
Regulation of the Target of Rapamycin and Other Phosphatidylinositol 3-Kinase-Related Kinases by Membrane Targeting

{kind=link}
{kind=link}
{kind=link}
Abstract
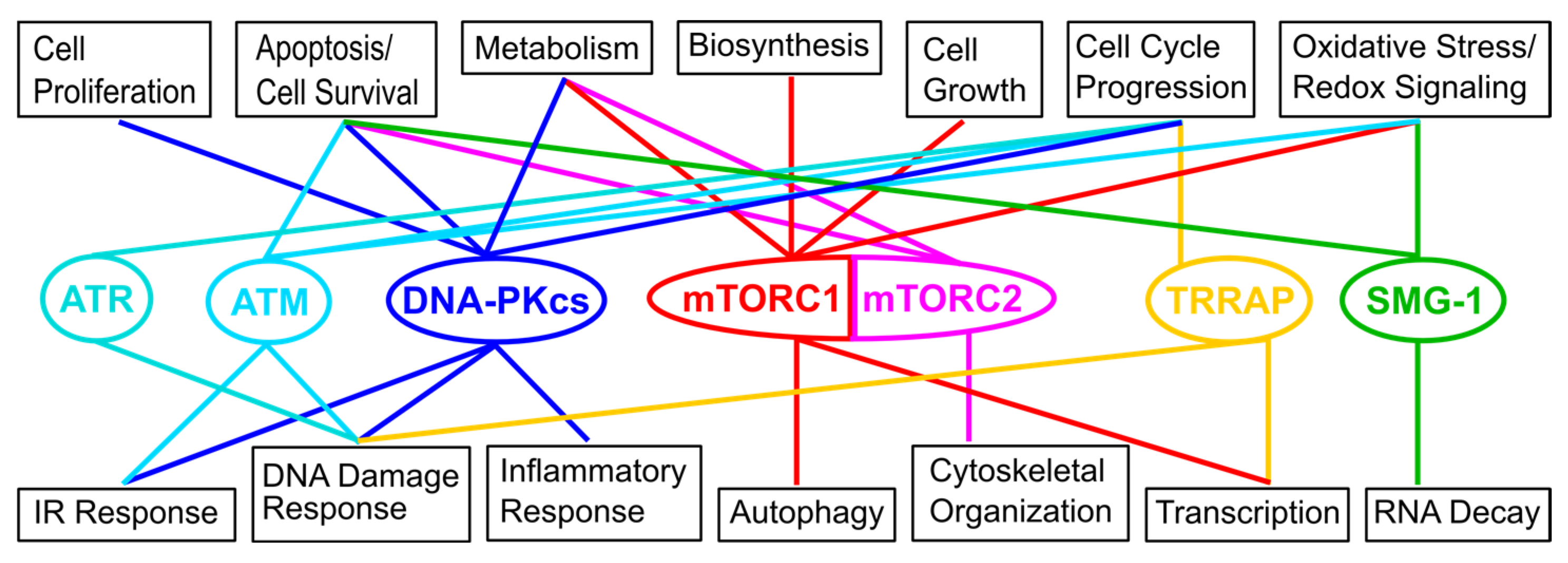
:1. Introduction
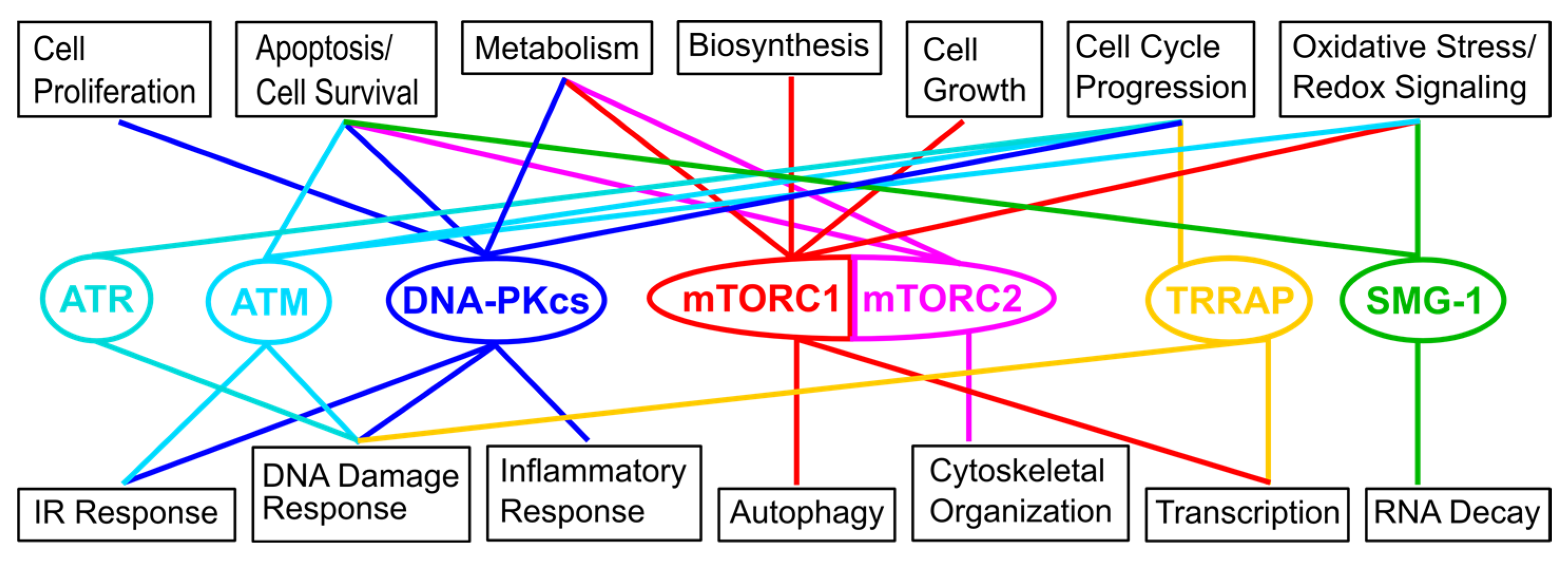
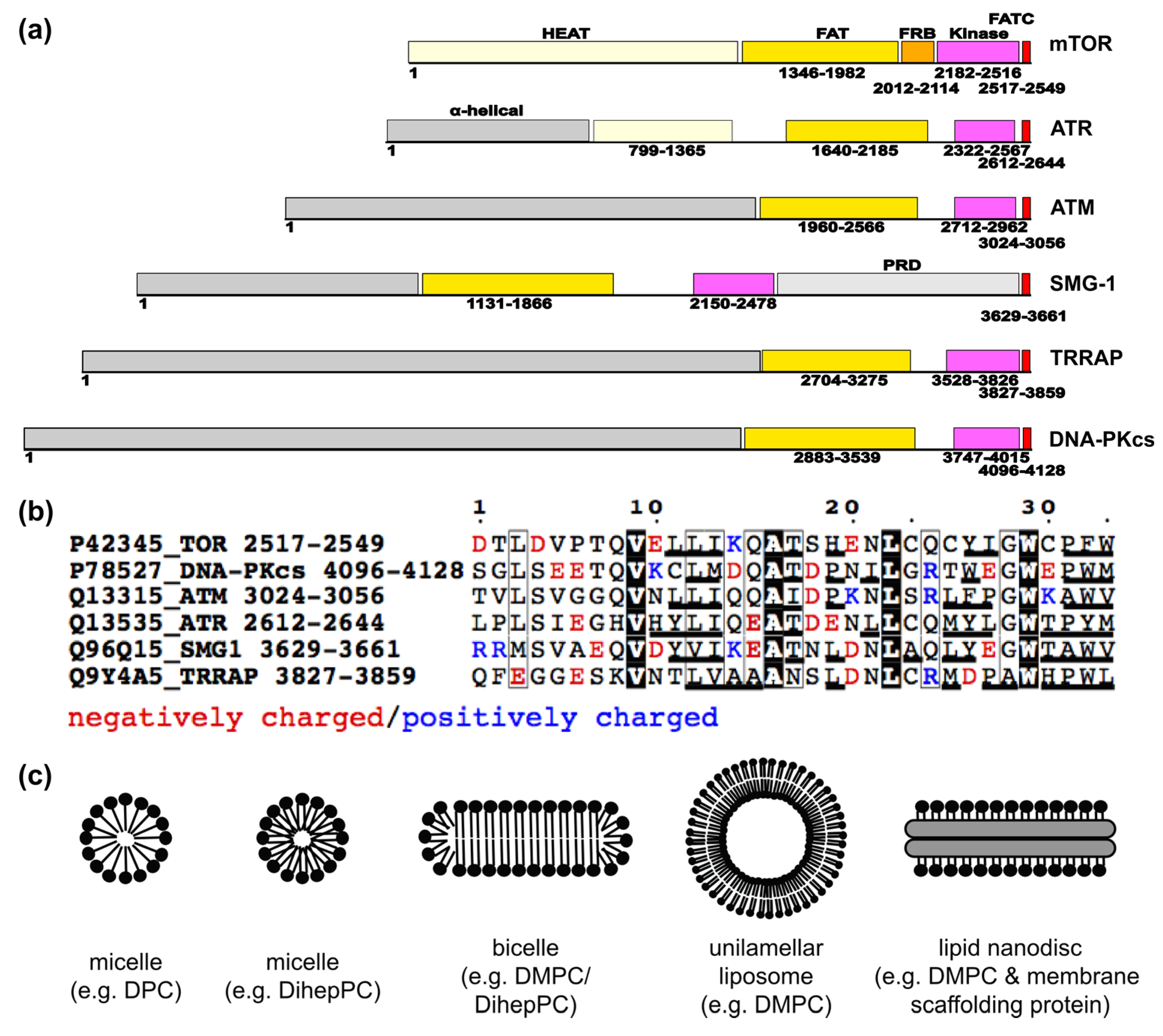
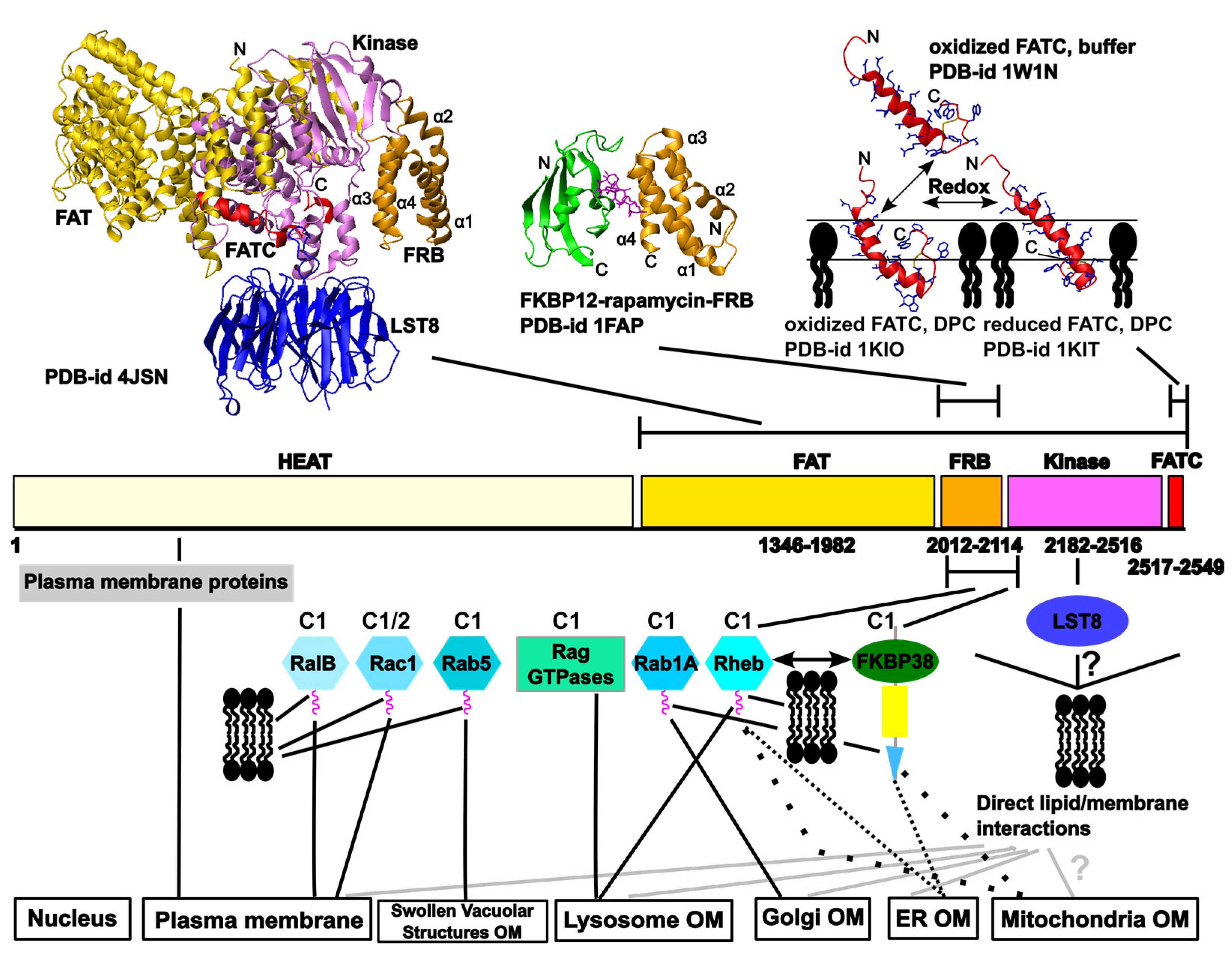
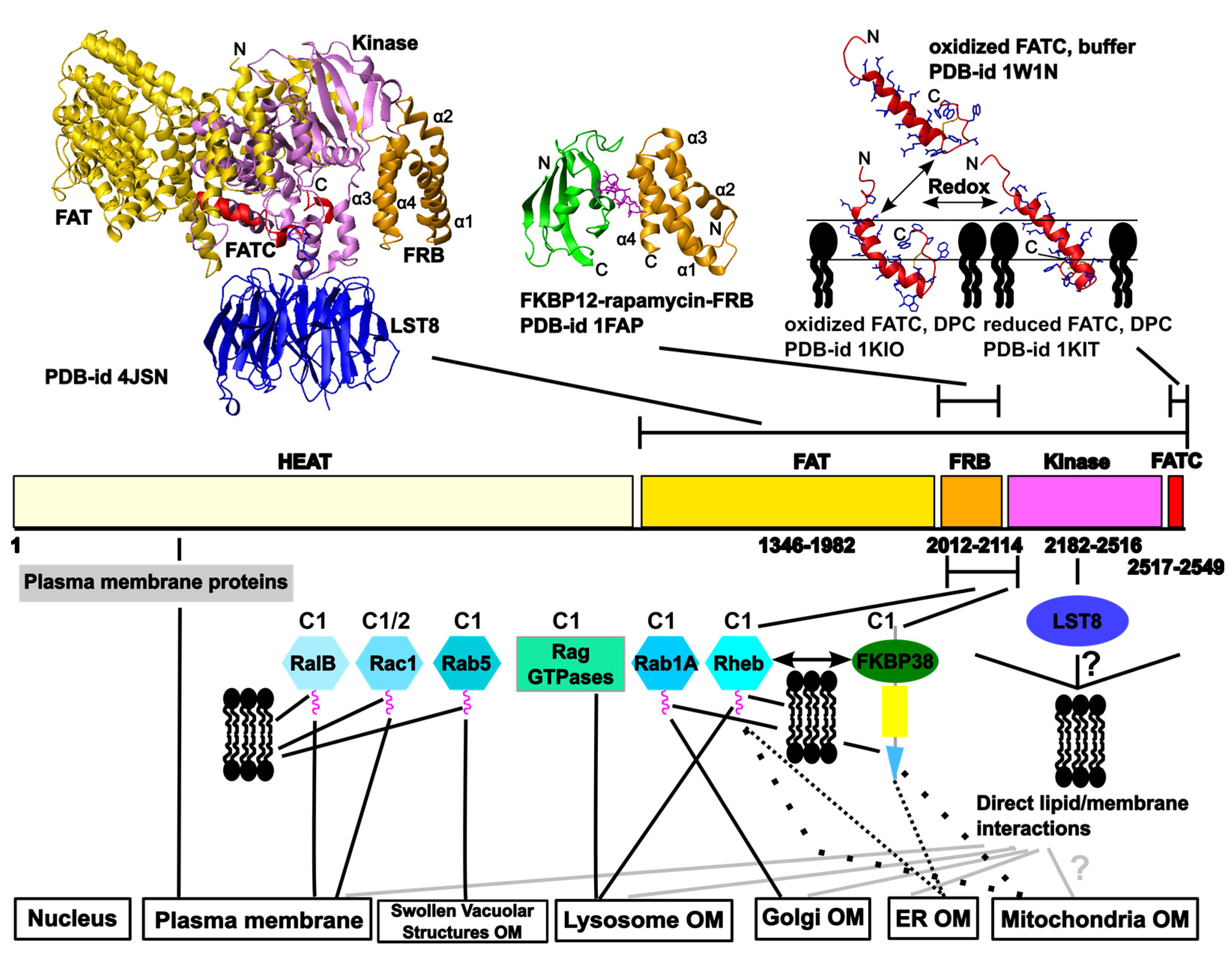
2. Overview of the Network of Interactions Mediating the Localization of the Target of Rapamycin (TOR) at Different Cellular Membranes
2.1. Regulation of TOR Membrane Association by GTPases
2.1.1 TOR Regulators that May Play a Role for the Localization to the Outer Membranes of the Endoplasmic Reticulum (ER), the Golgi Apparatus, and Mitochondria
2.1.2. The Localization of mTOR Complex 1 (mTORC1) at Lysosomes
2.1.3. Further GTPases that May Play a Role in TOR Membrane Targeting
2.2. Suggested Direct Lipid/Membrane Interactions of TOR Domains
2.2.1. The FATC Domain of TOR May Function as a Conditional, Redox-Sensitive Membrane Anchor
2.2.2. Lipid/ Membrane Interactions by the FKBP-Rapamycin Binding (FRB) Domain
2.3. Further Interactions with Membrane-Localized Proteins
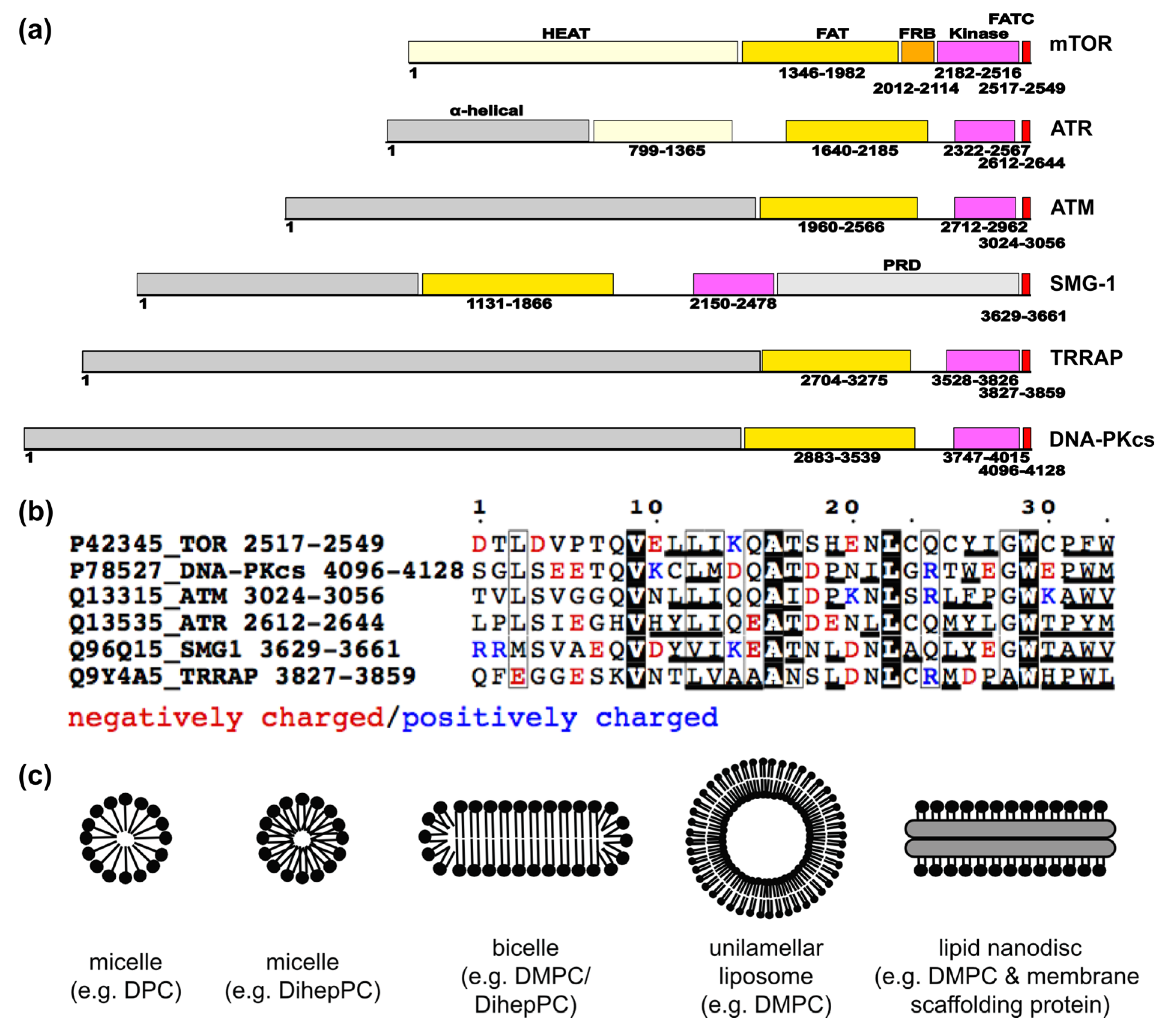
3. The Current Knowledge about Membrane Localization of Other PIKKs
4. Conclusions Regarding PIKK Activation at Different Cellular Membranes
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Keith, C.T.; Schreiber, S.L. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science 1995, 270, 50–51. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair 2009, 8, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Tichy, A.; Durisova, K.; Novotna, E.; Zarybnicka, L.; Vavrova, J.; Pejchal, J.; Sinkorova, Z. Phosphatidylinositol-3-kinase related kinases (PIKKs) in radiation-induced DNA damage. Mil. Med. Sci. Lett. 2012, 81, 177–187. [Google Scholar]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Aramburu, J.; Ortells, M.C.; Tejedor, S.; Buxade, M.; Lopez-Rodriguez, C. Transcriptional regulation of the stress response by mTOR. Sci. Signal 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Gradzka, I.; Sochanowicz, B.; Brzoska, K.; Wojciuk, G.; Sommer, S.; Wojewodzka, M.; Gasinska, A.; Degen, C.; Jahreis, G.; Szumiel, I. Cis-9,trans-11-conjugated linoleic acid affects lipid raft composition and sensitizes human colorectal adenocarcinoma HT-29 cells to X-radiation. Biochim. Biophys. Acta 2013, 1830, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Ditch, S.; Paull, T.T. The ATM protein kinase and cellular redox signaling: Beyond the DNA damage response. Trends Biochem. Sci. 2012, 37, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Shen, Y.; Jiang, N.; Fei, X.; Mi, J. Emerging roles of DNA-PK besides DNA repair. Cell Signal. 2011, 23, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.; Ralser, M. ATM is a redox sensor linking genome stability and carbon metabolism. Sci. Signal 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Naura, A.S.; Errami, Y.; Zerfaoui, M.; Kim, H.; Kim, J.G.; Abd Elmageed, Z.Y.; Abdel-Mageed, A.B.; Giardina, C.; Beg, A.A.; et al. Phosphorylation of p50 NF-kappaB at a single serine residue by DNA-dependent protein kinase is critical for VCAM-1 expression upon TNF treatment. J. Biol. Chem. 2010, 285, 41152–41160. [Google Scholar] [CrossRef] [PubMed]
- Lucero, H.; Gae, D.; Taccioli, G.E. Novel localization of the DNA-PK complex in lipid rafts: A putative role in the signal transduction pathway of the ionizing radiation response. J. Biol. Chem. 2003, 278, 22136–22143. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Lee, K.J.; Fattah, K.R.; Lin, Y.F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. AKT promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. MCR 2012, 10, 945–957. [Google Scholar] [CrossRef]
