
A Set of Fluorescent Protein-Based Markers for Major Vesicle Coat Proteins in Yeast


Abstract
1. Introduction
2. Materials and Methods
2.1. Yeast Culturing and Fluorescence Microscopy
2.2. Yeast Strain Construction
2.3. Plasmid Construction
2.3.1. GFP- or 2mCherry-Tagged Coat Proteins
2.3.2. 2GFP-Tagged AP-2 Subunits
2.3.3. mNeonGreen-Tagged COPII Subunits
2.3.4. mCherry-Tagged Sso2 (Plasma Membrane Marker)
2.3.5. GFP- or mCherry-Tagged Vps10
2.3.6. GFP- or mCherry-Tagged Sna2
2.4. Western Blot
2.5. Spot Assay
2.6. Mating Assay
3. Results
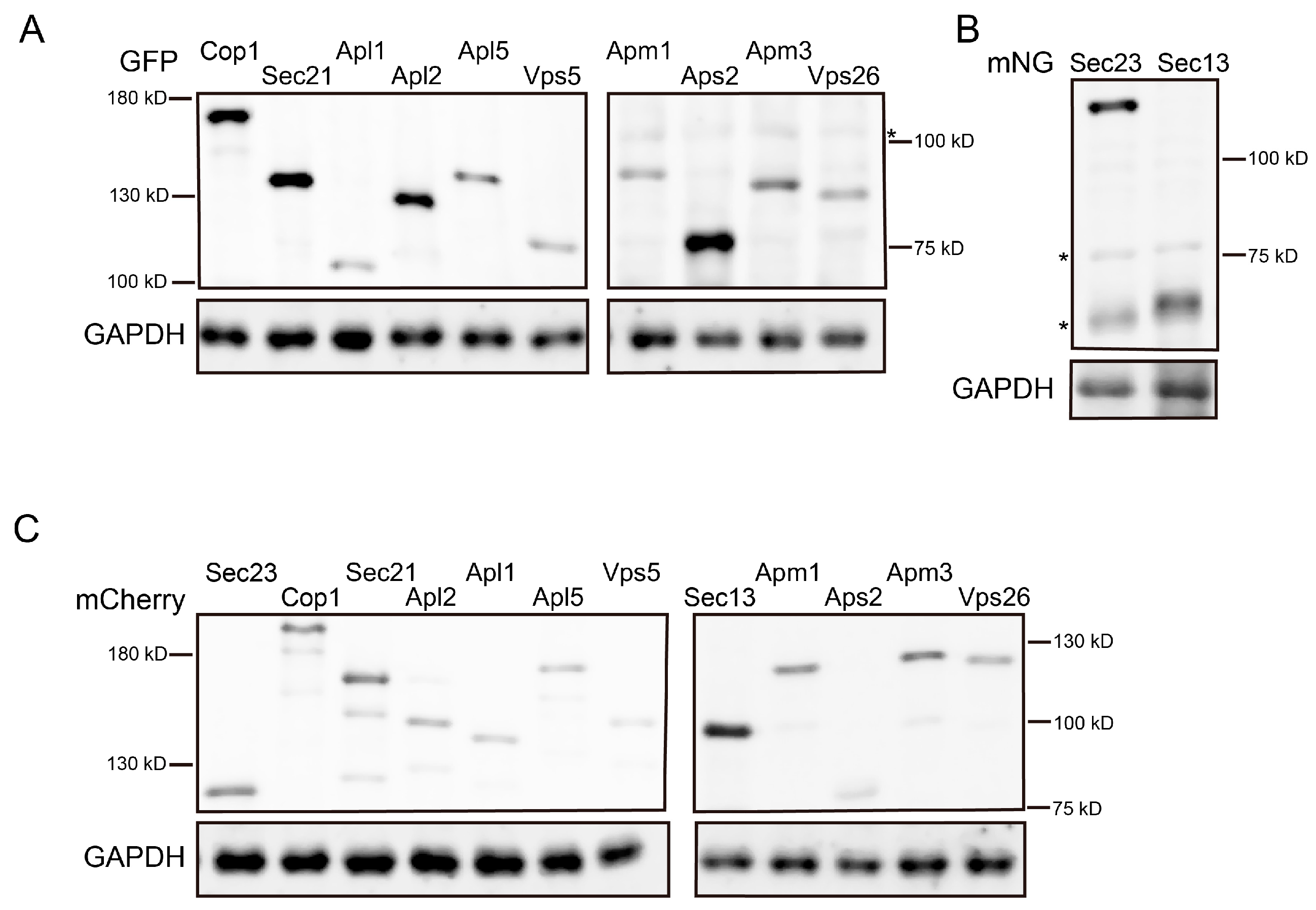
3.1. Visualization of All Six Coat Protein Complexes
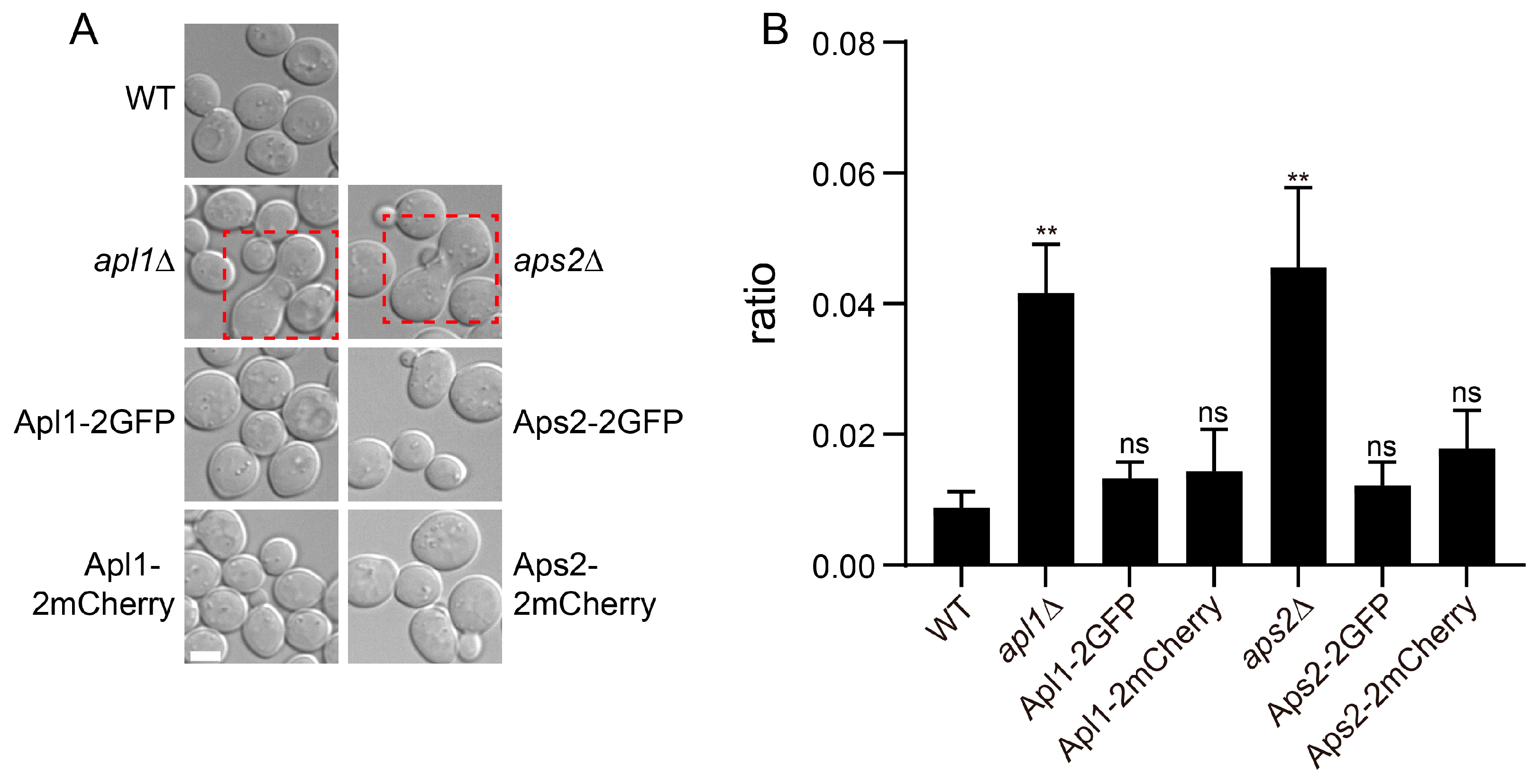
3.2. Functionality of Coat Protein Constructs
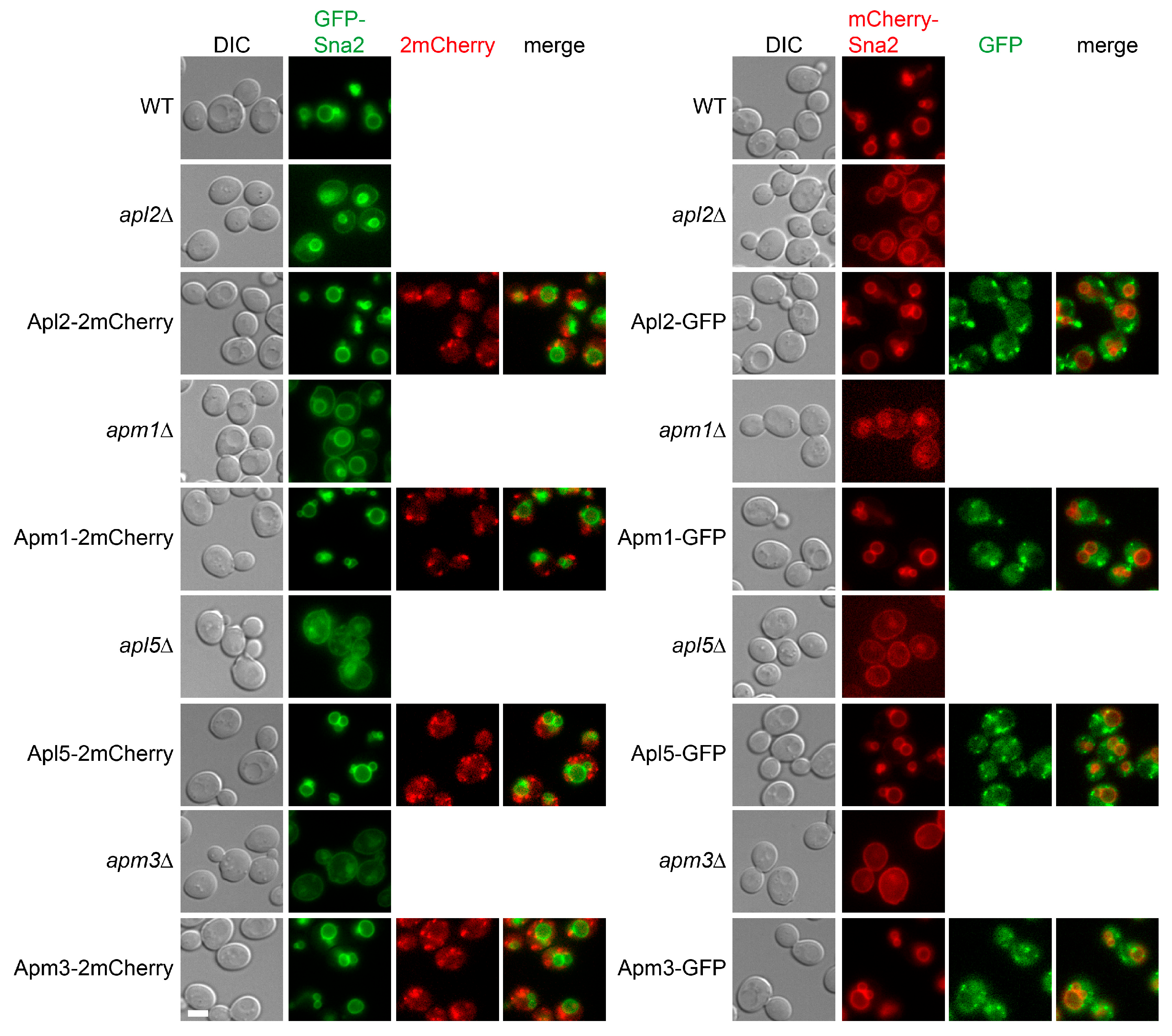
3.3. Co-Localization Between Subunits of the Same Coat Complex
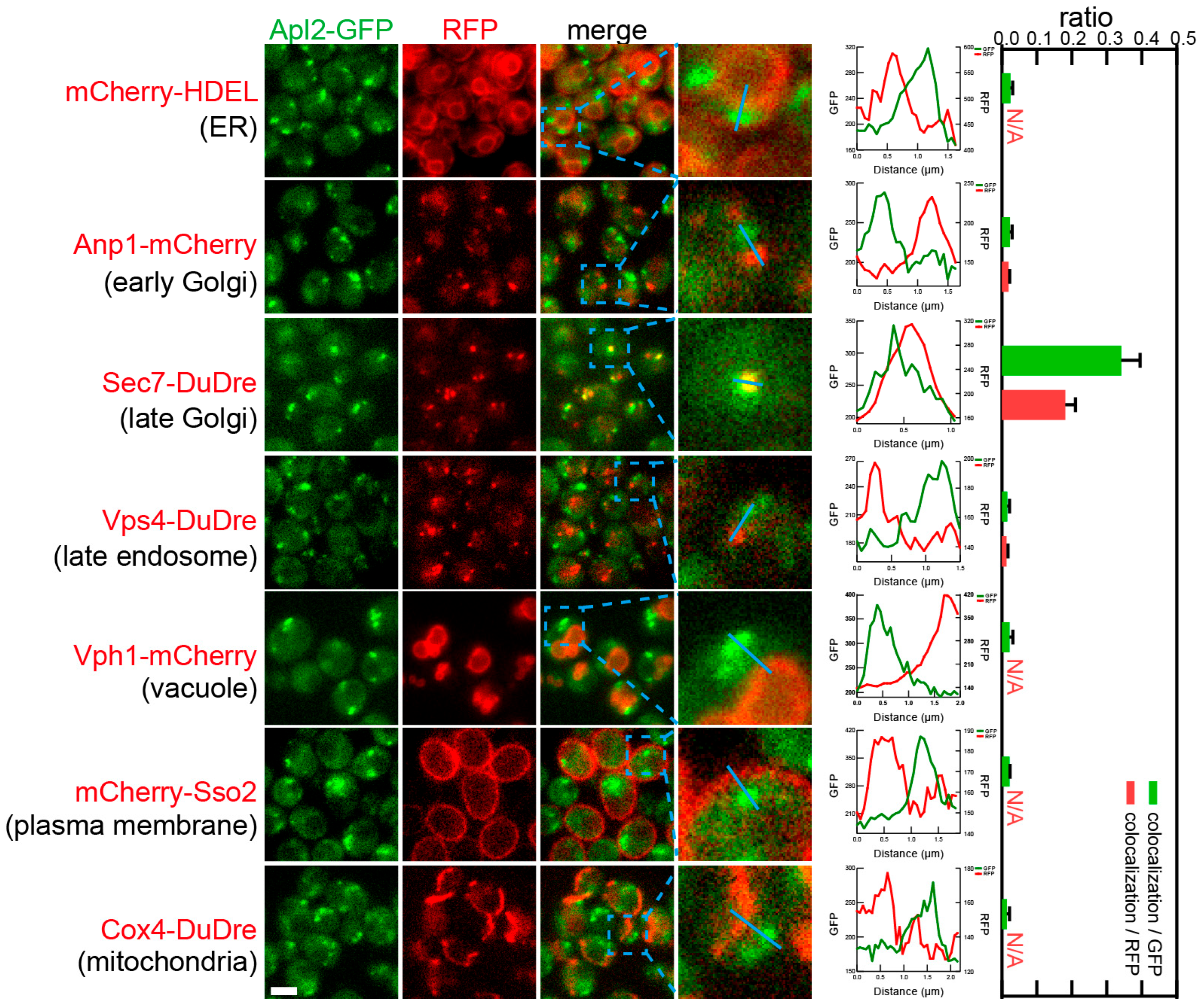
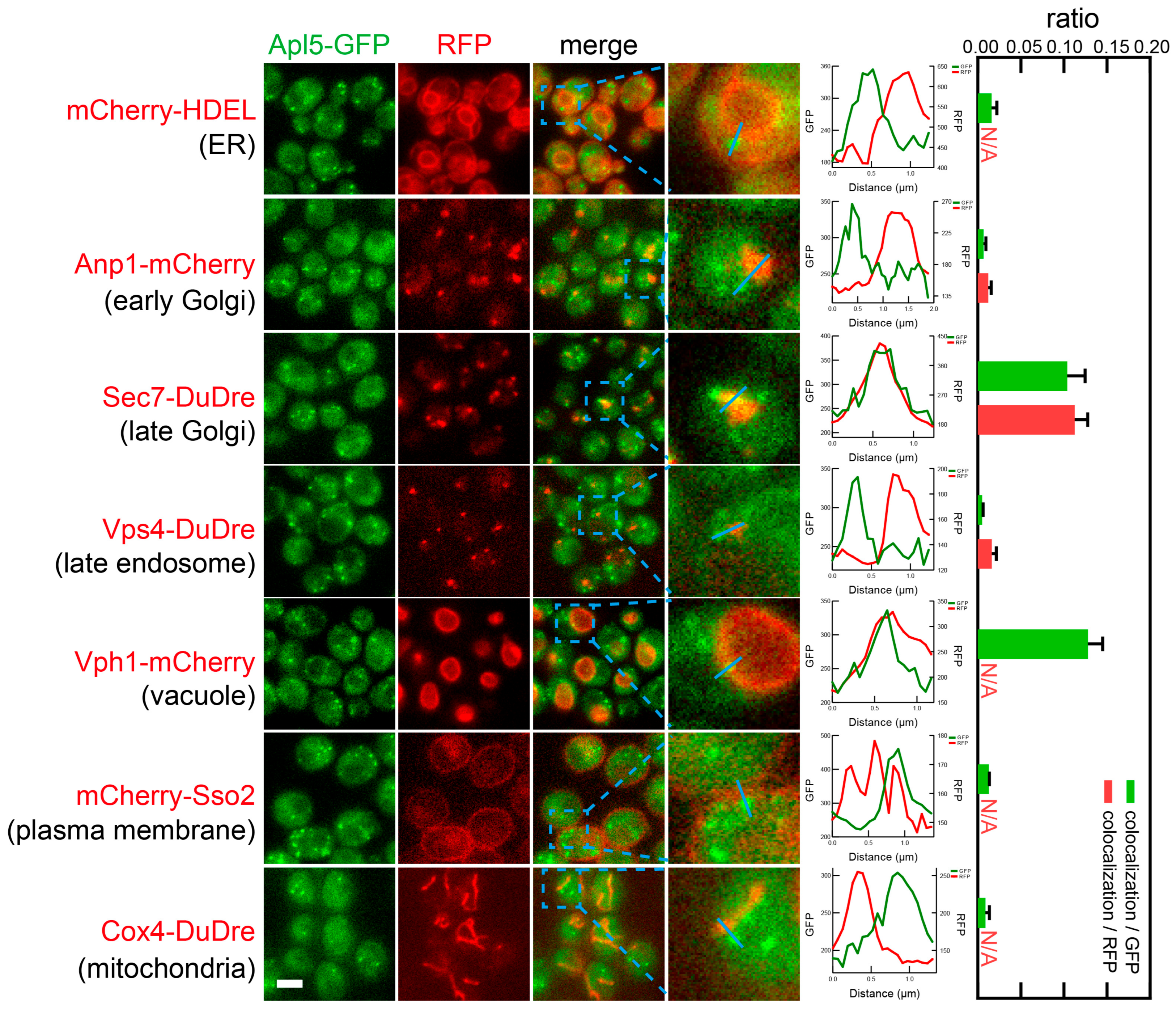
3.4. Identification of the Subcellular Localizations of the Coat Complexes by Co-Localization Analysis with Organelle Markers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| COPII | Coat protein II |
| COPI | Coat protein I |
| AP | Adaptor protein |
| ER | Endoplasmic reticulum |
| PM | Plasma membrane |
| mNG | mNeonGreen |
Appendix A

References
- Downes, K.W.; Zanetti, G. Mechanisms of COPII coat assembly and cargo recognition in the secretory pathway. Nat. Rev. Mol. Cell Biol. 2025, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sanger, A.; Hirst, J.; Davies, A.K.; Robinson, M.S. Adaptor protein complexes and disease at a glance. J. Cell Sci. 2019, 132, jcs222992. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Hierro, A. Retromer. Curr. Biol. 2017, 27, R687–R689. [Google Scholar] [CrossRef] [PubMed]
