
Interactions of N-Mannich Bases of Pyrrolo[3,4-c]pyrrole with Artificial Models of Cell Membranes and Plasma Proteins, Evaluation of Anti-Inflammatory and Antioxidant Activity

 ,
,  ,
,  , ,
, ,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Biological Evaluation
2.1.1. Cell Line and Culture Conditions
2.1.2. Tested Compounds
2.1.3. Lipoxygenase Inhibition Assay
2.1.4. Experimental Design
- Protective properties against ROS or NO (RNS) formation
- Scavenging exogenous ROS or NO (RNS)
2.1.5. DCF-DA Assay
2.1.6. Griess Assay
2.1.7. MDA Assay
2.1.8. FHA Assay
2.1.9. Statistical Analysis
2.2. Interactions with Artificial Models of Cell Membranes
2.2.1. Chemicals
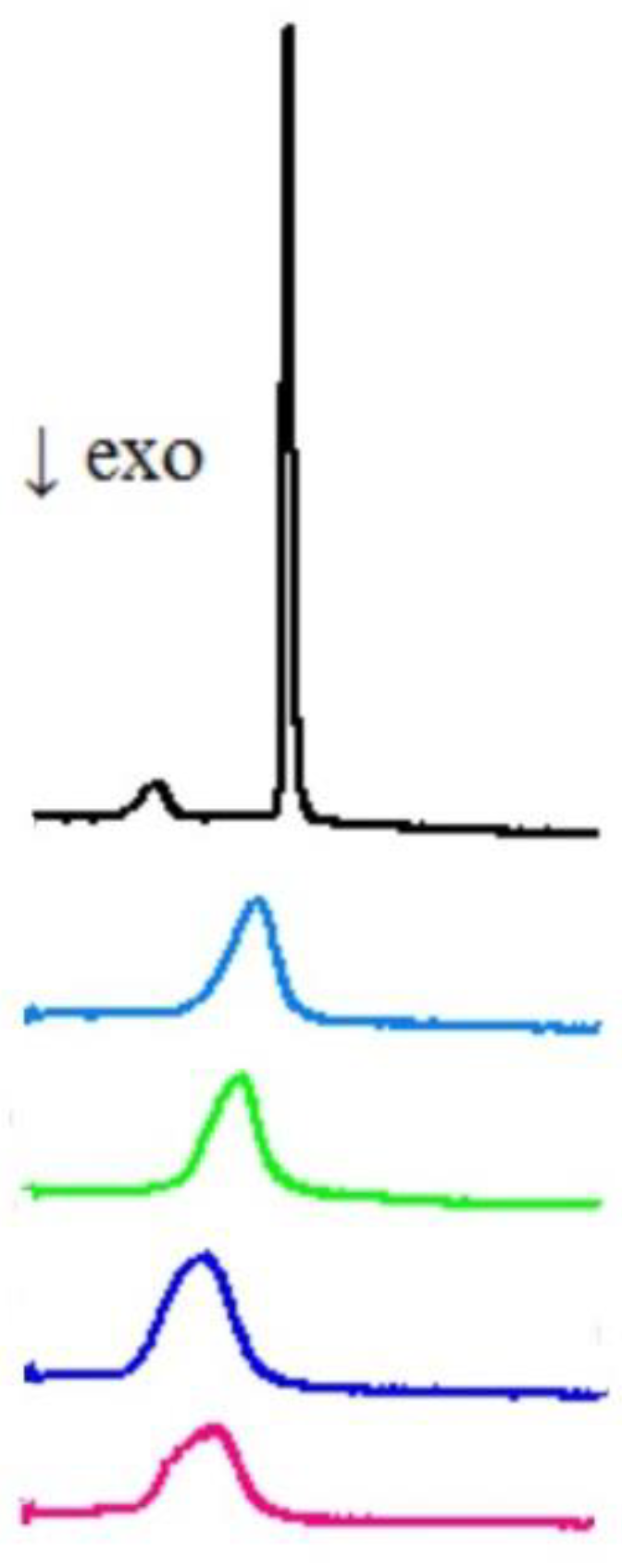
2.2.2. Differential Scanning Calorimetry (DSC)
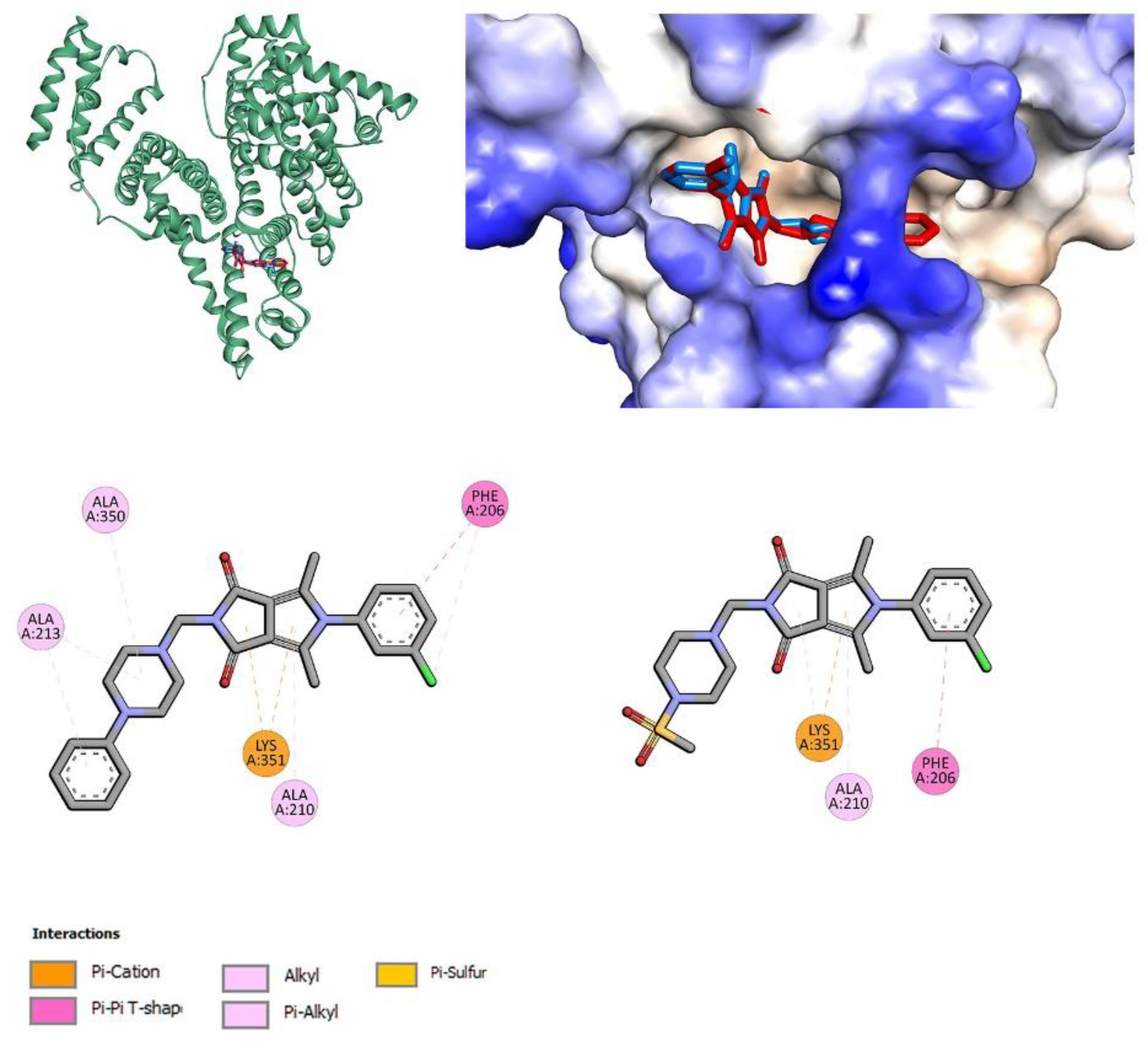
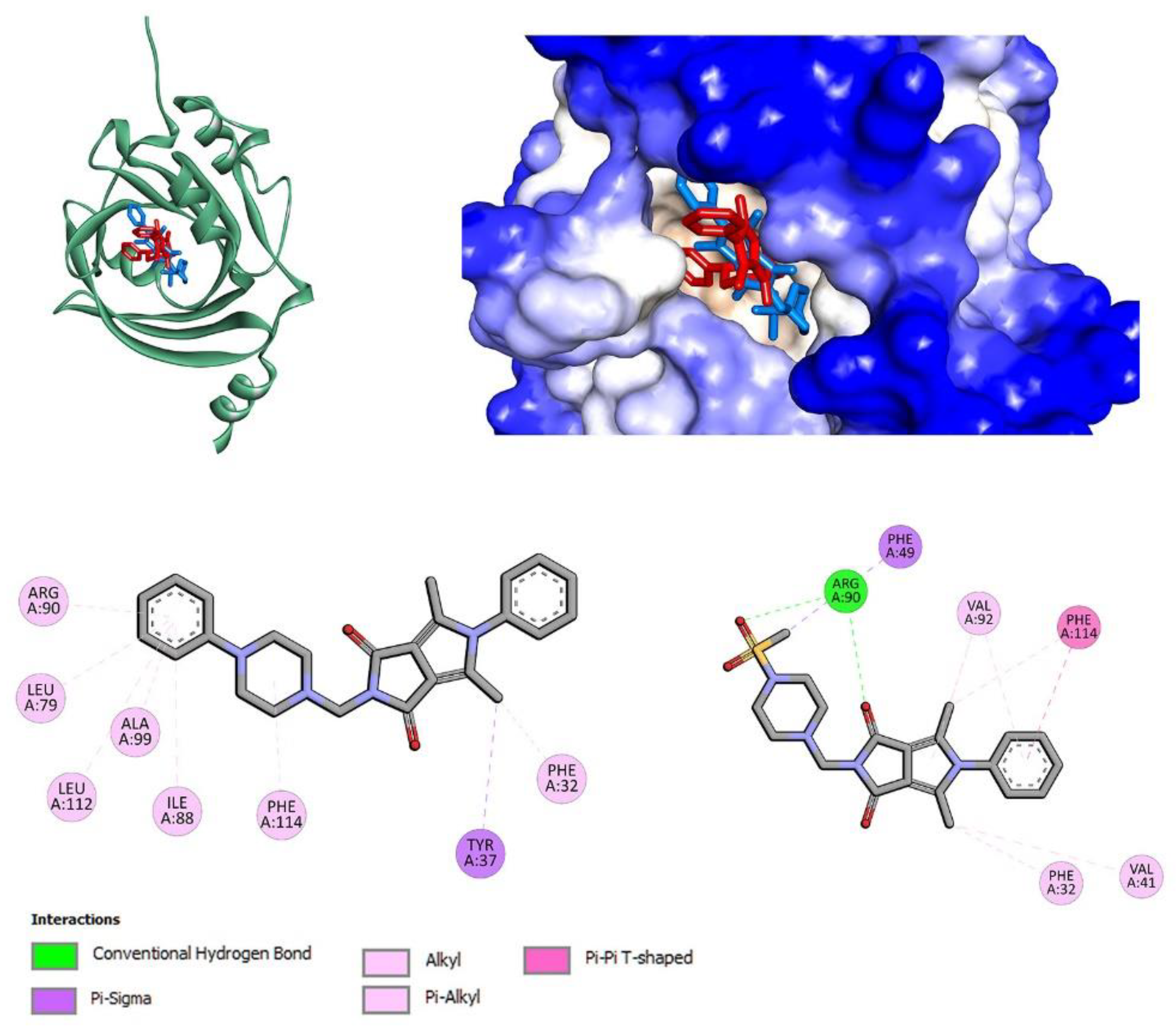
2.3. Molecular Docking Studies
2.4. Spectroscopic Studies
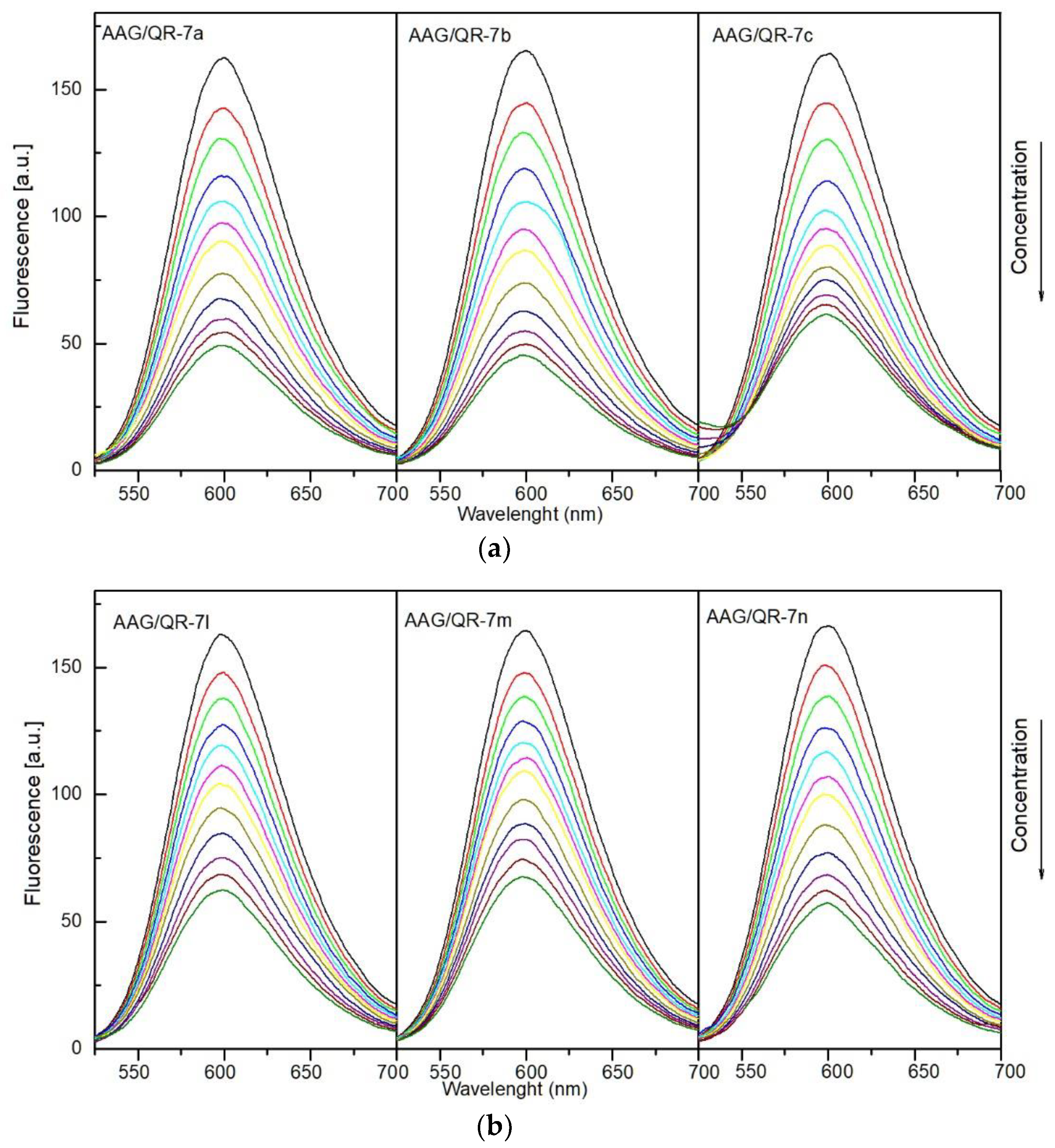
2.4.1. Fluorescence Quenching
2.4.2. Circular Dichroism Spectroscopy
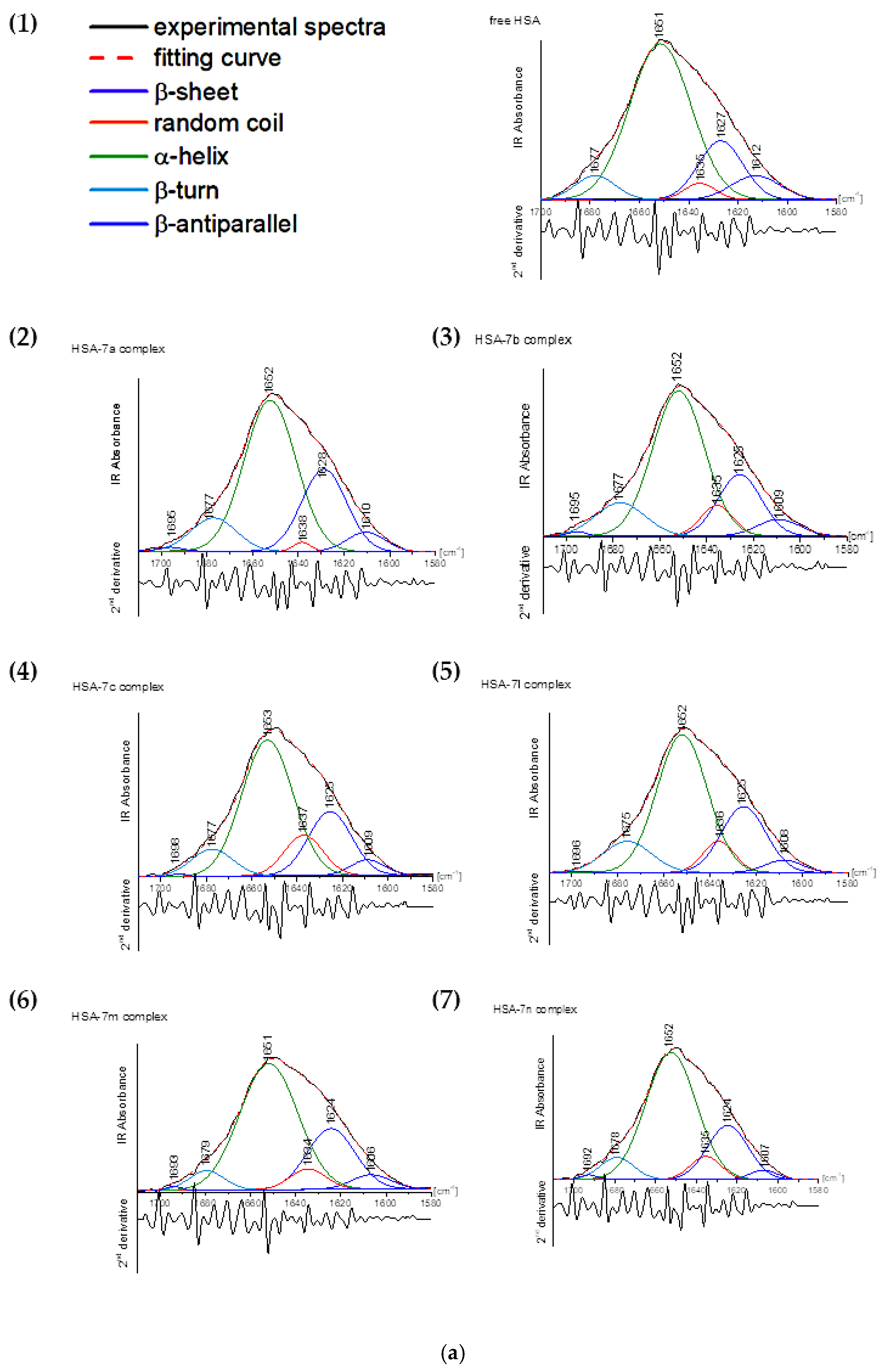
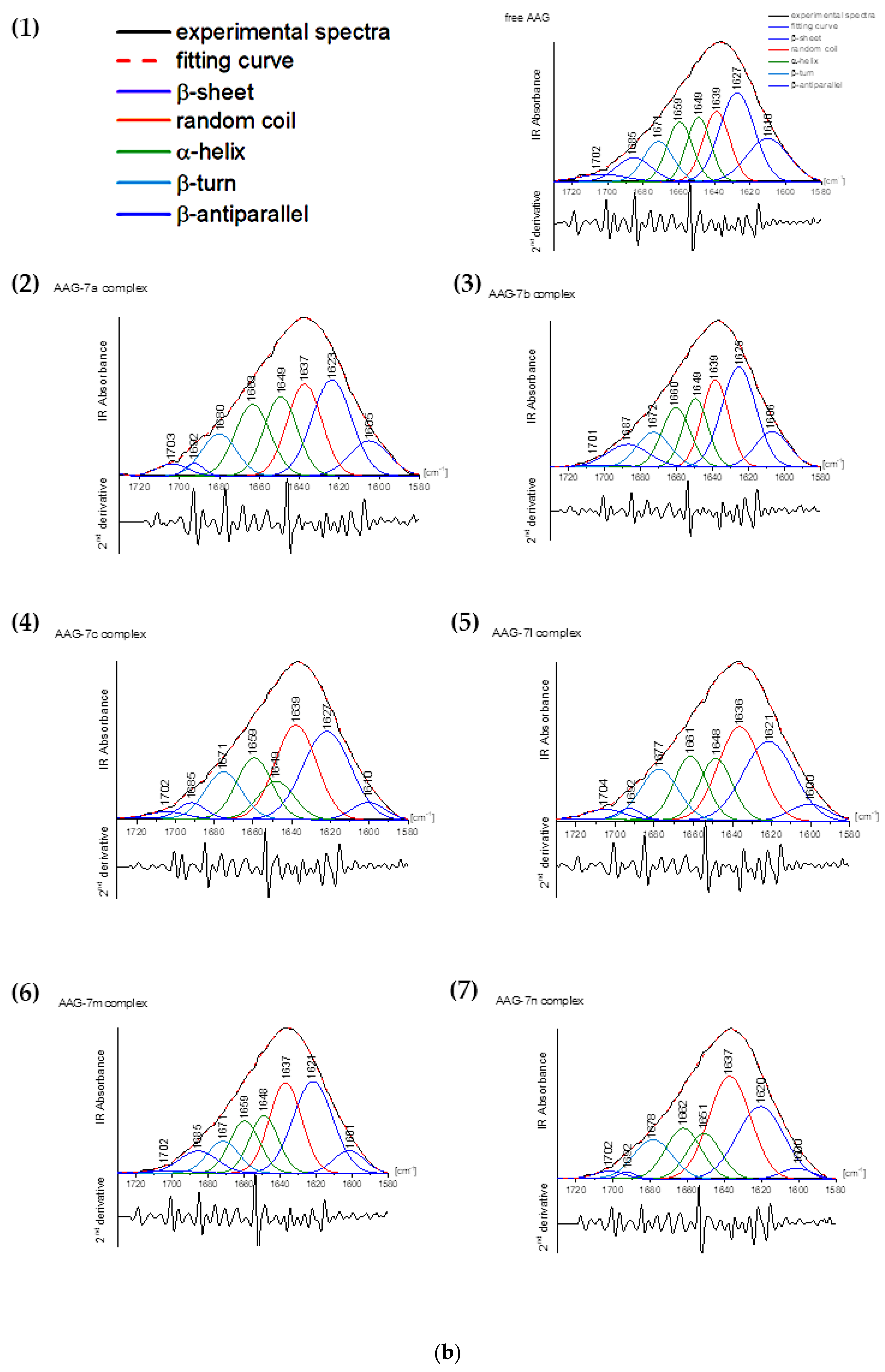
2.4.3. FT-IR Measurements
2.5. Computational Investigations
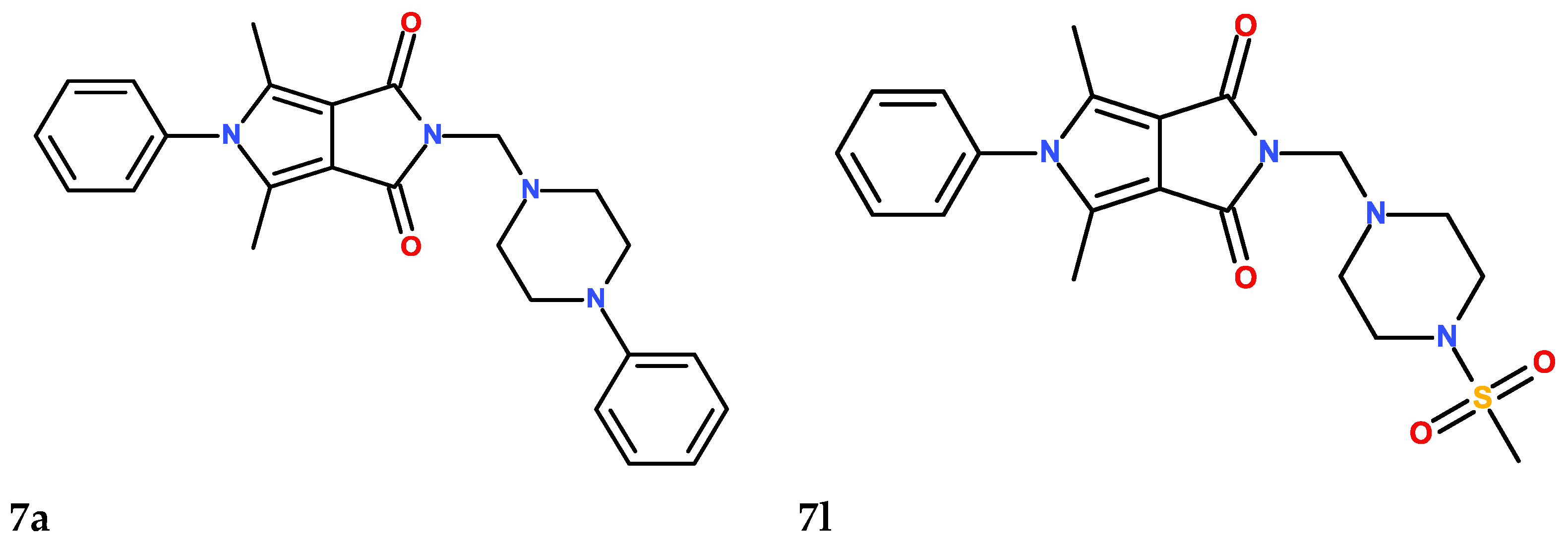
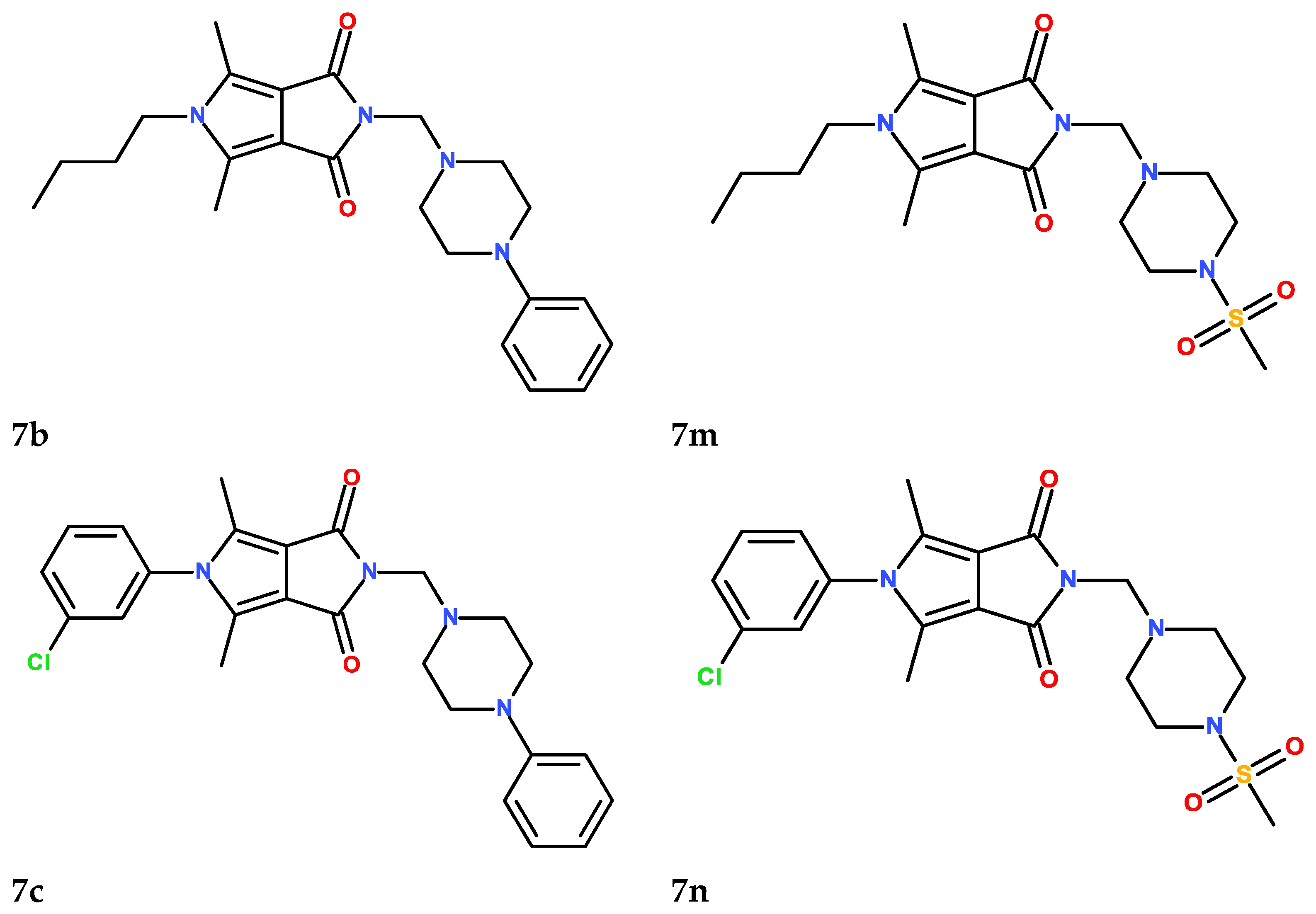
2.6. Chemistry
3. Results
3.1. 15-Lipoxygenase (15-LOX) Inhibition Studies
3.1.1. In Vitro 15-LOX Inhibition Assay
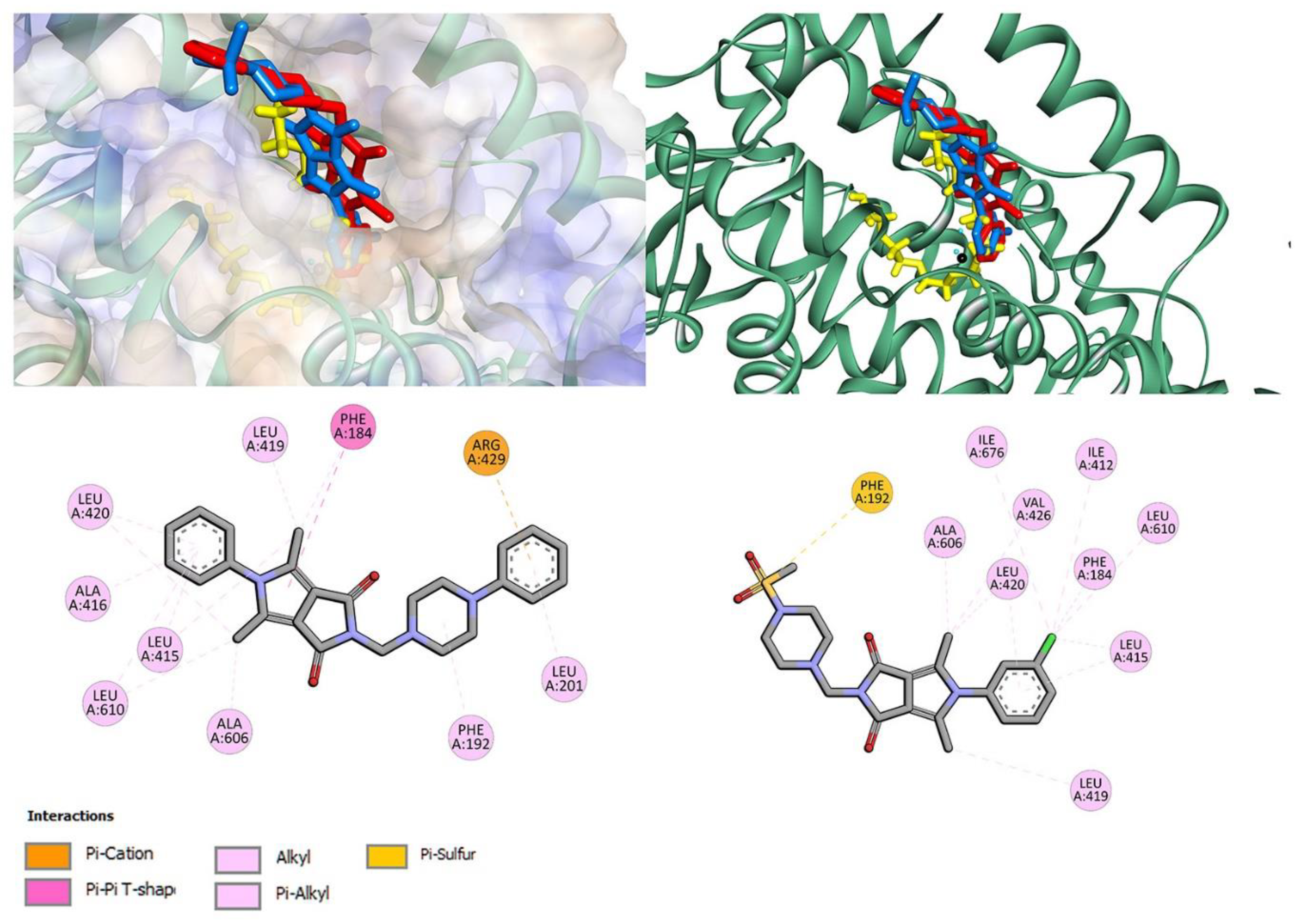
3.1.2. Molecular Docking Study
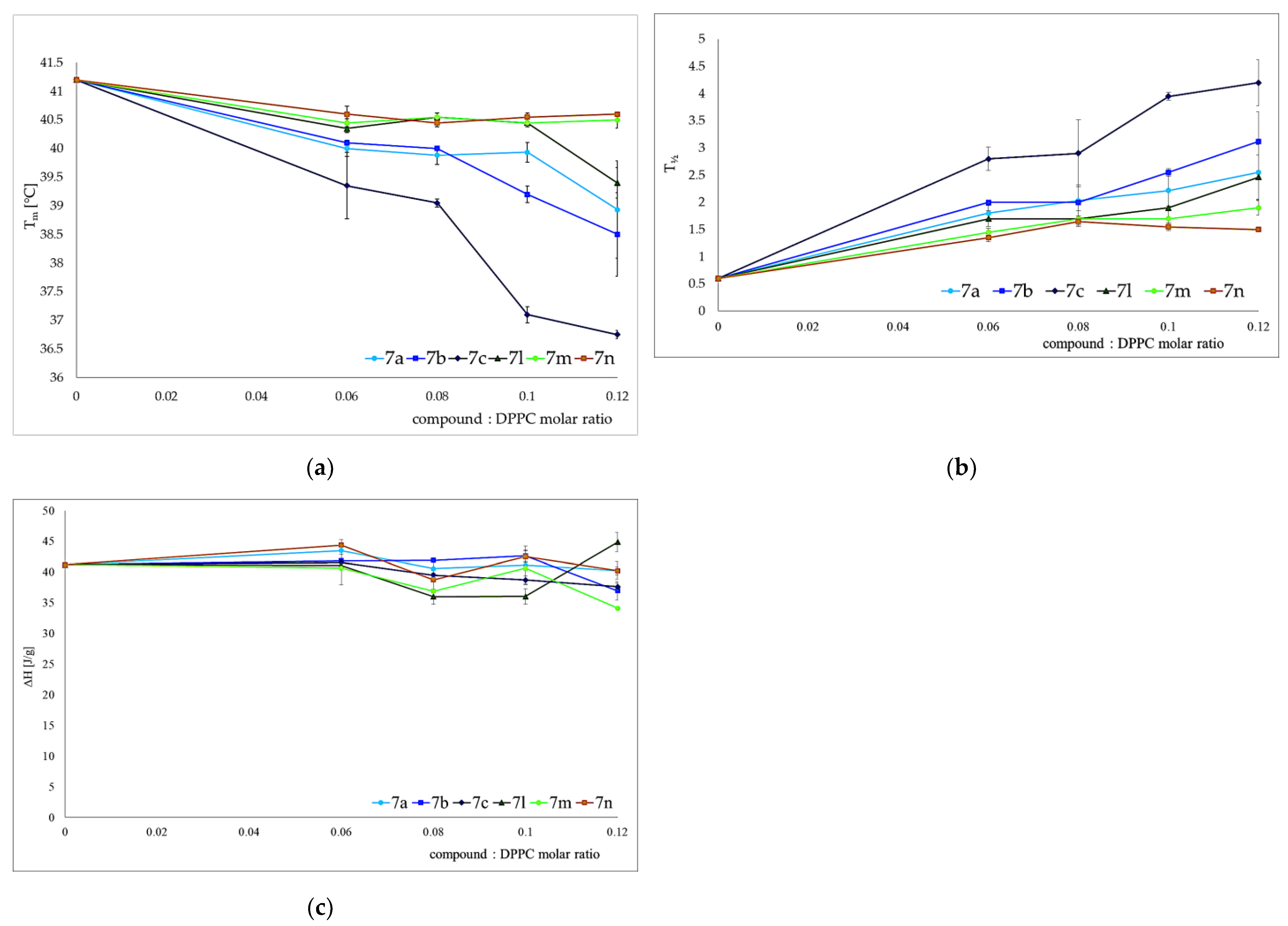
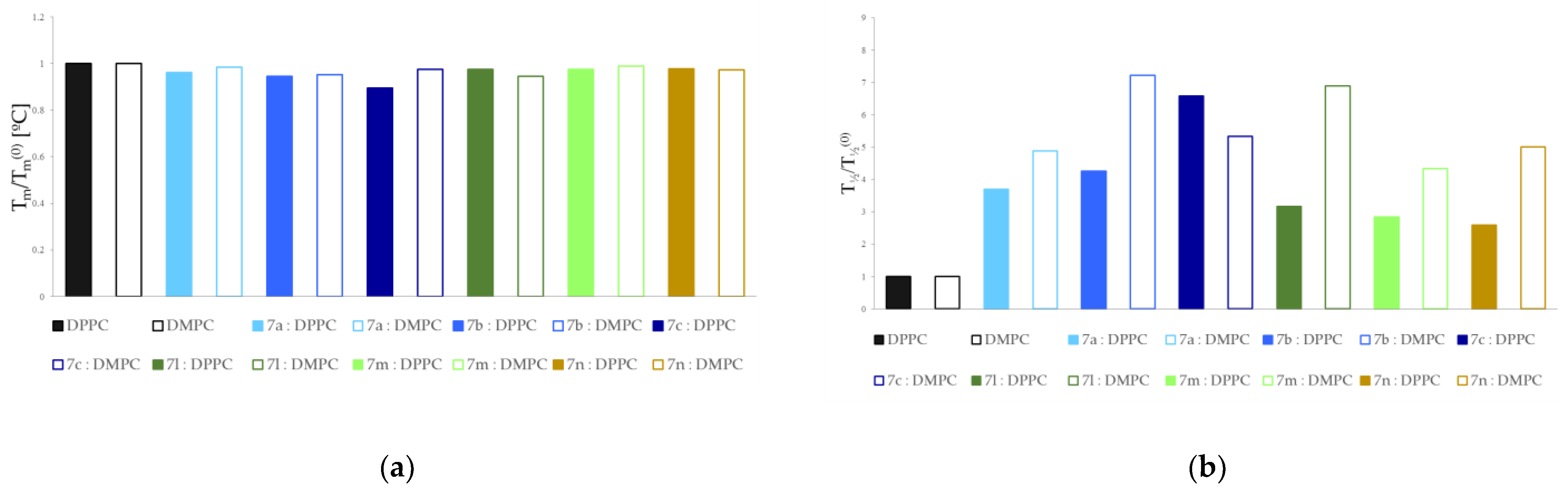
3.2. Interactions with Artificial Models of Cell Membranes
3.3. Antioxidant Activity within Cells
3.4. Human Serum Albumin (HSA) and Alpha-1-Acid Glycoprotein (AAG) Ligand-Binding Assay
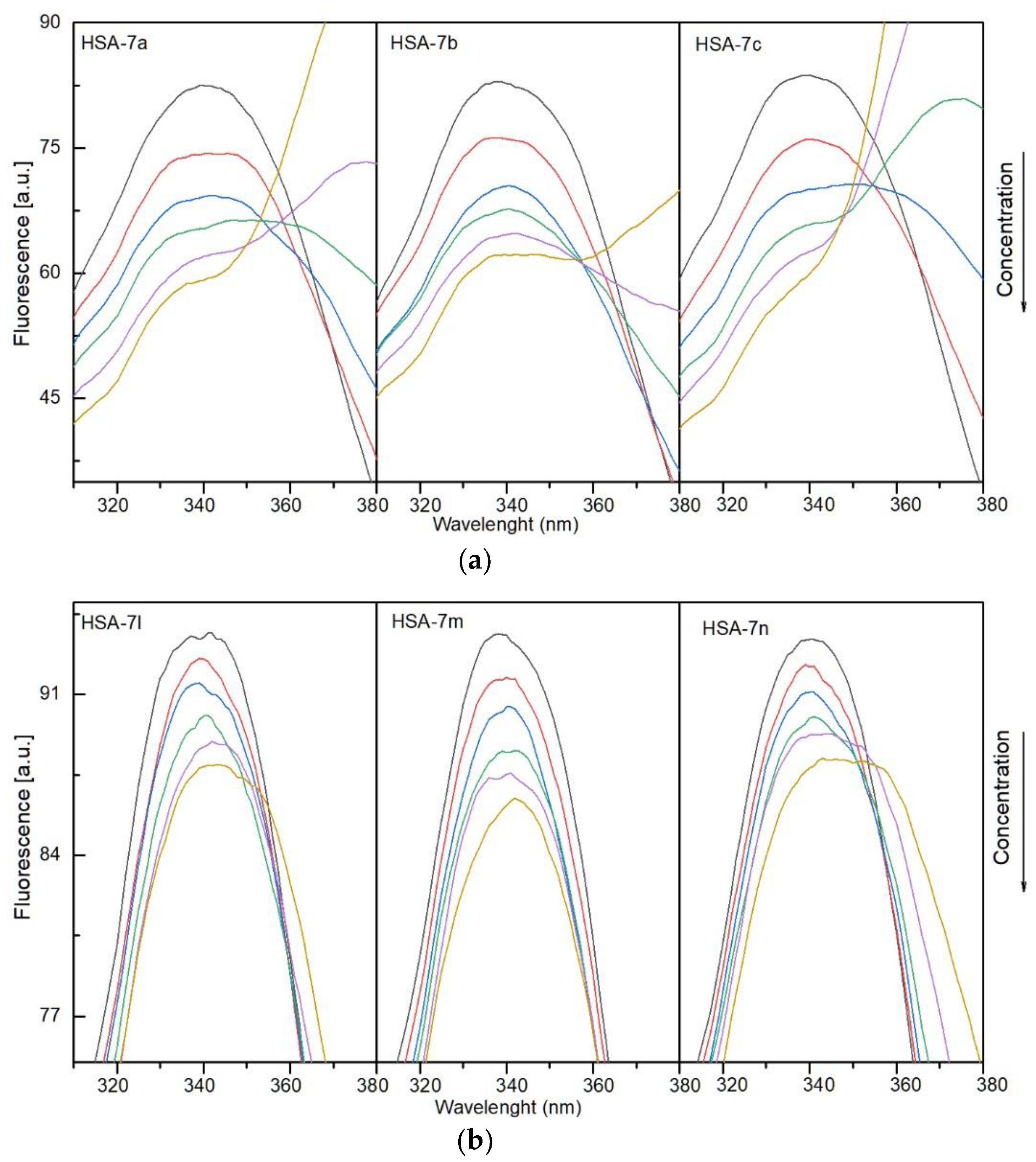
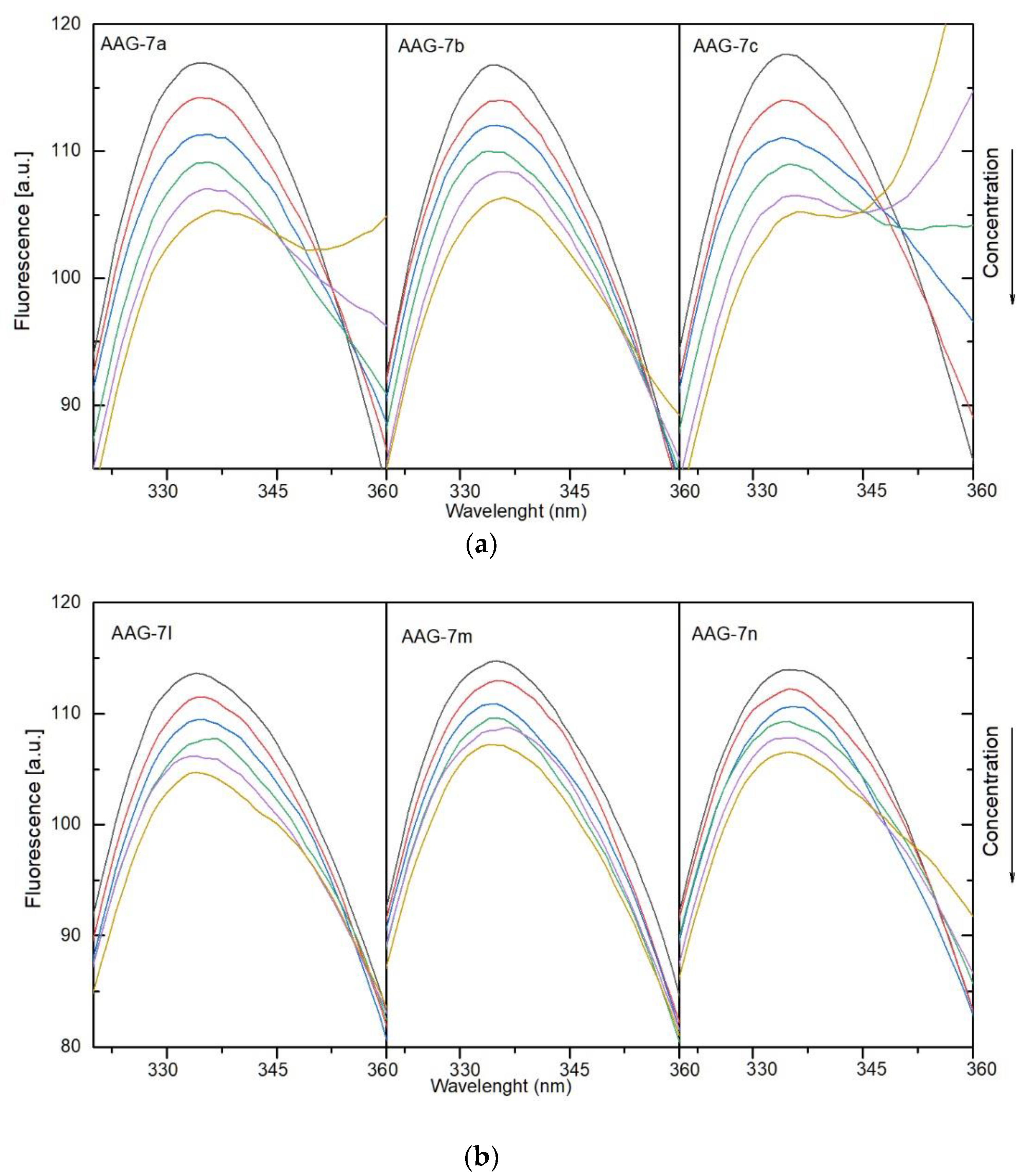
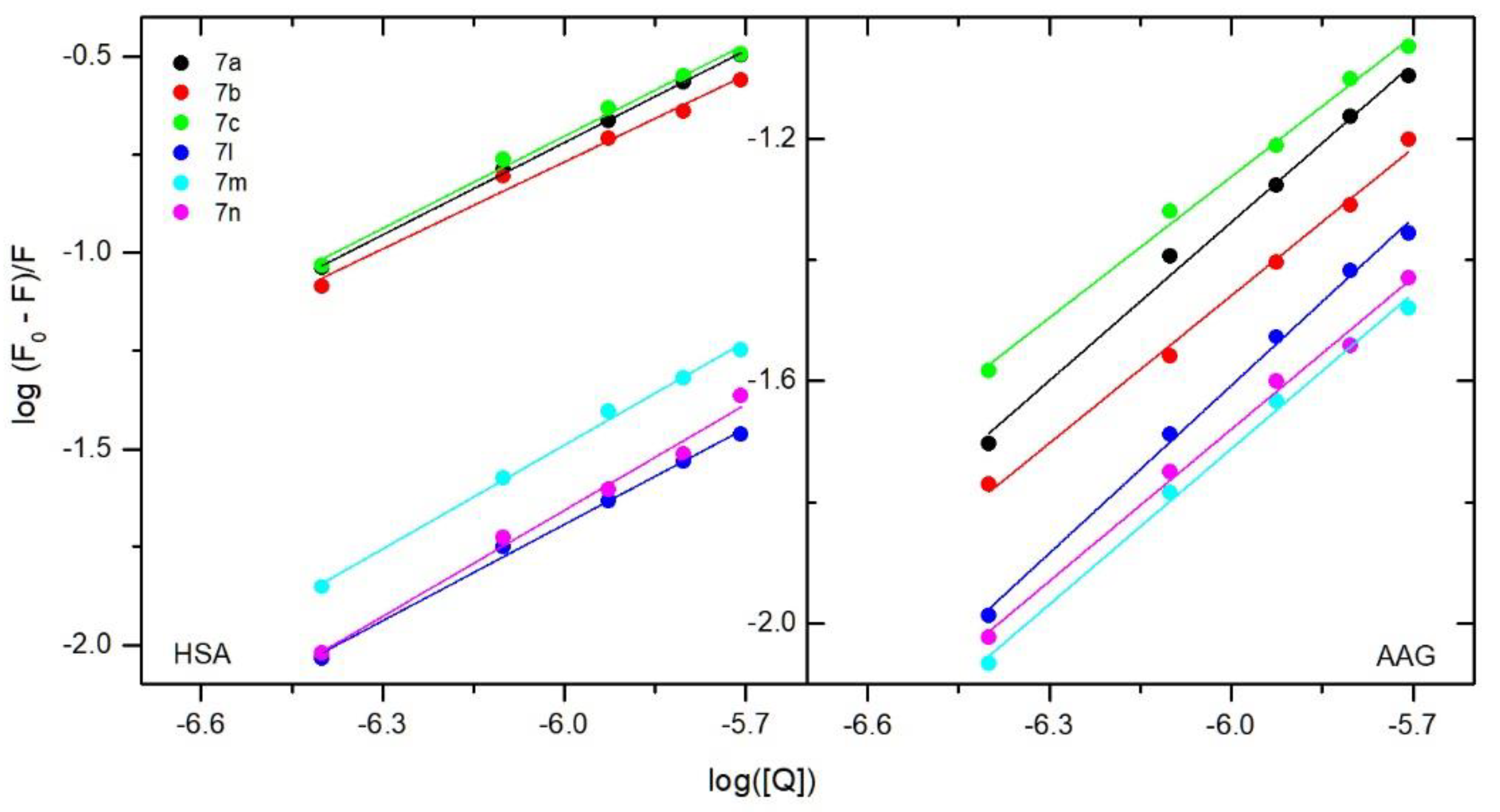
3.4.1. Fluorescence Quenching, Binding Constants, Site Markers and Thermodynamic Studies
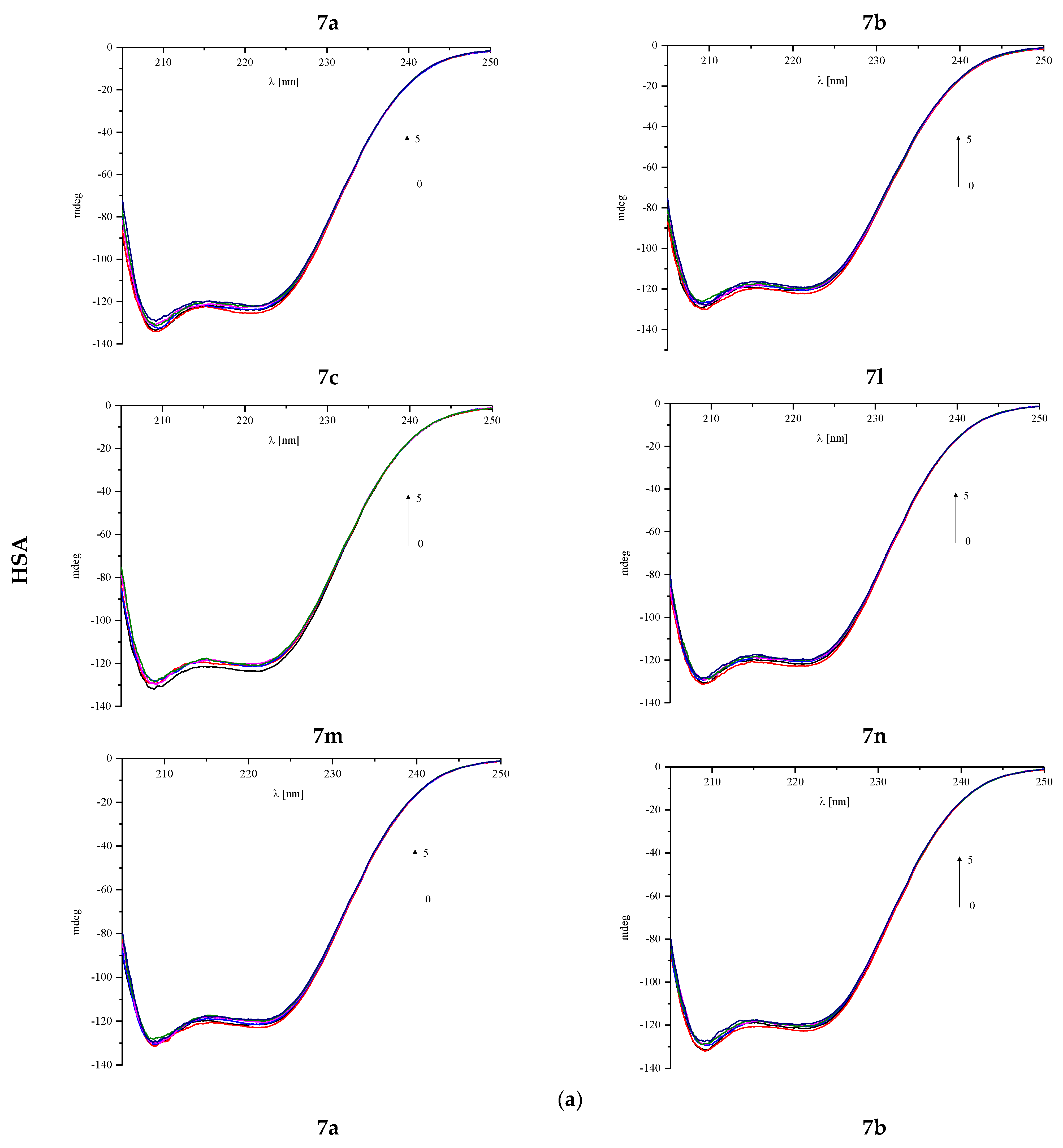
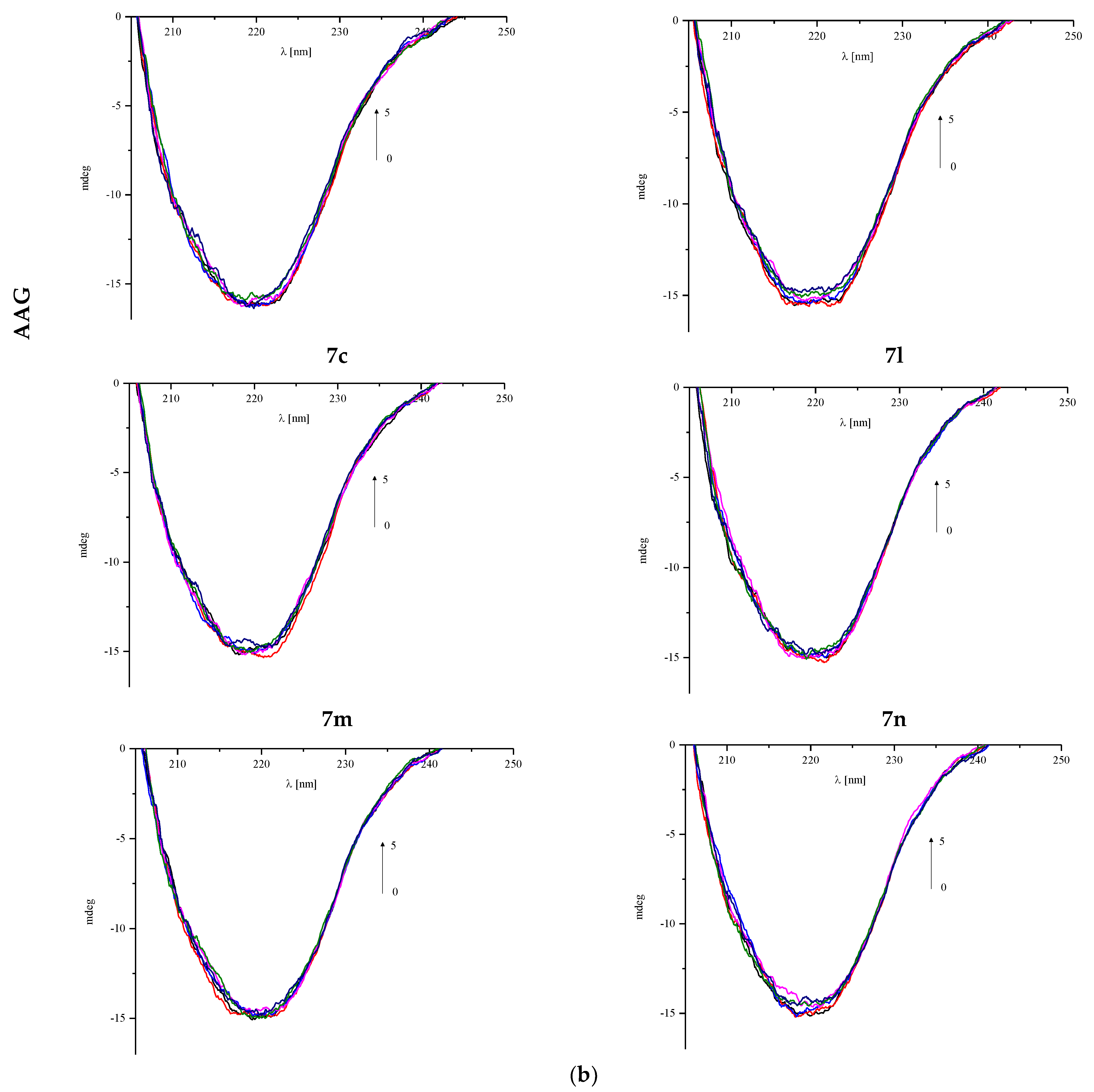
3.4.2. Circular Dichroism Spectra
3.4.3. Fourier-Transform Infrared Spectroscopic Measurements
3.4.4. Site Markers and Molecular Docking Studies
3.5. In Silico ADME, Physicochemical and Drug-Likeness Predictions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lucio, M.; Lima, J.L.F.C.; Reis, S. Drug-Membrane Interactions: Significance for Medicinal Chemistry. Curr. Med. Chem. 2010, 17, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Bunea, A.I.; Harloff-Helleberg, S.; Taboryski, R.; Nielsen, H.M. Membrane Interactions in Drug Delivery: Model Cell Membranes and Orthogonal Techniques. Adv. Colloid Interface Sci. 2020, 281, 102177. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leite, C.; Nunes, C.; Reis, S. Interaction of Nonsteroidal Anti-Inflammatory Drugs with Membranes: In Vitro Assessment and Relevance for Their Biological Actions. Prog. Lipid Res. 2013, 52, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.J.; Nicolson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Escribá, P.V.; González-Ros, J.M.; Goñi, F.M.; Kinnunen, P.K.J.; Vigh, L.; Sánchez-Magraner, L.; Fernández, A.M.; Busquets, X.; Horváth, I.; Barceló-Coblijn, G. Membranes: A Meeting Point for Lipids, Proteins and Therapies. J. Cell. Mol. Med. 2008, 12, 829–875. [Google Scholar] [CrossRef]
- Nathan, C. Points of Control in Inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef]
- Leuti, A.; Fazio, D.; Fava, M.; Piccoli, A.; Oddi, S.; Maccarrone, M. Bioactive Lipids, Inflammation and Chronic Diseases. Adv. Drug Deliv. Rev. 2020, 159, 133–169. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of Inflammation: The Beginning Programs the End. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef]
- Marnett, L.J. Cyclooxygenase Mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 545–552. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J.; Hancock, A.B. Perspective Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2006, 50, 1425–1441. [Google Scholar] [CrossRef]
- Cashman, J.N. The Mechanisms of Action of NSAIDs in Analgesia. Drugs 1996, 52, 13–23. [Google Scholar] [CrossRef]
- Omar, Y.M.; Abdel-Moty, S.G.; Abdu-Allah, H.H.M. Further Insight into the Dual COX-2 and 15-LOX Anti-Inflammatory Activity of 1,3,4-Thiadiazole-Thiazolidinone Hybrids: The Contribution of the Substituents at 5th Positions Is Size Dependent. Bioorg. Chem. 2020, 97, 103657. [Google Scholar] [CrossRef]