- Watters, D.; Khanna, K.K.; Beamish, H.; Birrell, G.; Spring, K.; Kedar, P.; Gatei, M.; Stenzel, D.; Hobson, K.; Kozlov, S.; et al. Cellular localisation of the ataxia-telangiectasia (ATM) gene product and discrimination between mutated and normal forms. Oncogene 1997, 14, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Zhan, Q.; el-Deiry, W.S.; Carrier, F.; Jacks, T.; Walsh, W.V.; Plunkett, B.S.; Vogelstein, B.; Fornace, A.J., Jr. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992, 71, 587–597. [Google Scholar] [CrossRef]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Jeggo, P.A. Clinical impact of ATR checkpoint signalling failure in humans. Cell Cycle 2003, 2, 194–195. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in seckel syndrome. Nat. Genet. 2003, 33, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, M.; Ijspeert, H.; Verkaik, N.S.; Turul, T.; Wiegant, W.W.; Morotomi-Yano, K.; Mari, P.O.; Tezcan, I.; Chen, D.J.; Zdzienicka, M.Z.; et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits artemis activation and nonhomologous end-joining. J. Clin. Investig. 2009, 119, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, N.; Platzer, M.; Rosenthal, A.; Dork, T.; Bendix, R.; Skawran, B.; Stuhrmann, M.; Wegner, R.D.; Sperling, K.; Banin, S.; et al. Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum. Mol. Genet. 1999, 8, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.Y.; Greiner, T.C.; Weisenburger, D.D.; Chan, W.C.; Vose, J.M.; Smith, L.M.; Armitage, J.O.; Mayer, R.A.; Pike, B.L.; Collins, F.S.; et al. Oligonucleotide microarrays demonstrate the highest frequency of atm mutations in the mantle cell subtype of lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 5372–5377. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.Y.; Pyun, B.J.; Seo, H.R.; Jin, Y.B.; Lee, H.J.; Lee, Y.J.; Lee, Y.S. Inhibition of snail1-DNA-PKcs protein-protein interface sensitizes cancer cells and inhibits tumor metastasis. J. Biol. Chem. 2013, 288, 32506–32516. [Google Scholar] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Houghton, P.J. The mTOR pathway negatively controls atm by up-regulating miRNAs. Proc. Natl. Acad. Sci. USA 2013, 110, 11869–11874. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Jacinto, E.; Hall, M.N. TOR signalling in bugs, brain and brawn. Nat. Rev. Mol. Cell Biol. 2003, 4, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Soulard, A.; Hall, M.N. Snapshot: mTOR signaling. Cell 2007, 129. [Google Scholar] [CrossRef] [PubMed]
- Dann, S.G.; Selvaraj, A.; Thomas, G. mTOR complex1-S6K1 signaling: At the crossroads of obesity, diabetes and cancer. Trends Mol. Med. 2007, 13, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Valjent, E.; Fisone, G. mTORC1 signaling in parkinson’s disease and L-DOPA-induced dyskinesia: A sensitized matter. Cell Cycle 2010, 9, 2713–2718. [Google Scholar] [CrossRef] [PubMed]