- Dacks, J.B.; Robinson, M.S. Outerwear through the ages: Evolutionary cell biology of vesicle coats. Curr. Opin. Cell Biol. 2017, 47, 108–116. [Google Scholar] [CrossRef]
- Barlow, L.D.; Dacks, J.B.; Wideman, J.G. From all to (nearly) none: Tracing adaptin evolution in Fungi. Cell. Logist. 2014, 4, e28114. [Google Scholar] [CrossRef]
- Bethune, J.; Wieland, F.T. Assembly of COPI and COPII Vesicular Coat Proteins on Membranes. Annu. Rev. Biophys. 2018, 47, 63–83. [Google Scholar] [CrossRef]
- Taylor, R.J.; Tagiltsev, G.; Briggs, J.A.G. The structure of COPI vesicles and regulation of vesicle turnover. FEBS Lett. 2023, 597, 819–835. [Google Scholar] [CrossRef]
- Tu, Y.; Seaman, M.N.J. Navigating the Controversies of Retromer-Mediated Endosomal Protein Sorting. Front. Cell Dev. Biol. 2021, 9, 658741. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, Z.T.; Tang, S.W.; Zhao, B.S.; Li, H.; Song, J.Z.; Li, D.; Xie, Z. A Validated Set of Fluorescent-Protein-Based Markers for Major Organelles in Yeast (Saccharomyces cerevisiae). mBio 2019, 10, e01691-19. [Google Scholar] [CrossRef]
- Day, K.J.; Casler, J.C.; Glick, B.S. Budding Yeast Has a Minimal Endomembrane System. Dev. Cell 2018, 44, 56–72.e4. [Google Scholar] [CrossRef]
- Liu, C.Y.; Zhu, J.; Xie, Z.P. Visualizing Yeast Organelles with Fluorescent Protein Markers. JoVE J. Vis. Exp. 2022, 182, 1–15. [Google Scholar] [CrossRef]