- Omar, Y.M.; Abdu-Allah, H.H.M.; Abdel-Moty, S.G. Synthesis, Biological Evaluation and Docking Study of 1,3,4-Thiadiazole-Thiazolidinone Hybrids as Anti-Inflammatory Agents with Dual Inhibition of COX-2 and 15-LOX. Bioorg. Chem. 2018, 80, 461–471. [Google Scholar] [CrossRef]
- Guo, H.; Verhoek, I.C.; Prins, G.G.H.; Van Der Vlag, R.; Van Der Wouden, P.E.; Van Merkerk, R.; Quax, W.J.; Olinga, P.; Hirsch, A.K.H.; Dekker, F.J. Novel 15-Lipoxygenase-1 Inhibitor Protects Macrophages from Lipopolysaccharide-Induced Cytotoxicity. J. Med. Chem. 2019, 62, 4624–4637. [Google Scholar] [CrossRef]
- Vane, J.R.; Botting, R.M. Mechanism of Action of Nonsteroidal Anti-Inflammatory Drugs. Am. J. Med. 1998, 104, 25–85. [Google Scholar] [CrossRef]
- Soll, A.H.; McCarthy, D. NSAID-Related Gastrointestinal Complications. Clin. Cornerstone 1999, 1, 42–56. [Google Scholar] [CrossRef]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse Effects of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs, Aspirin and Coxibs) on Upper Gastrointestinal Tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar] [CrossRef]
- Wallace, J.L. NSAID Gastropathy and Enteropathy: Distinct Pathogenesis Likely Necessitates Distinct Prevention Strategies. Br. J. Pharmacol. 2012, 165, 67–74. [Google Scholar] [CrossRef]
- Wallace, J.L.; Devchand, P.R. Emerging Roles for Cyclooxygenase-2 in Gastrointestinal Mucosal Defense. Br. J. Pharmacol. 2005, 145, 275–282. [Google Scholar] [CrossRef]
- Laine, L. Gastrointestinal Effects of NSAIDs and Coxibs. J. Pain Symptom Manag. 2003, 25, 32–40. [Google Scholar] [CrossRef]
- Cannon, C.P.; Cannon, P.J. Physiology. COX-2 Inhibitors and Cardiovascular Risk. Science 2012, 336, 1386–1387. [Google Scholar] [CrossRef] [PubMed]
- Dogné, J.-M.; Supuran, C.T.; Pratico, D. Adverse Cardiovascular Effects of the Coxibs. J. Med. Chem. 2005, 48, 2251–2257. [Google Scholar] [CrossRef]
- Redzicka, A.; Szczukowski, Ł.; Kochel, A.; Wiatrak, B.; Gębczak, K.; Czyżnikowska, Ż. COX-1/COX-2 Inhibition Activities and Molecular Docking Study of Newly Designed and Synthesized pyrrolo[3,4-C]pyrrole Mannich Bases. Bioorg. Med. Chem. 2019, 27, 3918–3928. [Google Scholar] [CrossRef] [PubMed]
- Gedawy, E.M.; Kassab, A.E.; El Kerdawy, A.M. Design, Synthesis and Biological Evaluation of Novel Pyrazole Sulfonamide Derivatives as Dual COX-2/5-LOX Inhibitors. Eur. J. Med. Chem. 2020, 189, 112066. [Google Scholar] [CrossRef] [PubMed]
- Jacob, P.J.; Manju, S.L. Identification and Development of Thiazole Leads as COX-2/5-LOX Inhibitors through in-Vitro and in-Vivo Biological Evaluation for Anti-Inflammatory Activity. Bioorg. Chem. 2020, 100, 103882. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Awad, S.M.; Said, A.M.; Mahgoub, S.; Taha, H.; Ahmed, N.M. Design, Synthesis, Molecular Modelling and Biological Evaluation of Novel 3-(2-Naphthyl)-1-Phenyl-1H-Pyrazole Derivatives as Potent Antioxidants and 15-Lipoxygenase Inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 847. [Google Scholar] [CrossRef]
- Chen, W.; Xu, Q.; Ma, X.; Mo, J.; Lin, G.; He, G.; Chu, Z.; Li, J. Synthesis and Biological Evaluation of N-(Benzene Sulfonyl)acetamide Derivatives as Anti-Inflammatory and Analgesic Agents with COX-2/5-LOX/ TRPV1 Multifunctional Inhibitory Activity. Bioorg. Med. Chem. Lett. 2023, 80, 129101. [Google Scholar] [CrossRef]
- Burdon, C.; Mann, C.; Cindrova-Davies, T.; Ferguson-Smith, A.C.; Burton, G.J. Oxidative Stress and the Induction of Cyclooxygenase Enzymes and Apoptosis in the Murine Placenta. Placenta 2007, 28, 724–733. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, Oxidative Stress and Inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- Maciążek-Jurczyk, M.; Morak-Młodawska, B.; Jeleń, M.; Kopeć, W.; Szkudlarek, A.; Owczarzy, A.; Kulig, K.; Rogóż, W.; Pożycka, J. The Influence of Oxidative Stress on Serum Albumin Structure as a Carrier of Selected Diazaphenothiazine with Potential Anticancer Activity. Pharmaceuticals 2021, 14, 285. [Google Scholar] [CrossRef] [PubMed]
- Szczukowski, Ł.; Redzicka, A.; Wiatrak, B.; Krzyżak, E.; Marciniak, A.; Gębczak, K.; Gębarowski, T.; Świątek, P. Design, Synthesis, Biological Evaluation and in Silico Studies of Novel pyrrolo[3,4-D]pyridazinone Derivatives with Promising Anti-Inflammatory and Antioxidant Activity. Bioorg. Chem. 2020, 102, 104035. [Google Scholar] [CrossRef]
- Li, Y.; He, W.; Liu, J.; Sheng, F.; Hu, Z.; Chen, X. Binding of the Bioactive Component Jatrorrhizine to Human Serum Albumin. Biochim. Biophys. Acta-Gen. Subj. 2005, 1722, 15–21. [Google Scholar] [CrossRef]