- Young, D.A.; Nickerson-Nutter, C.L. mTOR—Beyond transplantation. Curr. Opin. Pharmacol. 2005, 5, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino acids and mtorc1: From lysosomes to disease. Trends Mol. Med. 2012, 18, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Shortt, J.; Martin, B.P.; Newbold, A.; Hannan, K.M.; Devlin, J.R.; Baker, A.J.; Ralli, R.; Cullinane, C.; Schmitt, C.A.; Reimann, M.; et al. Combined inhibition of PI3K-related DNA damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood 2013, 121, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Cornu, M.; Albert, V.; Hall, M.N. mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev. 2013, 23, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Masse, I.; Molin, L.; Mouchiroud, L.; Vanhems, P.; Palladino, F.; Billaud, M.; Solari, F. A novel role for the SMG-1 kinase in lifespan and oxidative stress resistance in caenorhabditis elegans. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Romanow, W.J.; Geisen, C.; Otterness, D.M.; Mercurio, F.; Wang, H.G.; Dalton, W.S.; Abraham, R.T. A protective role for the human SMG-1 kinase against tumor necrosis factor-alpha-induced apoptosis. J. Biol. Chem. 2008, 283, 13174–13184. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Yamashita, A.; Kashima, I.; Ogata, K.; Ishiura, S.; Ohno, S. Distant N- and C-terminal domains are required for intrinsic kinase activity of SMG-1, a critical component of nonsense-mediated mRNA decay. J. Biol. Chem. 2007, 282, 7799–7808. [Google Scholar] [CrossRef] [PubMed]
- Unno, A.; Takada, I.; Takezawa, S.; Oishi, H.; Baba, A.; Shimizu, T.; Tokita, A.; Yanagisawa, J.; Kato, S. TRRAP as a hepatic coactivator of LXR and FXR function. Biochem. Biophys. Res. Commun. 2005, 327, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Herceg, Z.; Wang, Z.Q. Rendez-vous at Mitosis: TRRAPed in the chromatin. Cell Cycle 2005, 4, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Robert, F.; Hardy, S.; Nagy, Z.; Baldeyron, C.; Murr, R.; Dery, U.; Masson, J.Y.; Papadopoulo, D.; Herceg, Z.; Tora, L. The transcriptional histone acetyltransferase cofactor TRRAP associates with the MRN repair complex and plays a role in DNA double-strand break repair. Mol. Cell Biol. 2006, 26, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Bosotti, R.; Isacchi, A.; Sonnhammer, E.L. FAT: A novel domain in PIK-related kinases. Trends Biochem. Sci. 2000, 25, 225–227. [Google Scholar] [CrossRef]
- Perry, J.; Kleckner, N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell 2003, 112, 151–155. [Google Scholar] [CrossRef]
- Groves, M.R.; Barford, D. Topological characteristics of helical repeat proteins. Curr. Opin. Struct. Biol. 1999, 9, 383–389. [Google Scholar] [CrossRef]
- Knutson, B.A. Insights into the domain and repeat architecture of target of rapamycin. J. Struct. Biol. 2010, 170, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Baretic, D.; Williams, R.L. PIKKs-the solenoid nest where partners and kinases meet. Curr. Opin. Struct. Biol. 2014, 29, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Mordes, D.A.; Glick, G.G.; Zhao, R.; Cortez, D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008, 22, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Hara, K.; Inoue, H.; Kawa, Y.; Tokunaga, C.; Hidayat, S.; Yoshino, K.; Kuroda, Y.; Yonezawa, K. Carboxyl-terminal region conserved among phosphoinositide-kinase-related kinases is indispensable for mTOR function in vivo and in vitro. Genes Cells 2000, 5, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Hoke, S.M.; Irina Mutiu, A.; Genereaux, J.; Kvas, S.; Buck, M.; Yu, M.; Gloor, G.B.; Brandl, C.J. Mutational analysis of the C-terminal FATC domain of saccharomyces cerevisiae Tra1. Curr. Genet. 2010, 56, 447–465. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 2006, 281, 15741–15746. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef] [PubMed]
- Sommer, L.A.; Schaad, M.; Dames, S.A. NMR- and circular dichroism-monitored lipid binding studies suggest a general role for the fatc domain as membrane anchor of phosphatidylinositol 3-kinase-related kinases (PIKK). J. Biol. Chem. 2013, 288, 20046–20063. [Google Scholar] [CrossRef] [PubMed]