- Li, H.; Song, J.Z.; He, C.W.; Xie, M.X.; Zhang, Z.T.; Zhou, Y.; Li, X.J.; Cui, L.; Zhu, J.; Gong, Q.Q.; et al. Temporal dissection of the roles of Atg4 and ESCRT in autophagosome formation in yeast. Cell Death Differ. 2024, 32, 866–879. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Z.; Li, H.; Yang, H.Y.; Liu, R.; Zhang, W.T.; He, T.L.; Xie, M.X.; Chen, C.; Cui, L.; Wu, S.A.; et al. Recruitment of Atg1 to the phagophore by Atg8 orchestrates autophagy machineries. Nat. Struct. Mol. Biol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Z.; Feng, Y.H.; Sergevnina, V.; Zhu, J.; Li, H.; Xie, Z.P. Assessing the Presence of Phosphoinositides on Autophagosomal Membrane in Yeast by Live Cell Imaging. Microorganisms 2024, 12, 1458. [Google Scholar] [CrossRef] [PubMed]
- He, C.W.; Cui, X.F.; Ma, S.J.; Xu, Q.; Ran, Y.P.; Chen, W.Z.; Mu, J.X.; Li, H.; Zhu, J.; Gong, Q.Q.; et al. Membrane recruitment of Atg8 by Hfl1 facilitates turnover of vacuolar membrane proteins in yeast cells approaching stationary phase. BMC Biol. 2021, 19, 117. [Google Scholar] [CrossRef]
- Brachmann, C.B.; Davies, A.; Cost, G.J.; Caputo, E.; Li, J.C.; Hieter, P.; Boeke, J.D. Designer deletion strains derived from S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 1998, 14, 115–132. [Google Scholar] [CrossRef]
- Finger, F.P.; Novick, P. Synthetic interactions of the post-Golgi sec mutations of Saccharomyces cerevisiae. Genetics 2000, 156, 943–951. [Google Scholar] [CrossRef]
