circulating Tumor DNA as a Non-Invasive Tool for Monitoring the Progression of Colorectal Cancer. PLoS ONE 2016, 11, e0159708. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Bouché, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: The AGEO RASANC prospective multicenter study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Galvano, A.; Taverna, S.; Badalamenti, G.; Incorvaia, L.; Castiglia, M.; Barraco, N.; Passiglia, F.; Fulfaro, F.; Beretta, G.; Duro, G.; et al. Detection of RAS mutations in circulating tumor DNA: A new weapon in an old war against colorectal cancer. A systematic review of literature and meta-analysis. Ther. Adv. Med. Oncol. 2019, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Cervantes, A.; Ciardiello, F.; De Luca, A.; Pinto, C. The liquid biopsy in the management of colorectal cancer patients: Current applications and future scenarios. Cancer Treat. Rev. 2018, 70, 1–8. [Google Scholar] [CrossRef]
- Wan, J.C.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Vidal, J.; Bellosillo, B.; Santos Vivas, C.; García-Alfonso, P.; Carrato, A.; Cano, M.T.; García-Carbonero, R.; Élez, E.; Losa, F.; Massutí, B.; et al. Ultra-selection of metastatic colorectal cancer patients using next-generation sequencing to improve clinical efficacy of anti-EGFR therapy. Ann. Oncol. 2019, 30, 439–446. [Google Scholar] [CrossRef]
- Holdhoff, M.; Schmidt, K.; Donehower, R.; Diaz, L.A., Jr. Analysis of circulating tumor DNA to confirm somatic KRAS mutations. J. Natl. Cancer Inst. 2009, 101, 1284–1285. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; El Messaoudi, S.; Mollevi, C.; Raoul, J.L.; Guimbaud, R.; Pezet, D.; Artru, P.; Assenat, E.; Borg, C.; Mathonnet, M.; et al. Clinical utility of circulating DNA analysis for rapid detection of actionable mutations to select metastatic colorectal patients for anti-EGFR treatment. Ann. Oncol. 2017, 28, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Tsui, D.W.; Diaz, L.A., Jr. Detection of somatic RAS mutations in circulating tumor DNA from metastatic colorectal cancer patients: Are we ready for clinical use? Ann. Oncol. 2018, 29, 1083–1084. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Khan, K.H.; Cunningham, D.; Werner, B.; Vlachogiannis, G.; Spiteri, I.; Heide, T.; Mateos, J.F.; Vatsiou, A.; Lampis, A.; Damavandi, M.D.; et al. Longitudinal liquid biopsy and mathematical modeling of clonal evolution forecast time to treatment failure in the PROSPECT-C phase II colorectal cancer clinical trial. Cancer Discov. 2018, 8, 1270–1285. [Google Scholar] [CrossRef]
- Vandeputte, C.; Kehagias, P.; El Housni, H.; Ameye, L.; Laes, J.F.; Desmedt, C.; Sotiriou, C.; Deleporte, A.; Puleo, F.; Geboes, K.; et al. Circulating tumor DNA in early response assessment and monitoring of advanced colorectal cancer treated with a multi- kinase inhibitor. Oncotarget 2018, 9, 17756–17769. [Google Scholar] [CrossRef]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for Patients With RAS and BRAF Wild-Type Metastatic Colorectal Cancer With Acquired Resistance to First-line Cetuximab and Irinotecan: A Phase 2 Single-Arm Clinical Trial. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef]