- Gouet, P.; Courcelle, E.; Stuart, D.I.; Metoz, F. ESPript: Analysis of multiple sequence alignments in POSTScript. Bioinformatics 1999, 15, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Chen, J.; Schreiber, S.L.; Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Dames, S.A.; Mulet, J.M.; Rathgeb-Szabo, K.; Hall, M.N.; Grzesiek, S. The solution structure of the FATC domain of the protein kinase target of rapamycin suggests a role for redox-dependent structural and cellular stability. J. Biol. Chem. 2005, 280, 20558–20564. [Google Scholar] [CrossRef] [PubMed]
- Dames, S.A. Structural basis for the association of the redox-sensitive target of rapamycin FATC domain with membrane-mimetic micelles. J. Biol. Chem. 2010, 285, 7766–7775. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide protein data bank. Nat. Struct. Biol. 2003, 10. [Google Scholar] [CrossRef] [PubMed]
- Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Mugler, A.; Bailey, A.G.; Takahashi, K.; ten Wolde, P.R. Membrane clustering and the role of rebinding in biochemical signaling. Biophys. J. 2012, 102, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Mugler, A.; Tostevin, F.; Ten Wolde, P.R. Spatial partitioning improves the reliability of biochemical signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 5927–5932. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, D.; Walther, T.C. TORC2 plasma membrane localization is essential for cell viability and restricted to a distinct domain. Mol. Biol. Cell 2009, 20, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Drenan, R.M.; Liu, X.; Bertram, P.G.; Zheng, X.F. FKBP12-rapamycin-associated protein or mammalian target of rapamycin (FRAP/mTOR) localization in the endoplasmic reticulum and the Golgi apparatus. J. Biol. Chem. 2004, 279, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.; Schneider, U.; Howald, I.; Schmidt, A.; Hall, M.N. HEAT repeats mediate plasma membrane localization of Tor2p in yeast. J. Biol. Chem. 2000, 275, 37011–37020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shu, L.; Hosoi, H.; Murti, K.G.; Houghton, P.J. Predominant nuclear localization of mammalian target of rapamycin in normal and malignant cells in culture. J. Biol. Chem. 2002, 277, 28127–28134. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zheng, X.F. Endoplasmic reticulum and Golgi localization sequences for mammalian target of rapamycin. Mol. Biol. Cell 2007, 18, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Wu, C.C.; Kim, S.J.; Facchinetti, V.; Julien, L.A.; Finlan, M.; Roux, P.P.; Su, B.; Jacinto, E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010, 29, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- Schieke, S.M.; Phillips, D.; McCoy, J.P., Jr.; Aponte, A.M.; Shen, R.F.; Balaban, R.S.; Finkel, T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J. Biol. Chem. 2006, 281, 27643–27652. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Khanna, K.K.; Lavin, M.F. Defective radiation signal transduction in ataxia-telangiectasia cells. Int. J. Radiat. Biol. 2000, 76, 1025–1035. [Google Scholar] [PubMed]
- Odagaki, Y.; Clardy, J. Structural basis for peptidomimicry by the effector element of rapamycin. J. Am. Chem. Soc. 1997, 119, 10253–10254. [Google Scholar] [CrossRef]