- Zadorozhnii, P.V.; Kiselev, V.V.; Kharchenko, A.V. In Silico ADME Profiling of Salubrinal and Its Analogues. Futur. Pharmacol. 2022, 2, 160–197. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominny, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Revision D. 01; Gaussian. Inc.: Wallingford, CT, USA.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785. [Google Scholar] [CrossRef] [PubMed]
- Byler, D.M.; Susi, H. Examination of the Secondary Structure of Proteins by Deconvolved FTIR Spectra. Biopolymers 1986, 25, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xie, M.-X.; Kang, J.; Zheng, D. Studies on the Interaction of Total Saponins of Panax Notoginseng and Human Serum Albumin by Fourier Transform Infrared Spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2003, 59, 2747–2758. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Yu, S. Fourier Transform Infrared Spectroscopic Analysis of Protein Secondary Structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. [Google Scholar] [CrossRef]
- Shi, J.-H.; Pan, D.-Q.; Wang, X.-X.; Liu, T.-T.; Jiang, M.; Wang, Q. Characterizing the Binding Interaction between Antimalarial Artemether (AMT) and Bovine Serum Albumin (BSA): Spectroscopic and Molecular Docking Methods. J. Photochem. Photobiol. B Biol. 2016, 162, 14–23. [Google Scholar] [CrossRef]
- Kobe, M.J.; Neau, D.B.; Mitchell, C.E.; Bartlett, S.G.; Newcomer, M.E. The Structure of Human 15-Lipoxygenase-2 with a Substrate Mimic. J. Biol. Chem. 2014, 289, 8562. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Gilbert, N.C.; Ohler, A.; Armstrong, M.; Perry, S.; Kalyanaraman, C.; Yasgar, A.; Rai, G.; Simeonov, A.; Jadhav, A.; et al. Kinetic and Structural Investigations of Novel Inhibitors of Human Epithelial 15-Lipoxygenase-2. Bioorg. Med. Chem. 2021, 46, 116349. [Google Scholar] [CrossRef]
- Ng, C.H.; Rullah, K.; Aluwi, M.F.F.M.; Abas, F.; Lam, K.W.; Ismail, I.S.; Narayanaswamy, R.; Jamaludin, F.; Shaari, K. Synthesis and Docking Studies of 2,4,6-Trihydroxy-3-Geranylacetophenone Analogs as Potential Lipoxygenase Inhibitor. Molecules 2014, 19, 11645. [Google Scholar] [CrossRef]
- Mahdavi, M.; Shirazi, M.S.; Taherkhani, R.; Saeedi, M.; Alipour, E.; Moghadam, F.H.; Moradi, A.; Nadri, H.; Emami, S.; Firoozpour, L.; et al. Synthesis, Biological Evaluation and Docking Study of 3-Aroyl-1-(4-Sulfamoylphenyl)thiourea Derivatives as 15-Lipoxygenase Inhibitors. Eur. J. Med. Chem. 2014, 82, 308–313. [Google Scholar] [CrossRef] [PubMed]
- ElBordiny, H.S.; El-Miligy, M.M.; Kassab, S.E.; Daabees, H.; Abdelhamid Mohamed El-Hawash, S.; Mohamed Ali, W.A. Design, Synthesis, Biological Evaluation and Docking Studies of New 3-(4,5-Dihydro-1H-Pyrazol/isoxazol-5-Yl)-2-Phenyl-1H-Indole Derivatives as Potent Antioxidants and 15-Lipoxygenase Inhibitors. Eur. J. Med. Chem. 2018, 145, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Kumar Jain, M.; Min Wu, N. Effect of Small Molecules on the Dipalmitoyl Lecithin Liposomal Bilayer: III. Phase Transition in Lipid Bilayer. J. Membr. Biol 1977, 34, 157–201. [Google Scholar] [CrossRef]
- Chen, G.Z.; X.Z., H.; Xu, J.H.; Zneng, Z.Z.; Wang, Z.B. The Methods of Fluorescence Analysis, 2nd ed.; Science: Beijing, China, 1990. [Google Scholar]
- Lakowicz, J.R.; Weber, G. Quenching of Fluorescence by Oxygen. Probe for Structural Fluctuations in Macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, MA, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Ware, W.R. Oxygen Quenching of Fluorescence in Solution: An Experimental Study of the Diffusion Process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Mohammadnia, F.; Fatemi, M.H.; Taghizadeh, S.M. Study on the Interaction of Anti-Inflammatory Drugs with Human Serum Albumin Using Molecular Docking, Quantitative Structure–activity Relationship, and Fluorescence Spectroscopy. Luminescence 2020, 35, 266–273. [Google Scholar] [CrossRef]
- Dufour, C.; Dangles, O. Flavonoid-Serum Albumin Complexation: Determination of Binding Constants and Binding Sites by Fluorescence Spectroscopy. Biochim. Biophys. Acta-Gen. Subj. 2005, 1721, 164–173. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Bakheit, A.H.; Mohamed, M.S.; Eldehna, W.M.; Abdel-Aziz, H.A.; Attia, M.I. Synthesis and Biophysical Insights into the Binding of a Potent Anti-Proliferative Non-Symmetric Bis-Isatin Derivative with Bovine Serum Albumin: Spectroscopic and Molecular Docking Approaches. Appl. Sci. 2017, 7, 617. [Google Scholar] [CrossRef]
