
High-Throughput Proteomic Profiling of Nipple Aspirate Fluid from Breast Cancer Patients Compared with Non-Cancer Controls: A Step Closer to Clinical Feasibility


Abstract
1. Introduction
2. Experimental Section
2.1. Study Participants and Sample Collection
2.2. Sample Characterisation
2.3. Proteomic Analysis
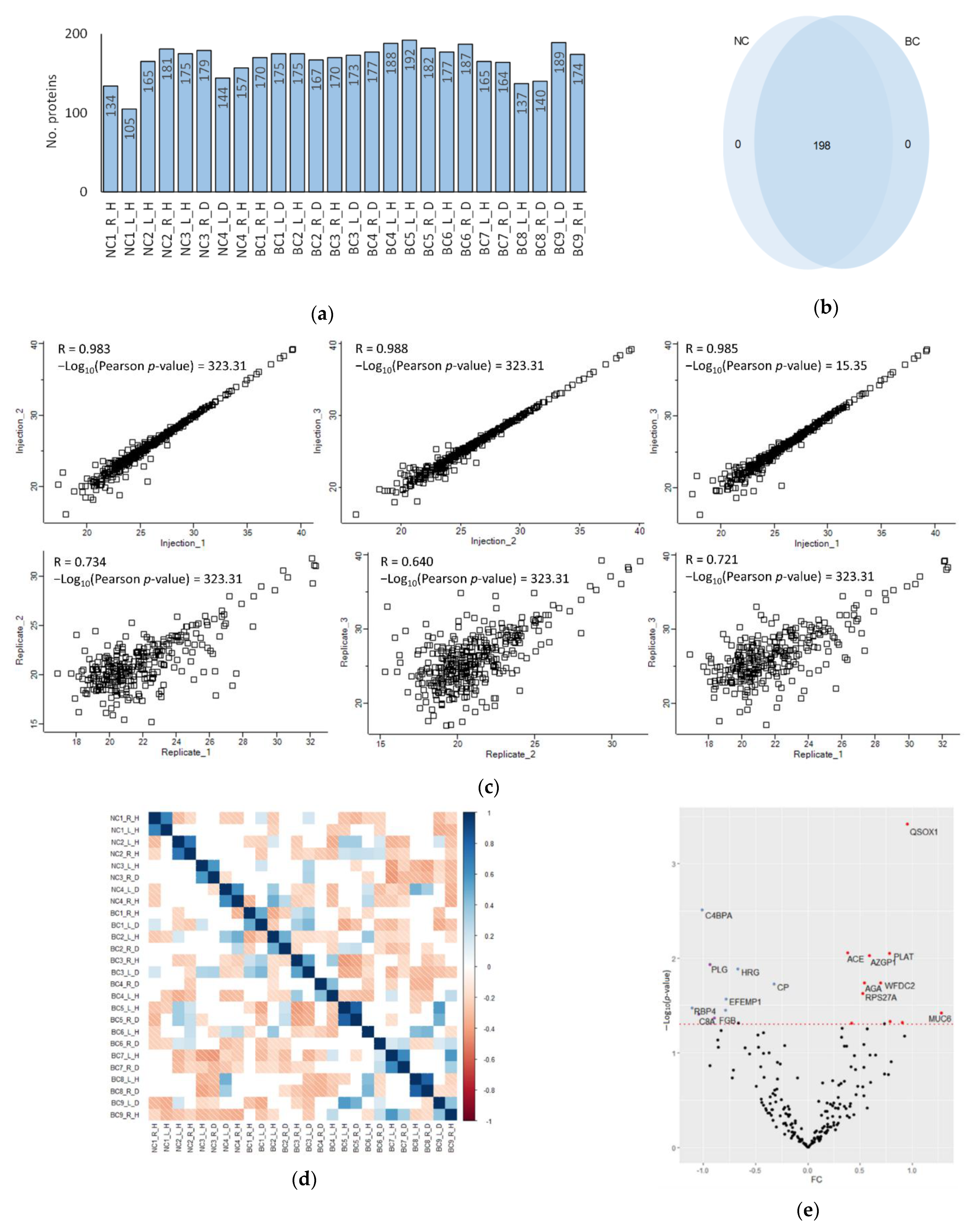
2.4. Data Analysis
3. Results
3.1. NAF Characterisation
3.2. A One-Dimensional Insight into the NAF Proteome
3.3. Quantification of Proteomic Pertubations in BC NAF Compared with NC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Porter, P. “Westernizing” women’s risks? Breast cancer in lower-income countries. N. Engl. J. Med. 2008, 358, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Swaminathan, R.; Brenner, H.; Chen, K.; Chia, K.S.; Chen, J.G.; Law, S.C.; Ahn, Y.O.; Xiang, Y.B.; Yeole, B.B.; et al. Cancer survival in Africa, Asia, and Central America: A population-based study. Lancet Oncol. 2010, 11, 165–173. [Google Scholar] [CrossRef]
- Lima, Z.S.; Ebadi, M.R.; Amjad, G.; Younesi, L. Application of Imaging Technologies in Breast Cancer Detection: A Review Article. Open Access Maced. J. Med. Sci. 2019, 7, 838–848. [Google Scholar] [CrossRef]
- Marmot, M.G.; Altman, D.G.; Cameron, D.A.; Dewar, J.A.; Thompson, S.G.; Wilcox, M. The benefits and harms of breast cancer screening: An independent review. Br. J. Cancer 2013, 108, 2205–2240. [Google Scholar] [CrossRef]
- Ho, P.J.; Bok, C.M.; Ishak, H.M.M.; Lim, L.Y.; Liu, J.; Wong, F.Y.; Chia, K.S.; Tan, M.H.; Chay, W.Y.; Hartman, M.; et al. Factors associated with false-positive mammography at first screen in an Asian population. PLoS ONE 2019, 14, e0213615. [Google Scholar] [CrossRef]
- Hellquist, B.N.; Czene, K.; Hjälm, A.; Nyström, L.; Jonsson, H. Effectiveness of population-based service screening with mammography for women ages 40 to 49 years with a high or low risk of breast cancer: Socioeconomic status, parity, and age at birth of first child. Cancer 2015, 121, 251–258. [Google Scholar] [CrossRef]
- Sharma, N.; McMahon, M.; Haigh, I.; Chen, Y.; Dall, B.J.G. The Potential Impact of Digital Breast Tomosynthesis on the Benign Biopsy Rate in Women Recalled within the UK Breast Screening Programme. Radiology 2019, 291, 310–317. [Google Scholar] [CrossRef]
- Skaane, P.; Bandos, A.I.; Gullien, R.; Eben, E.B.; Ekseth, U.; Haakenaasen, U.; Izadi, M.; Jebsen, I.N.; Jahr, G.; Krager, M.; et al. Prospective trial comparing full-field digital mammography (FFDM) versus combined FFDM and tomosynthesis in a population-based screening programme using independent double reading with arbitration. Eur. Radiol. 2013, 23, 2061–2071. [Google Scholar] [CrossRef]