- Veverka, V.; Crabbe, T.; Bird, I.; Lennie, G.; Muskett, F.W.; Taylor, R.J.; Carr, M.D. Structural characterization of the interaction of mtor with phosphatidic acid and a novel class of inhibitor: Compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene 2007, 27, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Crowell, K.J.; Chen, J.; Jung, D.; Chiang, G.G.; Sareth, S.; Abraham, R.T.; Pellecchia, M. The FRB domain of mTOR: NMR solution structure and inhibitor design. Biochemistry 2006, 45, 10294–10302. [Google Scholar] [CrossRef] [PubMed]
- Marz, A.M.; Fabian, A.K.; Kozany, C.; Bracher, A.; Hausch, F. Large FK506-binding proteins shape the pharmacology of rapamycin. Mol. Cell Biol. 2013, 33, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Saucedo, L.J.; Gao, X.; Chiarelli, D.A.; Li, L.; Pan, D.; Edgar, B.A. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 2003, 5, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Tee, A.R.; Blenis, J.; Proud, C.G. Analysis of mTOR signaling by the small G-proteins, Rheb and RhebL1. FEBS Lett. 2005, 579, 4763–4768. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; DeVries, B.; Stambolic, V. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem. Biophys. Res. Commun. 2006, 344, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Zhang, Y.J.; Wei, Y.H.; Cho, J.H.; Morris, L.E.; Wang, H.Y.; Zheng, X.F. Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell 2014, 26, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Calero, M.; Chen, C.Z.; Zhu, W.; Winand, N.; Havas, K.A.; Gilbert, P.M.; Burd, C.G.; Collins, R.N. Dual prenylation is required for Rab protein localization and function. Mol. Biol. Cell 2003, 14, 1852–1867. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.Q.; Ali, B.R.; Ramalho, J.S.; Godfrey, R.F.; Barral, D.C.; Hume, A.N.; Seabra, M.C. Membrane targeting of Rab GTPases is influenced by the prenylation motif. Mol. Biol. Cell 2003, 14, 1882–1899. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Ma, D.; Liu, A.; Shen, X.; Wang, Q.J.; Liu, Y.; Jiang, Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science 2007, 318, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fonseca, B.D.; Tang, H.; Liu, R.; Elia, A.; Clemens, M.J.; Bommer, U.A.; Proud, C.G. Re-evaluating the roles of proposed modulators of mammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem. 2008, 283, 30482–30492. [Google Scholar] [CrossRef] [PubMed]
- Uhlenbrock, K.; Weiwad, M.; Wetzker, R.; Fischer, G.; Wittinghofer, A.; Rubio, I. Reassessment of the role of FKBP38 in the Rheb/mTORC1 pathway. FEBS Lett. 2009, 583, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Bai, X.; Zou, H.; Lai, Y.; Jiang, Y. Rheb GTPase controls apoptosis by regulating interaction of FKBP38 with Bcl-2 and Bcl-XL. J. Biol. Chem. 2010, 285, 8621–8627. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Lai, Y.; Zhao, X.; Yan, G.; Ma, D.; Cardenes, N.; SHIVa, S.; Liu, Y.; Bai, X.; Jiang, Y.; et al. Regulation of mammalian target of rapamycin complex 1 by Bcl-2 and Bcl-XL proteins. J. Biol. Chem. 2013, 288, 28824–28830. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Guan, K.L. Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 2013, 38, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, T.; Hirose, E.; Nakashima, N.; Ii, M.; Nishimoto, T. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J. Biol. Chem. 2001, 276, 7246–7257. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.V.; Hall, M.N. Regulation of TOR by small GTPases. EMBO Rep. 2012, 13, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Salloum, D.; Medlin, P.S.; Saqcena, M.; Yellen, P.; Perrella, B.; Foster, D.A. Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1). J. Biol. Chem. 2011, 286, 25477–25486. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.D.; Chen, X.W.; Kaplan, R.E.; Saltiel, A.R.; Walker, C.L.; Reiner, D.J.; Der, C.J. Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol. Cell 2014, 53, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Falsetti, S.C.; Wang, D.A.; Peng, H.; Carrico, D.; Cox, A.D.; Der, C.J.; Hamilton, A.D.; Sebti, S.M. Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth. Mol. Cell Biol. 2007, 27, 8003–8014. [Google Scholar] [CrossRef] [PubMed]