- Isono, E.; Nishihara, K.; Saeki, Y.; Yashiroda, H.; Kamata, N.; Ge, L.Y.; Ueda, T.; Kikuchi, Y.; Tanaka, K.; Nakano, A.; et al. The assembly pathway of the 19S regulatory particle of the yeast 26S proteasome. Mol. Biol. Cell 2007, 18, 569–580. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.C.; Wu, C.F.; Cai, Z.Y.; Yao, W.J.; Yang, H.; Song, J.; Xie, X.K.; Zhang, L.Q.; Yi, C. Establishment of a yeast essential protein conditional-degradation library and screening for autophagy-regulating genes. Autophagy 2025, 21, 1578–1590. [Google Scholar] [CrossRef]
- Chapa-y-Lazo, B.; Allwood, E.G.; Smaczynska-de Rooij, I.I.; Snape, M.L.; Ayscough, K.R. Yeast Endocytic Adaptor AP-2 Binds the Stress Sensor Mid2 and Functions in Polarized Cell Responses. Traffic 2014, 15, 546–557. [Google Scholar] [CrossRef]
- Renard, H.F.; Demaegd, D.; Guerriat, B.; Morsomme, P. Efficient ER Exit and Vacuole Targeting of Yeast Sna2p Require Two Tyrosine-Based Sorting Motifs. Traffic 2010, 11, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.W.; Chuang, Y.S.; Li, M.; Seaman, M.N.J.; Emr, S.D. A bipartite sorting signal ensures specificity of retromer complex in membrane protein recycling. J. Cell Biol. 2019, 218, 2876–2886. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Okamoto, M.; Nakano, A. Contact of cis-Golgi with ER exit sites executes cargo capture and delivery from the ER. Nat. Commun. 2014, 5, 3653. [Google Scholar] [CrossRef]
- Ishii, M.; Suda, Y.; Kurokawa, K.; Nakano, A. COPI is essential for Golgi cisternal maturation and dynamics. J. Cell Sci. 2016, 129, 3251–3261. [Google Scholar] [CrossRef] [PubMed]
- Papanikou, E.; Day, K.J.; Austin, J.; Glick, B.S. COPI selectively drives maturation of the early Golgi. eLife 2015, 4, e13232. [Google Scholar] [CrossRef]