- Suryawanshi, V.D.; Walekar, L.S.; Gore, A.H.; Anbhule, P.V.; Kolekar, G.B. Spectroscopic Analysis on the Binding Interaction of Biologically Active Pyrimidine Derivative with Bovine Serum Albumin. J. Pharm. Anal. 2016, 6, 56–63. [Google Scholar] [CrossRef]
- Wani, T.A.; Bakheit, A.H.; Zargar, S.; Bhat, M.A.; Al-Majed, A.A. Molecular Docking and Experimental Investigation of New Indole Derivative Cyclooxygenase Inhibitor to Probe Its Binding Mechanism with Bovine Serum Albumin. Bioorg. Chem. 2019, 89, 103010. [Google Scholar] [CrossRef]
- Wani, T.A.; Bakheit, A.H.; Al-Majed, A.R.A.; Bhat, M.A.; Zargar, S. Study of the Interactions of Bovine Serum Albumin with the New Anti-Inflammatory Agent 4-(1,3-Dsioxo-1,3-Dihydro-2H-Isoindol-2-Yl)-N-[(4-Ethoxy-Phenyl) Methylidene]benzohydrazide Using a Multi-Spectroscopic Approach and Molecular Docking. Molecules 2017, 22, 1258. [Google Scholar] [CrossRef]
- Krzyżak, E.; Szkatuła, D.; Wiatrak, B.; Gębarowski, T.; Marciniak, A. Synthesis, Cyclooxygenases Inhibition Activities and Interactions with BSA of N-Substituted 1H-pyrrolo[3,4-C]pyridine-1,3(2H)-Diones Derivatives. Molecules 2020, 25, 2934. [Google Scholar] [CrossRef]
- Owczarzy, A.; Zięba, A.; Pożycka, J.; Kulig, K.; Rogóż, W.; Szkudlarek, A.; Maciążek-jurczyk, M. Spectroscopic Studies of Quinobenzothiazine Derivative in Terms of the in Vitro Interaction with Selected Human Plasma Proteins. Part 1. Molecules 2021, 26, 4776. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Terada, H. Estimation of State and Amount of Phenylalanine Residues in Proteins by Second Derivative Spectrophotometry. Biochim. Biophys. Acta-Protein Struct. 1979, 580, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Al-Qadiri, H.M.; Lin, M.; Rasco, B.A. Application of Mid-Infrared and Raman Spectroscopy to the Study of Bacteria. Food Bioprocess Technol. 2011, 4, 919–935. [Google Scholar] [CrossRef]
- Ojeda, J.J.; Dittrich, M. Fourier Transform Infrared Spectroscopy for Molecular Analysis of Microbial Cells. Methods Mol. Biol. 2012, 881, 187–211. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; McEwen, G.D.; Wu, Y.; Miller, C.D.; Zhou, A. Characterization and Analysis of Mycobacteria and Gram-Negative Bacteria and Co-Culture Mixtures by Raman Microspectroscopy, FTIR, and Atomic Force Microscopy. Anal. Bioanal. Chem. 2013, 405, 1577–1591. [Google Scholar] [CrossRef]
- Zohdi, V.; Whelan, D.R.; Wood, B.R.; Pearson, J.T.; Bambery, K.R.; Black, M.J. Importance of Tissue Preparation Methods in FTIR Micro-Spectroscopical Analysis of Biological Tissues: “Traps for New Users.”. PLoS ONE 2015, 10, e0116491. [Google Scholar] [CrossRef]
- Machovič, V.; Lapčák, L.; Havelcová, M.; Borecká, L.; Novotná, M.; Novotná, M.; Javůrková, I.; Langrová, I.; Hájková, S.; Brožová, A.; et al. Analysis of European Honeybee (Apis Mellifera) Wings Using ATR-FTIR and Raman Spectroscopy: A Pilot Study. Sci. Agric. Bohem. 2017, 48, 22–29. [Google Scholar] [CrossRef]
- Han, Y.; Han, L.; Yao, Y.; Li, Y.; Liu, X. Key Factors in FTIR Spectroscopic Analysis of DNA: The Sampling Technique, Pretreatment Temperature and Sample Concentration. Anal. Methods 2018, 10, 2436–2443. [Google Scholar] [CrossRef]
- Kochan, K.; Lai, E.; Richardson, Z.; Nethercott, C.; Peleg, A.Y.; Heraud, P.; Wood, B.R. Vibrational Spectroscopy as a Sensitive Probe for the Chemistry of Intra-Phase Bacterial Growth. Sensors 2020, 20, 3452. [Google Scholar] [CrossRef]
- Pakbin, B.; Zolghadr, L.; Rafiei, S.; Brück, W.M.; Brück, T.B. FTIR Differentiation Based on Genomic DNA for Species Identification of Shigella Isolates from Stool Samples. Sci. Rep. 2022, 12, 2780. [Google Scholar] [CrossRef]
- Svenson, J.; Brandsdal, B.O.; Stensen, W.; Svendsen, J.S. Albumin Binding of Short Cationic Antimicrobial Micropeptides and Its Influence on the in Vitro Bactericidal Effect. J. Med. Chem. 2007, 50, 3334–3339. [Google Scholar] [CrossRef]
- Wani, T.A.; Bakheit, A.H.; Abounassif, M.A.; Zargar, S. Study of Interactions of an Anticancer Drug Neratinib with Bovine Serum Albumin: Spectroscopic and Molecular Docking Approach. Front. Chem. 2018, 6, 47. [Google Scholar] [CrossRef]
- Zorzi, A.; Linciano, S.; Angelini, A. Non-Covalent Albumin-Binding Ligands for Extending the Circulating Half-Life of Small Biotherapeutics. Medchemcomm 2019, 10, 1068. [Google Scholar] [CrossRef]