- Ciatto, S.; Houssami, N.; Bernardi, D.; Caumo, F.; Pellegrini, M.; Brunelli, S.; Tuttobene, P.; Bricolo, P.; Fantò, C.; Valentini, M.; et al. Integration of 3D digital mammography with tomosynthesis for population breast-cancer screening (STORM): A prospective comparison study. Lancet Oncol. 2013, 14, 583–589. [Google Scholar] [CrossRef]
- Mannello, F.; Tonti, G.A.; Medda, V.; Simone, P.; Darbre, P.D. Analysis of aluminium content and iron homeostasis in nipple aspirate fluids from healthy women and breast cancer-affected patients. J. Appl. Toxicol. 2011, 31, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Anderson, K.E.; Nagamani, M.; Grady, J.J.; Lu, L.J. Dietary intake of lactose as a strong predictor for secretor status of nipple aspirate fluid in healthy premenopausal nonlactating women. Clin. Cancer Res. 2008, 14, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.S.; Pang, D.; Wang, F.; Xue, Y.W.; Gao, D.N.; Li, H.; Li, K.; Wang, B.Y.; Wang, D.; Li, H.Y. Nipple aspirate fluid collection, related factors and relationship between carcinoembryonic antigen in nipple aspirate fluid and breast diseases in women in Harbin, PRC. Cancer Epidemiol. Biomark. Prev. 2009, 18, 732–738. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morimoto, Y.; Conroy, S.M.; Franke, A.A.; Maskarinec, G. Nipple aspirate fluid producer status among premenopausal women in Hawaii. Breast J. 2012, 18, 504–505. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suijkerbuijk, K.P.; van der Wall, E.; Meijrink, H.; Pan, X.; Borel Rinkes, I.H.; Ausems, M.G.; van Diest, P.J. Successful oxytocin-assisted nipple aspiration in women at increased risk for breast cancer. Fam Cancer 2010, 9, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Gui, G.; Zhang, K.; Twelves, D.; Kliethermes, B.; Sauter, E.R. Proteins and carbohydrates in nipple aspirate fluid predict the presence of atypia and cancer in women requiring diagnostic breast biopsy. BMC Cancer 2012, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, N.L. Physiologic, biochemical, and cytologic aspects of nipple aspirate fluid. Breast Cancer Res. Treat. 1986, 8, 7–19. [Google Scholar] [CrossRef]
- Shaheed, S.-u.; Tait, C.; Kyriacou, K.; Linforth, R.; Salhab, M.; Sutton, C. Evaluation of nipple aspirate fluid as a diagnostic tool for early detection of breast cancer. Clin. Proteom. 2018, 15, 3. [Google Scholar] [CrossRef]