- Tojima, T.; Suda, Y.; Ishii, M.; Kurokawa, K.; Nakano, A. Spatiotemporal dissection of the trans-Golgi network in budding yeast. J. Cell Sci. 2019, 132, jcs231159. [Google Scholar] [CrossRef]
- Casler, J.C.; Johnson, N.; Krahn, A.H.; Pantazopoulou, A.; Day, K.J.; Glick, B.S. Clathrin adaptors mediate two sequential pathways of intra-Golgi recycling. J. Cell Biol. 2022, 221, e202103199. [Google Scholar] [CrossRef]
- Casler, J.C.; Papanikou, E.; Barrero, J.J.; Glick, B.S. Maturation-driven transport and AP-1-dependent recycling of a secretory cargo in the Golgi. J. Cell Biol. 2019, 218, 1582–1601. [Google Scholar] [CrossRef]
- Knafler, H.C.; Smaczynska-de, R., II; Walker, L.A.; Lee, K.K.; Gow, N.A.R.; Ayscough, K.R. AP-2-Dependent Endocytic Recycling of the Chitin Synthase Chs3 Regulates Polarized Growth in Candida albicans. mBio 2019, 10, e02421-18. [Google Scholar] [CrossRef]
- de Leon, N.; Hoya, M.; Curto, M.A.; Moro, S.; Yanguas, F.; Doncel, C.; Valdivieso, M.H. The AP-2 complex is required for proper temporal and spatial dynamics of endocytic patches in fission yeast. Mol. Microbiol. 2016, 100, 409–424. [Google Scholar] [CrossRef]
- Carroll, S.Y.; Stirling, P.C.; Stimpson, H.E.; Giesselmann, E.; Schmitt, M.J.; Drubin, D.G. A yeast killer toxin screen provides insights into a/b toxin entry, trafficking, and killing mechanisms. Dev. Cell 2009, 17, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Chapa, Y.L.B.; Ayscough, K.R. Apm4, the mu subunit of yeast AP-2 interacts with Pkc1, and mutation of the Pkc1 consensus phosphorylation site Thr176 inhibits AP-2 recruitment to endocytic sites. Commun. Integr. Biol. 2014, 7, e28522. [Google Scholar] [CrossRef]