- Dasari, A.; Morris, V.K.; Allegra, C.J.; Atreya, C.; Benson, A.B., 3rd; Boland, P.; Chung, K.; Copur, M.S.; Corcoran, R.B.; Deming, D.A.; et al. ctDNA applications and integration in colorectal cancer: An NCI Colon and Rectal-Anal Task Forces whitepaper. Nat. Rev. Clin. Oncol. 2020, 17, 757–770. [Google Scholar] [CrossRef]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | % of Patients (Total n = 45) | Characteristic | % of Patients (Total n = 45) |
|---|---|---|---|
| Median Age (years, range) | 67 (41–89) | Median Karnofsky Performance Status (KPS) score (range) | 100 (60–100) |
| Gender | Sidedness | ||
| Female | 36% (16) | Right | 22% (10) |
| Male | 64% (29) | Left | 78% (35) |
| Metastasis | Mutational status in tissue | ||
| Liver only | 24% (11) | BRAF mutation | 13% (6) |
| Liver and others | 67% (30) | RAS mutation | 34% (15) |
| Non-liver only | 9% (4) | RAS/BRAF wild type | 53% (24) |
| Tumor burden | Treatment | ||
| TNL | 4 (1–56) | FOLFOX-bevacizumab | 47% (21) |
| SMD | 122 mm (25–1687) | FOLFOX-panitumumab | 53% (24) |
| Primary Resection | Tumor markers | ||
| Yes | 73% (33) | Carcinoembryonic antigen (CEA) (median ng/mL, range) | 599 (0.62–5580) |
| No | 27% (12) | CA19.9 (median ng/mL, range) | 214 (1–19,032) |
| Covariate | Coef | HR | Se (Coef) | Z | p |
|---|---|---|---|---|---|
| CEA | 0.0001 | 1.0001 | 0.0002 | 0.5390 | 0.5899 |
| Tissue mutational status | 0.1897 | 1.2088 | 0.4957 | 0.3830 | 0.7020 |
| Plasmatic mutational status | −1.9731 | 0.1390 | 0.5912 | −3.3370 | 0.0008 |
| Sideness left vs. right | −0.0118 | 0.9883 | 0.5185 | −0.0230 | 0.9818 |
| KPS | −0.0743 | 0.9284 | 0.0281 | −2.6440 | 0.0082 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Formica, V.; Lucchetti, J.; Doldo, E.; Riondino, S.; Morelli, C.; Argirò, R.; Renzi, N.; Nitti, D.; Nardecchia, A.; Dell’Aquila, E.; et al. Clinical Utility of Plasma KRAS, NRAS and BRAF Mutational Analysis with Real Time PCR in Metastatic Colorectal Cancer Patients—The Importance of Tissue/Plasma Discordant Cases. J. Clin. Med. 2021, 10, 87. https://doi.org/10.3390/jcm10010087
Formica V, Lucchetti J, Doldo E, Riondino S, Morelli C, Argirò R, Renzi N, Nitti D, Nardecchia A, Dell’Aquila E, et al. Clinical Utility of Plasma KRAS, NRAS and BRAF Mutational Analysis with Real Time PCR in Metastatic Colorectal Cancer Patients—The Importance of Tissue/Plasma Discordant Cases. Journal of Clinical Medicine. 2021; 10(1):87. https://doi.org/10.3390/jcm10010087
Chicago/Turabian StyleFormica, Vincenzo, Jessica Lucchetti, Elena Doldo, Silvia Riondino, Cristina Morelli, Renato Argirò, Nicola Renzi, Daniele Nitti, Antonella Nardecchia, Emanuela Dell’Aquila, and et al. 2021. "Clinical Utility of Plasma KRAS, NRAS and BRAF Mutational Analysis with Real Time PCR in Metastatic Colorectal Cancer Patients—The Importance of Tissue/Plasma Discordant Cases" Journal of Clinical Medicine 10, no. 1: 87. https://doi.org/10.3390/jcm10010087
APA StyleFormica, V., Lucchetti, J., Doldo, E., Riondino, S., Morelli, C., Argirò, R., Renzi, N., Nitti, D., Nardecchia, A., Dell’Aquila, E., Ferroni, P., Guadagni, F., Palmieri, G., Orlandi, A., & Roselli, M. (2021). Clinical Utility of Plasma KRAS, NRAS and BRAF Mutational Analysis with Real Time PCR in Metastatic Colorectal Cancer Patients—The Importance of Tissue/Plasma Discordant Cases. Journal of Clinical Medicine, 10(1), 87. https://doi.org/10.3390/jcm10010087