- Mannello, F. New horizon for breast cancer biomarker discoveries: What might the liquid biopsy of nipple aspirate fluid hold? Proteom. Clin. Appl. 2017, 11. [Google Scholar] [CrossRef]
- Sharma, G.N.; Dave, R.; Sanadya, J.; Sharma, P.; Sharma, K.K. Various types and management of breast cancer: An overview. J. Adv. Pharm. Technol. Res. 2010, 1, 109–126. [Google Scholar]
- Brunoro, G.V.; Carvalho, P.C.; Ferreira, A.T.; Perales, J.; Valente, R.H.; de Moura Gallo, C.V.; Pagnoncelli, D.; Neves-Ferreira, A.G. Proteomic profiling of nipple aspirate fluid (NAF): Exploring the complementarity of different peptide fractionation strategies. J. Proteom. 2015, 117, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Shaheed, S.U.; Tait, C.; Kyriacou, K.; Mullarkey, J.; Burrill, W.; Patterson, L.H.; Linforth, R.; Salhab, M.; Sutton, C.W. Nipple aspirate fluid-A liquid biopsy for diagnosing breast health. Proteom. Clin. Appl. 2017, 11. [Google Scholar] [CrossRef]
- Geyer, P.E.; Kulak, N.A.; Pichler, G.; Holdt, L.M.; Teupser, D.; Mann, M. Plasma Proteome Profiling to Assess Human Health and Disease. Cell Syst. 2016, 2, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Chisanga, D.; Alessandro, R.; Ang, C.S.; Askenase, P.; Batagov, A.O.; Benito-Martin, A.; Camussi, G.; Clayton, A.; et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J. Extracell Vesicles 2017, 6, 1321455. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Zhang, L.; Shao, Z.M.; Beatty, P.; Sartippour, M.; Wang, H.J.; Elashoff, R.; Chang, H.; Brooks, M.N. The use of oxytocin in nipple fluid aspiration. Breast J. 2003, 9, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Brunoro, G.V.F.; Carvalho, P.C.; Barbosa, V.C.; Pagnoncelli, D.; De Moura Gallo, C.V.; Perales, J.; Zahedi, R.P.; Valente, R.H.; Neves-Ferreira, A. Differential proteomic comparison of breast cancer secretome using a quantitative paired analysis workflow. BMC Cancer 2019, 19, 365. [Google Scholar] [CrossRef]
- Chatterton, R.T.; Heinz, R.E.; Fought, A.J.; Ivancic, D.; Shappell, C.; Allu, S.; Gapstur, S.; Scholtens, D.M.; Gann, P.H.; Khan, S.A. Nipple Aspirate Fluid Hormone Concentrations and Breast Cancer Risk. Horm Cancer 2016, 7, 127–136. [Google Scholar] [CrossRef]