- Khalid, I.M.; Sharkh, S.E.A.; Samamarh, H.; Alfaqeeh, R.; Abuteir, M.M.; Darwish, S.M. Spectroscopic Characterization of the Interaction between Dopamine and Human Serum Albumin. Open J. Biophys. 2019, 9, 110–130. [Google Scholar] [CrossRef]
- Barth, A. Infrared Spectroscopy of Proteins. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef]
- Roy, A.S.; Tripathy, D.R.; Chatterjee, A.; Dasgupta, S. A Spectroscopic Study of the Interaction of the Antioxidant Naringin with Bovine Serum Albumin. J. Biophys. Chem. 2010, 1, 141–152. [Google Scholar] [CrossRef]
- Baldassarre, M.; Galeazzi, R.; Maggiore, B.; Tanfani, F.; Scirè, A. Bovine α1-Acid Glycoprotein, a Thermostable Version of Its Human Counterpart: Insights from Fourier Transform Infrared Spectroscopy and in Silico Modelling. Biochimie 2014, 102, 19–28. [Google Scholar] [CrossRef]
- Marciniak, A.; Kotynia, A.; Szkatuła, D.; Krzyżak, E. The 2-Hydroxy-3-(4-Aryl-1-Piperazinyl)propyl Phthalimide Derivatives as Prodrugs—Spectroscopic and Theoretical Binding Studies with Plasma Proteins. Int. J. Mol. Sci. 2022, 23, 7003. [Google Scholar] [CrossRef]
- Kopecký, V., Jr.; Ettrich, R.; Hofbauerová, K.; Baumruk, V. Vibrational Spectroscopy and Computer Modeling of Proteins: Solving Structure of α1-Acid Glycoprotein. Spectroscopy 2004, 18, 323–330. [Google Scholar] [CrossRef]
- Nishi, K.; Sakai, N.; Komine, Y.; Maruyama, T.; Halsall, H.B.; Otagiri, M. Structural and Drug-Binding Properties of α1-Acid Glycoprotein in Reverse Micelles. Biochim. Biophys. Acta-Proteins Proteom. 2002, 1601, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Maruyama, T.; Halsall, H.B.; Handa, T.; Otagiri, M. Binding of α1-Acid Glycoprotein to Membrane Results in a Unique Structural Change and Ligand Release. Biochemistry 2004, 43, 10513–10519. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural Basis of the Drug-Binding Specificity of Human Serum Albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, D.L.; Ravelli, R.B.G.; Mueller, U.; Skerra, A. The 1.8-Å Crystal Structure of α1-Acid Glycoprotein (Orosomucoid) Solved by UV RIP Reveals the Broad Drug-Binding Activity of This Human Plasma Lipocalin. J. Mol. Biol. 2008, 384, 393–405. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhang, H.; Liu, J.; Lau, C.W.; Liu, P.; Chen, Z.Y.; Lee, H.K.; Tipoe, G.L.; Ho, H.M.; Yao, X.; et al. Cyclooxygenase-2-Dependent Oxidative Stress Mediates Palmitate-Induced Impairment of Endothelium-Dependent Relaxations in Mouse Arteries. Biochem. Pharmacol. 2015, 91, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Roman, G. Mannich Bases in Medicinal Chemistry and Drug Design. Eur. J. Med. Chem. 2015, 89, 743–816. [Google Scholar] [CrossRef]
- Ma, L.; Xiao, Y.; Li, C.; Xie, Z.L.; Li, D.D.; Wang, Y.T.; Ma, H.T.; Zhu, H.L.; Wang, M.H.; Ye, Y.H. Synthesis and Antioxidant Activity of Novel Mannich Base of 1,3,4-Oxadiazole Derivatives Possessing 1,4-Benzodioxan. Bioorganic Med. Chem. 2013, 21, 6763–6770. [Google Scholar] [CrossRef]
- Szandruk-Bender, M.; Wiatrak, B.; Szczukowski, Ł.; Świątek, P.; Rutkowska, M.; Dzimira, S.; Merwid-Ląd, A.; Danielewski, M.; Szeląg, A. Oxadiazole Derivatives of Pyrrolo[3,4-D]pyridazinone Exert Antinociceptive Activity in the Tail-Flick and Formalin Test in Rodents and Reveal Reduced Gastrotoxicity. Int. J. Mol. Sci. 2020, 21, 9685. [Google Scholar] [CrossRef]
- Szandruk-Bender, M.; Merwid-Ląd, A.; Wiatrak, B.; Danielewski, M.; Dzimira, S.; Szkudlarek, D.; Szczukowski, Ł.; Świątek, P.; Szeląg, A. Novel 1,3,4-Oxadiazole Derivatives of Pyrrolo [3,4-d]Pyridazinone Exert Anti-Inflammatory Activity without Acute Gastrotoxicity in the Carrageenan-Induced Rat Paw Edema Test. J. Inflamm. Res. 2021, 2021, 5739–5756. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 [µM] |
|---|---|
| 7a | 12.63 (0.06) * |
| 7b | 10.95 (0.04) * |
| 7c | 12.47 (0.06) * |
| 7l | 12.81 (0.03) * |
| 7m | 12.73 (0.05) * |
| 7n | 14.07 (0.04) * |
| zileuton | 13.41 (0.04) |
| 15-LOX | |
|---|---|
| 7a | −9.7 |
| 7b | −8.6 |
| 7c | −9.0 |
| 7l | −8.5 |
| 7m | −7.3 |
| 7n | −8.8 |
| Compound | Tm (°C) | T1/2 |
|---|---|---|
| 7a | 22.65 +/− 0.17 | 1.10 +/− 0.12 |
| 7b | 21.90 +/− 0.18 | 1.63 +/− 0.10 |
| 7c | 22.40 +/− 0.08 | 1.20 +/− 0.12 |
| 7l | 21.73 +/− 0.38 | 1.55 +/− 0.29 |
| 7m | 22.75 +/− 0.13 | 0.98 +/− 0.05 |
| 7n | 22.38 +/− 0.15 | 1.13 +/− 0.15 |