- Saci, A.; Cantley, L.C.; Carpenter, C.L. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell 2011, 42, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Bridges, D.; Fisher, K.; Zolov, S.N.; Xiong, T.; Inoki, K.; Weisman, L.S.; Saltiel, A.R. Rab5 proteins regulate activation and localization of target of rapamycin complex 1. J. Biol. Chem. 2012, 287, 20913–20921. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, H.; Morigasaki, S.; Murayama, S.; Zeng, C.T.; Shiozaki, K. Rab-family GTPase regulates TOR complex 2 signaling in fission yeast. Curr. Biol. 2010, 20, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Sommer, L.A.; Meier, M.A.; Dames, S.A. A fast and simple method for probing the interaction of peptides and proteins with lipids and membrane-mimetics using GB1 fusion proteins and NMR spectroscopy. Protein Sci. 2012, 21, 1566–1570. [Google Scholar] [CrossRef] [PubMed]
- Sommer, L.A.; Dames, S.A. Characterization of residue-dependent differences in the peripheral membrane association of the FATC domain of the kinase “target of rapamycin” by NMR and CD spectroscopy. FEBS Lett. 2014, 588, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Sommer, L.A.; Janke, J.J.; Bennett, W.F.; Burck, J.; Ulrich, A.S.; Tieleman, D.P.; Dames, S.A. Characterization of the immersion properties of the peripheral membrane anchor of the FATC domain of the kinase “target of rapamycin” by nmr, oriented CD spectroscopy, and MD simulations. J. Phys Chem. B 2014, 118, 4817–4831. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.T.; Beal, P.A.; Comb, M.J.; Schreiber, S.L. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J. Biol. Chem. 2000, 275, 7416–7423. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Selvy, P.E.; Lavieri, R.R.; Lindsley, C.W.; Brown, H.A. Phospholipase D: Enzymology, functionality, and chemical modulation. Chem. Rev. 2011, 111, 6064–6119. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.H.; Kim, D.H.; Kim, I.S.; Kim, J.H.; Lee, M.N.; Lee, H.J.; Kim, J.H.; Jang, S.K.; Suh, P.G.; Ryu, S.H. PLD2 forms a functional complex with mTOR/raptor to transduce mitogenic signals. Cell Signal 2006, 18, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.A. Phosphatidic acid signaling to mTOR: Signals for the survival of human cancer cells. Biochim. Biophys. Acta 2009, 1791, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Fang, Y.; Yoon, M.S.; Zhang, C.; Roccio, M.; Zwartkruis, F.J.; Armstrong, M.; Brown, H.A.; Chen, J. Phospholipase d1 is an effector of Rheb in the mTOR pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 8286–8291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wendel, A.A.; Keogh, M.R.; Harris, T.E.; Chen, J.; Coleman, R.A. Glycerolipid signals alter mTOR complex 2 (mTORC2) to diminish insulin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Camargo, D.C.; Link, N.M.; Dames, S.A. The FKBP-rapamycin binding domain of human TOR undergoes strong conformational changes in the presence of membrane mimetics with and without the regulator phosphatidic acid. Biochemistry 2012, 51, 4909–4921. [Google Scholar] [CrossRef] [PubMed]