- Sauter, E.R.; Winn, J.N.; Dale, P.S.; Wagner-Mann, C. Nipple aspirate fluid color is associated with breast cancer. Cancer Detect. Prev. 2006, 30, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Mannello, F.; Tonti, G.A.; Canestrari, F. Nutrients and nipple aspirate fluid composition: The breast microenvironment regulates protein expression and cancer aetiology. Genes Nutr. 2008, 3, 77–85. [Google Scholar] [CrossRef][Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Moraes, L.A.; Ampomah, P.B.; Lim, L.H.K. Annexin A1 in inflammation and breast cancer: A new axis in the tumor microenvironment. Cell Adh. Migr. 2018, 12, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Shapiro, D.J. The immune system and inflammation in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, P.; Li, L.; Gu, H.; Zhang, X.; Zhang, L.; Wang, N.; Gan, L.; Wang, Q.; Zhang, W.; et al. CD59 Regulation by SOX2 Is Required for Epithelial Cancer Stem Cells to Evade Complement Surveillance. Stem Cell Rep. 2017, 8, 140–151. [Google Scholar] [CrossRef]
- Lal, I.; Dittus, K.; Holmes, C.E. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res. BCR 2013, 15, 207. [Google Scholar] [CrossRef]
- Harrison, P.; Cramer, E.M. Platelet alpha-granules. Blood Rev. 1993, 7, 52–62. [Google Scholar] [CrossRef]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.A.; Edwards, A.B.; Wittliff, J.L. Expression of urokinase-type plasminogen activator (uPA), its receptor (uPAR), and inhibitor (PAI-1) in human breast carcinomas and their clinical relevance. J. Clin. Lab. Anal. 2012, 26, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, M.P.; Kulasingam, V.; Sauter, E.R.; Kliethermes, B.; Diamandis, E.P. Nipple Aspirate Fluid Proteome of Healthy Females and Patients with Breast Cancer. Clin. Chem. 2010, 56, 848. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Subject | Pathology | Disease Breast | Grade | TNM Staging | Phenotype | Age | Pre/Post-Menopausal | Breast | NAF—Appearance | Conc. mg/mL | Total Protein (μg) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NC1 | Normal | N/A | N/A | N/A | N/A | 48 | Pre | Left | White, cloudy | 15.86 | 793.00 |
| Right | White, cloudy | 18.20 | 910.00 | ||||||||
| NC2 | Normal | N/A | N/A | N/A | N/A | 31 | Pre | Left | Light brown-waxy | 34.72 | 4999.49 |
| Right | Light brown-waxy | 33.30 | 4961.17 | ||||||||
| NC3 | Benign (phyllodes) | Right | N/A | N/A | N/A | 43 | Pre | Left | White, cloudy | 16.53 | 793.00 |
| Right | Light brown, cloudy | 21.50 | 1892.00 | ||||||||
| NC4 | Benign (duct ectasia) | Left | N/A | N/A | N/A | 59 | Post | Left | Terracotta, thick | 65.81 | 7831.75 |
| Right | Light brown, thick | 81.20 | 8039.20 | ||||||||
| BC1 | DCIS | Left | N/A | N/A | N/A | 62 | Post | Left | Yellow, cloudy | 26.80 | 750.00 |
| Right | Yellow, cloudy | 27.52 | 770.00 | ||||||||