- Leih, M.; Plemel, R.L.; West, M.; Angers, C.G.; Merz, A.J.; Odorizzi, G. Disordered hinge regions of the AP-3 adaptor complex promote vesicle budding from the late Golgi in yeast. J. Cell Sci. 2024, 137, jcs262234. [Google Scholar] [CrossRef]
- Schoppe, J.; Mari, M.; Yavavli, E.; Auffarth, K.; Cabrera, M.; Walter, S.; Frohlich, F.; Ungermann, C. AP-3 vesicle uncoating occurs after HOPS-dependent vacuole tethering. EMBO J. 2020, 39, e105117. [Google Scholar] [CrossRef]
- Angers, C.G.; Merz, A.J. HOPS interacts with Apl5 at the vacuole membrane and is required for consumption of AP-3 transport vesicles. Mol. Biol. Cell 2009, 20, 4563–4574. [Google Scholar] [CrossRef]
- Marquardt, L.; Taylor, M.; Kramer, F.; Schmitt, K.; Braus, G.H.; Valerius, O.; Thumm, M. Vacuole fragmentation depends on a novel Atg18-containing retromer-complex. Autophagy 2023, 19, 278–295. [Google Scholar] [CrossRef] [PubMed]
- Courtellemont, T.; De Leo, M.G.; Gopaldass, N.; Mayer, A. CROP: A retromer-PROPPIN complex mediating membrane fission in the endo-lysosomal system. EMBO J. 2022, 41, e109646. [Google Scholar] [CrossRef]
- Arlt, H.; Reggiori, F.; Ungermann, C. Retromer and the dynamin Vps1 cooperate in the retrieval of transmembrane proteins from vacuoles. J. Cell Sci. 2015, 128, 645–655. [Google Scholar] [CrossRef]
- Huh, W.K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef]