| Compound | Concentration [μM] | 24 h Incubation with Compounds [E/E0] | 1 h Incubation with Compounds [E/E0] | ||||
|---|---|---|---|---|---|---|---|
| With | With | ||||||
| H2O2 | H2O2 | ||||||
| Mean | SEM | p | Mean | SEM | p | ||
| H2O2 (incubated only 1 h) | 100 | 1.675 | 0.021 | * | 1.798 | 0.033 | * |
| 7a | 100 | 0.566 | 0.006 | * | 0.845 | 0.021 | * |
| 50 | 0.575 | 0.008 | * | 0.853 | 0.014 | * | |
| 10 | 0.669 | 0.003 | * | 0.888 | 0.012 | * | |
| 7b | 100 | 0.606 | 0.004 | * | 0.949 | 0.006 | * |
| 50 | 0.614 | 0.007 | * | 1.010 | 0.008 | ||
| 10 | 0.619 | 0.006 | * | 1.121 | 0.009 | * | |
| 7c | 100 | 0.544 | 0.003 | * | 0.939 | 0.004 | * |
| 50 | 0.551 | 0.002 | * | 1.091 | 0.004 | ||
| 10 | 0.575 | 0.005 | * | 1.226 | 0.034 | * | |
| 7l | 100 | 0.573 | 0.006 | * | 0.851 | 0.011 | * |
| 50 | 0.561 | 0.004 | * | 0.949 | 0.004 | * | |
| 10 | 0.544 | 0.008 | * | 1.029 | 0.003 | ||
| 7m | 100 | 0.567 | 0.009 | * | 1.286 | 0.087 | * |
| 50 | 0.557 | 0.011 | * | 1.244 | 0.023 | * | |
| 10 | 0.547 | 0.005 | * | 1.112 | 0.067 | * | |
| 7n | 100 | 0.631 | 0.009 | * | 1.418 | 0.055 | * |
| 50 | 0.515 | 0.006 | * | 1.418 | 0.023 | * | |
| 10 | 0.528 | 0.004 | * | 1.183 | 0.045 | * | |
| Compound | Concentration [μM] | 24 h Incubation with Compounds [E/E0] | 1 h Incubation with Compounds [E/E0] | ||||
|---|---|---|---|---|---|---|---|
| With | With | ||||||
| H2O2 | H2O2 | ||||||
| Mean | SEM | p | Mean | SEM | p | ||
| SIN-1 (incubated only 1 h) | 100 | 1.982 | 0.018 | * | 1.765 | 0.024 | * |
| 7a | 100 | 0.845 | 0.016 | * | 1.066 | 0.011 | * |
| 50 | 0.883 | 0.026 | * | 1.129 | 0.021 | ||
| 10 | 0.922 | 0.018 | * | 1.169 | 0.013 | ||
| 7b | 100 | 0.872 | 0.020 | * | 1.071 | 0.015 | |
| 50 | 0.832 | 0.038 | * | 1.114 | 0.033 | ||
| 10 | 0.832 | 0.028 | * | 1.229 | 0.023 | * | |
| 7c | 100 | 0.851 | 0.016 | * | 0.978 | 0.011 | |
| 50 | 0.840 | 0.010 | * | 0.964 | 0.005 | ||
| 10 | 0.834 | 0.010 | * | 0.906 | 0.005 | ||
| 7l | 100 | 0.977 | 0.014 | 0.995 | 0.009 | ||
| 50 | 0.902 | 0.012 | * | 0.931 | 0.004 | ||
| 10 | 0.842 | 0.039 | * | 0.853 | 0.022 | ||
| 7m | 100 | 0.912 | 0.013 | * | 0.923 | 0.011 | |
| 50 | 0.844 | 0.012 | * | 0.855 | 0.005 | ||
| 10 | 0.838 | 0.014 | * | 0.849 | 0.005 | ||
| 7n | 100 | 0.871 | 0.017 | * | 0.881 | 0.009 | |
| 50 | 0.802 | 0.008 | * | 0.813 | 0.007 | ||
| 10 | 0.790 | 0.008 | * | 0.801 | 0.034 | ||
| Compound | Concentration [μM] | 24 h Incubation with Compounds [E/E0] | 1 h Incubation with Compounds [E/E0] | ||||
|---|---|---|---|---|---|---|---|
| With H2O2 | With H2O2 | ||||||
| Mean | SEM | p | Mean | SEM | p | ||
| H2O2 (incubated only 1 h) | 100 | 1.542 | 0.043 | * | 1.432 | 0.053 | * |
| 7a | 100 | 0.578 | 0.012 | * | 0.951 | 0.021 | |
| 50 | 0.581 | 0.022 | * | 0.958 | 0.011 | ||
| 10 | 0.599 | 0.031 | * | 0.967 | 0.012 | ||
| 7b | 100 | 0.545 | 0.041 | * | 0.911 | 0.021 | |
| 50 | 0.580 | 0.022 | * | 0.935 | 0.023 | ||
| 10 | 0.595 | 0.027 | * | 0.945 | 0.021 | ||
| 7c | 100 | 0.554 | 0.028 | * | 0.974 | 0.024 | |
| 50 | 0.561 | 0.012 | * | 1.002 | 0.021 | ||
| 10 | 0.571 | 0.015 | * | 1.054 | 0.041 | ||
| 7l | 100 | 0.603 | 0.022 | * | 1.065 | 0.027 | |
| 50 | 0.589 | 0.027 | * | 1.024 | 0.024 | ||
| 10 | 0.528 | 0.043 | * | 0.824 | 0.018 | * | |
| 7m | 100 | 0.642 | 0.065 | * | 1.373 | 0.014 | * |
| 50 | 0.561 | 0.054 | * | 1.324 | 0.024 | * | |
| 10 | 0.501 | 0.061 | * | 1.209 | 0.034 | * | |
| 7n | 100 | 0.822 | 0.042 | * | 1.302 | 0.024 | * |
| 50 | 0.565 | 0.044 | * | 1.246 | 0.031 | * | |
| 10 | 0.550 | 0.057 | * | 1.045 | 0.033 | ||
| Compound | Concentration [μM] | 24 h Incubation with Compounds [E/E0] | 1 h Incubation with Compounds [E/E0] | ||||
|---|---|---|---|---|---|---|---|
| With | With | ||||||
| H2O2 | H2O2 | ||||||
| Mean | SEM | p | Mean | SEM | p | ||
| SIN-1 (incubated only 1 h) | 100 | 1.987 | 0.110 | * | 2.021 | 0.51 | * |
| 7a | 100 | 0.827 | 0.005 | * | 0.815 | 0.007 | * |
| 50 | 0.843 | 0.007 | * | 0.838 | 0.009 | * | |
| 10 | 0.887 | 0.008 | * | 0.898 | 0.010 | * | |
| 7b | 100 | 0.982 | 0.044 | * | 0.954 | 0.061 | * |
| 50 | 0.877 | 0.007 | * | 0.849 | 0.024 | * | |
| 10 | 0.852 | 0.009 | * | 0.824 | 0.026 | * | |
| 7c | 100 | 1.042 | 0.014 | 1.014 | 0.031 | ||
| 50 | 1.049 | 0.026 | 1.019 | 0.026 | |||
| 10 | 1.039 | 0.024 | 1.012 | 0.031 | |||
| 7l | 100 | 1.085 | 0.009 | 1.043 | 0.016 | ||
| 50 | 1.054 | 0.019 | 1.026 | 0.026 | |||
| 10 | 0.992 | 0.019 | 1.019 | 0.026 | |||
| 7m | 100 | 1.047 | 0.029 | 1.093 | 0.036 | ||