- Toschi, A.; Lee, E.; Xu, L.; Garcia, A.; Gadir, N.; Foster, D.A. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: Competition with rapamycin. Mol. Cell Biol. 2009, 29, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Zimmerberg, J.; Kozlov, M.M. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 2006, 7, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Kam, Y.; Exton, J.H. Role of phospholipase D1 in the regulation of mTOR activity by lysophosphatidic acid. FASEB J. 2004, 18, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Troya, S.; Florencio, F.J.; Crespo, J.L. Target of rapamycin and LST8 proteins associate with membranes from the endoplasmic reticulum in the unicellular green alga Chlamydomonas reinhardtii. Eukaryot. Cell 2008, 7, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Busse, R.A.; Scacioc, A.; Krick, R.; Perez-Lara, A.; Thumm, M.; Kuhnel, K. Characterization of PROPPIN-phosphoinositide binding and role of loop 6CD in PROPPIN-membrane binding. Biophys. J. 2015, 108, 2223–2234. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Oliveira, T.M.; Prouteau, M.; Leitner, A.; Karuppasamy, M.; Konstantinidou, G.; Rispal, D.; Eltschinger, S.; Robinson, G.C.; Thore, S.; et al. Molecular basis of the rapamycin insensitivity of target of rapamycin complex 2. Mol. Cell 2015, 58, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, D.; Piccolis, M.; Chiaruttini, N.; Riezman, I.; Riezman, H.; Roux, A.; Walther, T.C.; Loewith, R. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat. Cell Biol. 2012, 14, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Fadri, M.; Daquinag, A.; Wang, S.; Xue, T.; Kunz, J. The pleckstrin homology domain proteins Slm1 and Slm2 are required for actin cytoskeleton organization in yeast and bind phosphatidylinositol-4,5-bisphosphate and TORC2. Mol. Biol. Cell 2005, 16, 1883–1900. [Google Scholar] [CrossRef] [PubMed]
- Partovian, C.; Ju, R.; Zhuang, Z.W.; Martin, K.A.; Simons, M. Syndecan-4 regulates subcellular localization of mTOR Complex2 and Akt activation in a PKCalpha-dependent manner in endothelial cells. Mol. Cell 2008, 32, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.; Murakami, M.; Gao, Y.; Simons, M. Phosphatidylinositol-4,5-bisphosphate mediates the interaction of syndecan-4 with protein kinase C. Biochemistry 1999, 38, 15871–15877. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M.; Barrow, R.K.; Blackshaw, S.; Burnett, P.E.; Lai, M.M.; Field, M.E.; Bahr, B.A.; Kirsch, J.; Betz, H.; Snyder, S.H. Interaction of RAFT1 with gephyrin required for rapamycin-sensitive signaling. Science 1999, 284, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Pandey, P.; Sabatini, D.; Kumar, M.; Majumder, P.K.; Bharti, A.; Carmichael, G.; Kufe, D.; Kharbanda, S. Functional interaction between RAFT1/FRAP/mTOR and protein kinase cdelta in the regulation of cap-dependent initiation of translation. EMBO J. 2000, 19, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Kristof, A.S.; Marks-Konczalik, J.; Billings, E.; Moss, J. Stimulation of signal transducer and activator of transcription-1 (STAT1)-dependent gene transcription by lipopolysaccharide and interferon-gamma is regulated by mammalian target of rapamycin. J. Biol. Chem. 2003, 278, 33637–33644. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.; Kraeft, S.K.; Gounder, M.; Pandey, P.; Jin, S.; Yuan, Z.M.; Lees-Miller, S.P.; Weichselbaum, R.; Weaver, D.; Chen, L.B.; et al. Inactivation of DNA-dependent protein kinase by protein kinase Cdelta: Implications for apoptosis. Mol. Cell Biol. 1998, 18, 6719–6728. [Google Scholar] [PubMed]