- Habeler, G.; Natter, K.; Thallinger, G.G.; Crawford, M.E.; Kohlwein, S.D.; Trajanoski, Z. YPL.Db: The yeast protein localization database. Nucleic Acids Res. 2002, 30, 80–83. [Google Scholar] [CrossRef]
- Kals, M.; Natter, K.; Habeler, G.; Thallinger, G.; Trajanoski, Z.; Kohlwein, S.D. YPL.db—The yeast protein localization database. Yeast 2003, 20, S329. [Google Scholar]
- Breker, M.; Gymrek, M.; Moldavski, O.; Schuldiner, M. LoQAtE-Localization and Quantitation ATlas of the yeast proteomE. A new tool for multiparametric dissection of single-protein behavior in response to biological perturbations in yeast. Nucleic Acids Res. 2014, 42, D726–D730. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coat | Plasmid | Subunit |
|---|---|---|
| COPII | PSEC13-Sec13-mNG | Sec13 |
| Sec13-2mCherry | ||
| PSEC23-Sec23-mNG | Sec23 | |
| Sec23-2mCherry | ||
| COPI | Cop1-GFP | Cop1/α-COP |
| Cop1-2mCherry | ||
| Sec21-GFP | Sec21/γ-COP | |
| Sec21-2mCherry | ||
| AP-1 | Apl2-GFP | Apl2/β1 |
| Apl2-2mCherry | ||
| Apm1-GFP | Apm1/μ1 | |
| Apm1-2mCherry | ||
| AP-2 | Apl1-2GFP | Apl1/β2 |
| Apl1-2mCherry | ||
| Aps2-2GFP | Aps2/σ2 | |
| Aps2-2mCherry | ||
| AP-3 | Apl5-GFP | Apl5/δ |
| Apl5-2mCherry | ||
| Apm3-GFP | Apm3/μ3 | |
| Apm3-2mCherry | ||
| retromer | Vps26-GFP | Vps26 |
| Vps26-2mCherry | ||
| Vps5-GFP | Vps5 | |
| Vps5-2mCherry |
| Coat | Location Reported | Location Observed in This Study | Potential Function |
|---|---|---|---|
| COPII | ER, early Golgi [23] | ER, early Golgi, transport vesicles (possible) | Transport from the ER to cis-Golgi [23] |