| 50 | 1.054 | 0.006 | 1.046 | 0.013 | |||
| 10 | 1.033 | 0.005 | 1.033 | 0.012 | |||
| 7n | 100 | 1.180 | 0.007 | * | 1.048 | 0.017 | |
| 50 | 1.040 | 0.022 | 1.040 | 0.032 | |||
| 10 | 1.033 | 0.030 | 1.031 | 0.04 | |||
| Quenching | Binding | Thermodynamic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T [K] | Ksv × 105 [dm3·mol−1] | kq × 1013 [dm3·mol−1·s−1] | logKb | Kb × 103 [dm3·mol−1] | n | ΔG° [kJmol−1] | ΔH° [kJmol−1] | ΔS° [Jmol−1 K−1] | |
| 7a | 297 303 308 | 1.60 ± 0.10 1.23 ± 0.18 0.63 ± 0.06 | 1.60 1.23 0.63 | 3.98 ± 0.09 2.93 ± 0.21 2.44 ± 0.30 | 9.12 0.85 0.27 | 0.79 ± 0.02 0.65 ± 0.05 0.61 ± 0.07 | −22.22 | −245.67 | −752.33 |
| 7b | 297 303 308 | 1.36 ± 0.18 1.13 ± 0.13 0.99 ± 0.14 | 1.36 1.13 0.99 | 3.66 ± 0.19 2.91 ± 0.22 2.49 ± 0.23 | 4.57 0.79 0.31 | 0.73 ± 0.05 0.62 ± 0.04 0.56 ± 0.03 | −20.62 | −187.54 | −562.03 |
| 7c | 297 303 308 | 1.63 ± 0.13 1.58 ± 0.10 1.33 ± 0.11 | 1.63 1.58 1.33 | 3.99 ± 0.13 3.69 ± 0.14 3.18 ± 0.14 | 9.77 4.90 1.51 | 0.78 ± 0.04 0.74 ± 0.02 0.66 ± 0.02 | −22.95 | −127.23 | −351.10 |
| 7l | 297 303 308 | 1.73 ± 0.10 1.09 ± 0.09 0.77 ± 0.08 | 1.73 1.09 0.77 | 3.22 ± 0.19 2.90 ± 0.21 2.54 ± 0.22 | 1.66 0.79 0.35 | 0.82 ± 0.03 0.62 ± 0.07 0.79 ± 0.04 | −18.41 | −109.07 | −295.15 |
| 7m | 297 303 308 | 0.29 ± 0.02 0.19 ± 0.01 0.09 ± 0.01 | 0.29 0.19 0.09 | 3.80 ± 0.17 3.07 ± 0.30 2.47 ± 0.32 | 6.31 1.18 0.30 | 0.88 ± 0.03 0.79 ± 0.05 0.75 ± 0.08 | −21.63 | −211.64 | −639.76 |
| 7n | 297 303 308 | 0.21 ± 0.01 0.18 ± 0.02 0.11 ± 0.01 | 0.21 0.18 0.11 | 3.75 ± 0.23 3.00 ± 0.24 2.35 ± 0.19 | 5.62 1.12 0.22 | 0.91 ± 0.05 0.78 ± 0.07 0.70 ± 0.04 | −21.47 | −222.00 | −675.18 |
| Quenching | Binding | Thermodynamic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T [K] | Ksv × 104 [dm3·mol−1] | kq × 1012 [dm3·mol−1·s−1] | logKb | Kb × 103 [dm3·mol−1] | n | ΔG° [kJmol−1] | ΔH° [kJmol−1] | ΔS° [Jmol−1 K−1] | |
| 7a | 297 303 308 | 4.08 ± 0.20 3.44 ± 0.23 2.97 ± 0.17 | 4.08 3.44 2.97 | 3.89 ± 0.24 3.24 ± 0.20 2.73 ± 0.28 | 7.76 1.74 0.53 | 0.87 ± 0.04 0.77 ± 0.04 0.69 ± 0.05 | −22.18 | −186.27 | −552.71 |
| 7b | 297 303 308 | 3.08 ± 0.12 2.13 ± 0.17 1.77 ± 0.24 | 3.08 2.13 1.77 | 3.41 ± 0.21 2.65 ± 0.25 2.00 ± 0.32 | 1.57 0.45 0.10 | 0.81 ± 0.04 0.71 ± 0.04 0.61 ± 0.05 | −19.44 | −224.21 | −689.48 |
| 7c | 297 303 308 | 4.52 ± 0.31 4.19 ± 0.33 3.25 ± 0.39 | 4.52 4.19 3.25 | 3.39 ± 0.18 2.65 ± 0.21 2.31 ± 0.30 | 9.77 4.90 1.51 | 0.76 ± 0.03 0.65 ± 0.04 0.60 ± 0.07 | −19.05 | −175.20 | −525.78 |
| 7l | 297 303 308 | 2.50 ± 0.05 2.06 ± 0.10 1.16 ± 0.18 | 2.50 2.06 1.16 | 3.93 ± 0.18 3.31 ± 0.09 2.44 ± 0.31 | 8.32 2.04 0.28 | 0.92 ± 0.03 0.82 ± 0.02 0.69 ± 0.06 | −20.18 | −233.19 | −717.16 |
| 7m | 297 303 308 | 1.69 ± 0.10 1.45 ± 0.17 0.96 ± 0.11 | 1.69 1.45 0.96 | 3.42 ± 0.20 2.37 ± 0.29 2.00 ± 0.23 | 2.63 0.23 0.10 | 0.86 ± 0.03 0.68 ± 0.05 0.65 ± 0.04 | −19.00 | −229.09 | −707.36 |
| 7n | 297 303 308 | 1.83 ± 0.10 1.67 ± 0.09 1.40 ± 0.08 | 1.83 1.67 1.40 | 3.33 ± 0.23 3.06 ± 0.24 2.76 ± 0.21 | 2.14 1.15 0.56 | 0.84 ± 0.04 0.80 ± 0.04 0.75 ± 0.03 | −19.02 | −91.74 | −244.84 |
| Site Marker | LogKb | |||||
|---|---|---|---|---|---|---|
| 7a | 7b | 7c | 7l | 7m | 7n | |
| - | 3.98 ± 0.09 | 3.66 ± 0.19 | 3.99 ± 0.13 | 3.22 ± 0.19 | 3.80 ± 0.17 | 3.75 ± 0.23 |
| HSA+PHB (site I) | 3.00 ± 0.22 | 2.47 ± 0.20 | 3.20 ± 0.47 | 2.49 ± 0.17 | 3.07 ± 0.05 | 2.90 ± 0.40 |
| HSA+IBP (site II) | 2.64 ± 0.07 | 2.71 ± 0.32 | 3.05 ± 0.36 | 2.13 ± 0.30 | 3.19 ± 0.16 | 2.51 ± 0.27 |
| HSA: Analyzed Compound Molar Ratio | 7a | 7b | 7c | 7l | 7m | 7n |
|---|---|---|---|---|---|---|
| 1:0 | 69.1% | 67.2% | 68.0% | 67.6% | 67.7% | 67.6% |
| 1:0.5 | 68.5% | 66.7% | 67.2% | 67.3% | 67.0% | 66.9% |
| 1:1 | 68.7% | 66.4% | 66.9% | 66.6% | 66.7% | 66.4% |
| 1:2 | 68.1% | 66.3% | 66.6% | 66.3% | 66.5% | 66.5% |
| 1:5 | 68.0% | 65.8% | 66.8% | 66.4% | 66.2% | 66.3% |
| AAG: Analyzed Compound Molar Ratio | % α-Helix | % β-Sheet | % α-Helix | % β-Sheet |
|---|---|---|---|---|
| 7a | 7b | |||
| 1:0 | 24.3% | 35.3% | 22.8% | 35.1% |
| 1:0.5 | 24.0% | 34.6% | 22.9% | 35.8% |
| 1:1 | 24.0% | 35.8% | 22.4% | 36.0% |
| 1:2 | 23.8% | 35.6% | 22.0% | 35.8% |
| 1:5 | 23.7% | 35.9% | 21.9% | 36.1% |
| 7c | 7l | |||
| 1:0 | 22.0% | 35.9% | 21.7% | 36.6% |
| 1:0.5 | 21.6% | 35.7% | 21.4% | 36.0% |
| 1:1 | 21.9% | 36.9% | 21.4% | 36.5% |
| 1:2 | 21.9% | 36.8% | 21.3% | 36.4% |
| 1:5 | 21.3% | 36.2% | 21.5% | 37.1% |
| 7m | 7n | |||