- Majumder, P.K.; Mishra, N.C.; Sun, X.; Bharti, A.; Kharbanda, S.; Saxena, S.; Kufe, D. Targeting of protein kinase C delta to mitochondria in the oxidative stress response. Cell Growth Differ. 2001, 12, 465–470. [Google Scholar] [PubMed]
- Watters, D.; Kedar, P.; Spring, K.; Bjorkman, J.; Chen, P.; Gatei, M.; Birrell, G.; Garrone, B.; Srinivasa, P.; Crane, D.I.; et al. Localization of a portion of extranuclear ATM to peroxisomes. J. Biol. Chem. 1999, 274, 34277–34282. [Google Scholar] [CrossRef] [PubMed]
- Morita, A.; Tanimoto, K.; Murakami, T.; Morinaga, T.; Hosoi, Y. Mitochondria are required for ATM activation by extranuclear oxidative stress in cultured human hepatoblastoma cell line Hep G2 cells. Biochem. Biophys. Res. Commun. 2014, 443, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tie, Y.; Tian, C.; Xing, G.; Song, Y.; Zhu, Y.; Sun, Z.; He, F. CKIP-1 recruits nuclear ATM partially to the plasma membrane through interaction with ATM. Cell Signal 2006, 18, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bao, S.; Furumai, R.; Kucera, K.S.; Ali, A.; Dean, N.M.; Wang, X.F. Protein phosphatase 5 is required for ATR-mediated checkpoint activation. Mol. Cell Biol. 2005, 25, 9910–9919. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Wang, L.; Armstrong, D.L.; Rossie, S. Activated Rac1 GTPase translocates protein phosphatase 5 to the cell membrane and stimulates phosphatase activity in vitro. J. Biol. Chem. 2010, 285, 3872–3882. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente van Bentem, S.; Vossen, J.H.; Vermeer, J.E.; de Vroomen, M.J.; Gadella, T.W., Jr.; Haring, M.A.; Cornelissen, B.J. The subcellular localization of plant protein phosphatase 5 isoforms is determined by alternative splicing. Plant Physiol. 2003, 133, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Wenzel, E.M.; Stenmark, H. ER-endosome contact sites: Molecular compositions and functions. EMBO J. 2015, 34, 1848–1858. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.D.; Smith, E.A.; LeGros, M.A.; Larabell, C.A.; Ryan, M.T. Analysis of ER-mitochondria contacts using correlative fluorescence microscopy and soft X-ray tomography of mammalian cells. J. Cell Sci. 2015, 128, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Liou, J.; Emr, S.D. Molecular mechanisms of inter-organelle ER-PM contact sites. Curr. Opin. Cell Biol. 2015, 35, 123–130. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, M.A.; Rega, L.R. Endoplasmic reticulum-Golgi complex membrane contact sites. Curr. Opin. Cell Biol. 2015, 35, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Cicco, M.; Rahim, M.S.A.; Dames, S.A. Regulation of the Target of Rapamycin and Other Phosphatidylinositol 3-Kinase-Related Kinases by Membrane Targeting. Membranes 2015, 5, 553-575. https://doi.org/10.3390/membranes5040553
De Cicco M, Rahim MSA, Dames SA. Regulation of the Target of Rapamycin and Other Phosphatidylinositol 3-Kinase-Related Kinases by Membrane Targeting. Membranes. 2015; 5(4):553-575. https://doi.org/10.3390/membranes5040553
Chicago/Turabian StyleDe Cicco, Maristella, Munirah S. Abd Rahim, and Sonja A. Dames. 2015. "Regulation of the Target of Rapamycin and Other Phosphatidylinositol 3-Kinase-Related Kinases by Membrane Targeting" Membranes 5, no. 4: 553-575. https://doi.org/10.3390/membranes5040553
APA StyleDe Cicco, M., Rahim, M. S. A., & Dames, S. A. (2015). Regulation of the Target of Rapamycin and Other Phosphatidylinositol 3-Kinase-Related Kinases by Membrane Targeting. Membranes, 5(4), 553-575. https://doi.org/10.3390/membranes5040553