| BC2 | DCIS | Right | N/A | N/A | N/A | 57 | Post | Left | Light brown, cloudy | 63.25 | 1138.46 |
| Right | Light brown, cloudy | 28.04 | 504.79 | ||||||||
| BC3 | IC (ductal) | Left | 1 | G1 T3 N? | ER+ve/HER2−ve | 59 | Post | Left | White, cloudy | 3.28 | 522.00 |
| Right | Olive green, thick | 15.82 | 1725.00 | ||||||||
| BC4 | IC (ductal) | Right | 3 | G3 pT2 pN0 | ER+ve/HER2−ve | 50 | Post | Left | Light brown, cloudy | 35.14 | 1687.00 |
| Right | Light brown, cloudy | 22.74 | 637.00 | ||||||||
| BC5 | IC (ductal) | Right | 3 | G3 T1c or T2 N0? | ER+ve/HER2−ve | 86 | Post | Left | Light brown, cloudy | 58.81 | 11,643.43 |
| Right | Light brown, cloudy | 69.48 | 8893.00 | ||||||||
| BC6 | IC (ductal) | Right | 1 | pT2 | ND | 48 | Pre | Left | White, cloudy | 14.65 | 1743.25 |
| Right | Light brown, cloudy | 42.30 | 3342.02 | ||||||||
| BC7 | IC (ductal) | Right | ND | pT1c N1 | ND | 47 | Pre | Left | Yellow, translucent | 35.46 | 921.96 |
| Right | Red, opaque | 53.80 | 699.40 | ||||||||
| BC8 | IC (ductal) | Right | 1 | G1 ypT1c ypN2 | Tubular | 44 | Pre | Left | White, opaque | 39.72 | 993.00 |
| Right | White, opaque | 22.13 | 774.55 | ||||||||
| BC9 | IC (ductal) | Left | 2 | G2 pT1c | NST | 47 | ND | Left | Yellow-brown, thick | 46.91 | 3471.64 |
| Right | Yellow-brown, thick | 62.97 | 7493.91 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, A.L.; Shaheed, S.u.; Sutton, C.W. High-Throughput Proteomic Profiling of Nipple Aspirate Fluid from Breast Cancer Patients Compared with Non-Cancer Controls: A Step Closer to Clinical Feasibility. J. Clin. Med. 2021, 10, 2243. https://doi.org/10.3390/jcm10112243
George AL, Shaheed Su, Sutton CW. High-Throughput Proteomic Profiling of Nipple Aspirate Fluid from Breast Cancer Patients Compared with Non-Cancer Controls: A Step Closer to Clinical Feasibility. Journal of Clinical Medicine. 2021; 10(11):2243. https://doi.org/10.3390/jcm10112243
Chicago/Turabian StyleGeorge, Amy L., Sadr ul Shaheed, and Chris W. Sutton. 2021. "High-Throughput Proteomic Profiling of Nipple Aspirate Fluid from Breast Cancer Patients Compared with Non-Cancer Controls: A Step Closer to Clinical Feasibility" Journal of Clinical Medicine 10, no. 11: 2243. https://doi.org/10.3390/jcm10112243
APA StyleGeorge, A. L., Shaheed, S. u., & Sutton, C. W. (2021). High-Throughput Proteomic Profiling of Nipple Aspirate Fluid from Breast Cancer Patients Compared with Non-Cancer Controls: A Step Closer to Clinical Feasibility. Journal of Clinical Medicine, 10(11), 2243. https://doi.org/10.3390/jcm10112243