| 1:0 | 21.5% | 36.4% | 21.1% | 36.2% |
| 1:0.5 | 21.2% | 36.5% | 21.4% | 37.1% |
| 1:1 | 20.9% | 35.6% | 20.6% | 36.0% |
| 1:2 | 20.9% | 36.6% | 20.6% | 36.9% |
| 1:5 | 20.9% | 37.0% | 21.2% | 36.9% |
| Amide I Ingredient Related to Structure | Free HSA | Complex HSA with Compound | |||||
|---|---|---|---|---|---|---|---|
| 7a | 7b | 7c | 7l | 7m | 7n | ||
| β-sheet (1610–1640 cm−1) | 25.42 | 29.47 | 25.75 | 26.39 | 26.42 | 28.18 | 25.69 |
| random coil (1640–1650 cm−1) | 3.42 | 2.35 | 7.24 | 8.07 | 7.69 | 6.04 | 6.84 |
| α-helix (1650–1665 cm−1) | 64.55 | 56.83 | 56.41 | 56.53 | 54.09 | 59.77 | 59.64 |
| β-turn (1666–1673 cm−1) | 6.50 | 10.75 | 10.05 | 8.71 | 11.69 | 5.53 | 7.11 |
| β-antiparallel (1675–1695 cm−1) | 0.11 | 0.60 | 0.55 | 0.30 | 0.11 | 0.48 | 0.72 |
| Amide I Ingredient Related to Structure | Free AAG | Complex AAG with Compound | |||||
|---|---|---|---|---|---|---|---|
| 7a | 7b | 7c | 7l | 7m | 7n | ||
| β-sheet (1610–1640 cm−1) | 40.21 | 33.28 | 36.88 | 32.89 | 30.55 | 34.85 | 28.73 |
| random coil (1640–1650 cm−1) | 15.33 | 20.28 | 18.62 | 25.63 | 26.13 | 23.57 | 31.41 |
| α-helix (1650–1665 cm−1) | 25.02 | 30.06 | 26.95 | 25.09 | 26.63 | 25.80 | 25.92 |
| β-turn (1666–1673 cm−1) | 9.64 | 11.40 | 9.20 | 11.74 | 12.51 | 8.12 | 11.40 |
| β-antiparallel (1675–1695 cm−1) | 9.79 | 3.99 | 8.37 | 4.66 | 4.19 | 7.67 | 2.54 |
| Compound | HSA | AAG | ||
|---|---|---|---|---|
| IIA (PHB) | IIA-IIB (IBU) | IIIA (IBU) | ||
| 7a | −6.7 | −8.9 | −5.6 | −9.7 |
| 7b | −8.4 | −8.2 | −6.0 | −8.1 |
| 7c | −5.6 | −8.9 | −1.9 | −9.2 |
| 7l | −6.9 | −7.6 | −7.3 | −8.6 |
| 7m | −8.2 | −7.4 | −6.3 | −8.3 |
| 7n | −7.7 | −8.3 | −7.1 | −8.5 |
| Compound | Physicochemical Properties—Lipinski’s Rule of Five (Ro5) | ||||
|---|---|---|---|---|---|
| #H-Bond Acceptors | #H-Bond Donors | Log Po/w (MLOGP) | MW [g/mol] | #Violations | |
| 7a | 3 | 0 | 3.38 | 414.50 | 0 |
| 7b | 3 | 0 | 2.91 | 394.51 | 0 |
| 7c | 3 | 0 | 3.85 | 448.94 | 0 |
| 7l | 6 | 0 | 1.33 | 416.49 | 0 |
| 7m | 6 | 0 | 0.82 | 396.50 | 0 |
| 7n | 6 | 0 | 1.82 | 450.94 | 0 |
| Compound | Pharmacokinetics | |||
|---|---|---|---|---|
| GI Absorption | BBB Permeability | P-gp Substrate | Water Solubility | |
| 7a | High | Yes | Yes | Poorly soluble |
| 7b | High | Yes | No | Moderately soluble |
| 7c | High | Yes | No | Poorly soluble |
| 7l | High | No | No | Moderately soluble |
| 7m | High | No | No | Soluble |
| 7n | High | No | No | Moderately soluble |
| Compound | Drug-Likeness | |||
|---|---|---|---|---|
| Lipinski | Veber | Bioavailability Score | TPSA [Å2] | |
| 7a | Yes, 0 violation | Yes | 0.55 | 50.48 |
| 7b | Yes, 0 violation | Yes | 0.55 | 50.48 |
| 7c | Yes, 0 violation | Yes | 0.55 | 50.48 |
| 7l | Yes, 0 violation | Yes | 0.55 | 93.00 |
| 7m | Yes, 0 violation | Yes | 0.55 | 93.00 |
| 7n | Yes, 0 violation | Yes | 0.55 | 93.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczukowski, Ł.; Maniewska, J.; Wiatrak, B.; Jawień, P.; Krzyżak, E.; Kotynia, A.; Marciniak, A.; Janeczek, M.; Redzicka, A. Interactions of N-Mannich Bases of Pyrrolo[3,4-c]pyrrole with Artificial Models of Cell Membranes and Plasma Proteins, Evaluation of Anti-Inflammatory and Antioxidant Activity. Membranes 2023, 13, 349. https://doi.org/10.3390/membranes13030349
Szczukowski Ł, Maniewska J, Wiatrak B, Jawień P, Krzyżak E, Kotynia A, Marciniak A, Janeczek M, Redzicka A. Interactions of N-Mannich Bases of Pyrrolo[3,4-c]pyrrole with Artificial Models of Cell Membranes and Plasma Proteins, Evaluation of Anti-Inflammatory and Antioxidant Activity. Membranes. 2023; 13(3):349. https://doi.org/10.3390/membranes13030349
Chicago/Turabian StyleSzczukowski, Łukasz, Jadwiga Maniewska, Benita Wiatrak, Paulina Jawień, Edward Krzyżak, Aleksandra Kotynia, Aleksandra Marciniak, Maciej Janeczek, and Aleksandra Redzicka. 2023. "Interactions of N-Mannich Bases of Pyrrolo[3,4-c]pyrrole with Artificial Models of Cell Membranes and Plasma Proteins, Evaluation of Anti-Inflammatory and Antioxidant Activity" Membranes 13, no. 3: 349. https://doi.org/10.3390/membranes13030349
APA StyleSzczukowski, Ł., Maniewska, J., Wiatrak, B., Jawień, P., Krzyżak, E., Kotynia, A., Marciniak, A., Janeczek, M., & Redzicka, A. (2023). Interactions of N-Mannich Bases of Pyrrolo[3,4-c]pyrrole with Artificial Models of Cell Membranes and Plasma Proteins, Evaluation of Anti-Inflammatory and Antioxidant Activity. Membranes, 13(3), 349. https://doi.org/10.3390/membranes13030349