| COPI | Early Golgi [25] | Early Golgi, late Golgi | Transport from the Golgi to ER, transport between Golgi stacks [7]; essential for Golgi cisternal maturation and dynamics [24,25] |
| AP-1 | Late Golgi [27] | Late Golgi, transport vesicles | Transport from the late Golgi to endosome [10]; essential for Golgi cisternal maturation [27] |
| AP-2 | Plasma membrane [30], bud neck [29] | Plasma membrane, bud neck | Transport from plasma membrane to endosome [29,30] |
| AP-3 | Late Golgi [10,33], vacuole [34,35] | Late Golgi, vacuole, transport vesicles | Transport from the late Golgi to vacuole [10,33] |
| retromer | Late endosome [38], vacuole [37,38] | Late endosome, vacuole; transport vesicles (possible) | Transport from endosome to Golgi [8] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, X.-F.; Zhang, Z.-T.; Zhu, J.; Cui, L.; Xie, Z. A Set of Fluorescent Protein-Based Markers for Major Vesicle Coat Proteins in Yeast. Membranes 2025, 15, 209. https://doi.org/10.3390/membranes15070209
Cui X-F, Zhang Z-T, Zhu J, Cui L, Xie Z. A Set of Fluorescent Protein-Based Markers for Major Vesicle Coat Proteins in Yeast. Membranes. 2025; 15(7):209. https://doi.org/10.3390/membranes15070209
Chicago/Turabian StyleCui, Xue-Fei, Zheng-Tan Zhang, Jing Zhu, Li Cui, and Zhiping Xie. 2025. "A Set of Fluorescent Protein-Based Markers for Major Vesicle Coat Proteins in Yeast" Membranes 15, no. 7: 209. https://doi.org/10.3390/membranes15070209
APA StyleCui, X.-F., Zhang, Z.-T., Zhu, J., Cui, L., & Xie, Z. (2025). A Set of Fluorescent Protein-Based Markers for Major Vesicle Coat Proteins in Yeast. Membranes, 15(7), 209. https://doi.org/10.3390/membranes15070209