1. Introduction
Lipid nanoparticles (LNP) have emerged as ideal carriers for the delivery of genetic drugs based on nucleic acids directed at gene editing, silencing, or protein expression [
1,
2]. In addition, LNP have been used for the development of vaccines, which has resulted in the great success against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), where the viral spike protein mRNA was encapsulated into the particles [
3,
4,
5,
6]. The key advantage of the use of nucleic acid encapsulated LNP is that the DNA or RNA cargo is protected from the activity of circulating degrading enzymes present in body fluids. The main scaffold components of LNP are phospholipids that, due to their amphiphilic characteristics, assemble into lipid bilayers forming vesicles by a spontaneous process, entropically driven [
7]. This phospholipid membrane acts as a barrier between the vesicle lumen and the external environment protecting their internal cargo. In some instances, particularly for use as vaccines, phospholipid nanoparticles contain cholesterol to increase the stability of the vesicle and are decorated with polyethylene glycol for integrity preservation [
3].
Loading nucleic acids into LNP is not a trivial process. The addition of the potential cargo during the formation of the liposomes only resulted in equilibrium with the external substrate concentration, which is not very efficient for therapeutic goals. A great advance was the introduction of an ionizable phospholipid that is positively charged at low pHs and neutral at physiological pH conditions. Thus, the nucleic acids are loaded into liposomes at low pH, increasing the internal concentration of the substrate [
2,
8]. In spite of this advancement, the next step is how to direct LNP to specific target cells. In this regard, the incorporation of surface molecules on the vesicles has been shown to direct the particles to specific cell types, such as hepatocytes [
1]. However, a major challenge for the in vivo delivery of LNP is to avoid their capture by macrophages due to the higher capacity of these cells to ingest foreign particles. Another major question that remains to be answered concerns the mechanisms of LNP uptake by cells, the intracellular fate of the vesicles, and the transport of the nucleic acid cargo into the cytosol to carry out its biological activity.
As indicated above, the main components of LNP are phospholipids, particularly phosphatidylcholine, which has been coined the helper phospholipid to differentiate from other phospholipid components. In this regard, phospholipids have been considered as inert elements [
9]. However, it could be argued that these natural molecules could play an independent role in modulating cellular responses [
10]. Indeed, we have reported that liposomes made exclusively with palmitoyl oleoyl phosphatidylserine (POPS) induced robust activation of the innate immune response in peritoneal cells in vivo [
11]. In this report, we expand our prior observations by presenting data demonstrating the potential effect of phosphatidylcholine modulating the inflammatory response by macrophages, both in vivo and ex vivo models.
2. Materials and Methods
2.1. Animals
CD-1 (Crl:CD1 (ICR)) mice were acquired from Charles River and maintained under specific, pathogen–free conditions at the University of California, San Diego Animal Facility (La Jolla, CA, USA). Experiments were performed on 9-week-old male mice. All animal procedures were approved by the UCSD Institutional Animal Care and Use Committee, protocol number: S07177.
2.2. Cell Isolation and Culture
Peritoneal cells were collected by lavage. Briefly, 5 mL of serum-free phenol-red free RPMI1640 were injected into the peritoneal cavity of CD-1 mice, and after gentle massage of the peritoneum to dislodge any loosely attached cells, fluid was collected. Cell suspensions were immediately centrifuged for 10 min at 350× g and resuspended in PBS without Ca2+/Mg2+ supplemented with 0.5% BSA for counting.
J774A.1 macrophages. Cells were obtained from ATCC (TIB-67, ATCC, Manassas, VA, USA) and maintained in RPMI1640 with L-glutamine and penicillin/streptomycin and supplemented with 10% FBS.
Bone marrow cells were obtained from the femur and tibia of male CD-1 mice (9 weeks old). The bone marrow was flushed out with cold PBS supplemented with 2% heat-inactivated FBS using a 22-gauge needle. Single-cell suspensions were collected after passing through a 22-gauge needle 4–6 times to dissociate cell clumps. Cells were then passed through a 70-µm cell strainer to eliminate residual clumps and centrifuged at 500× g for 8 min at 4 °C. Cells were treated with red blood cell lysis buffer (eBioscience, San Diego, CA, USA), washed, and centrifuged as described above. Isolated bone marrow cells were resuspended (2 × 106 cells/mL) in RPMI1640 with L-glutamine and penicillin/streptomycin and supplemented with 10% FBS and 10 ng/mL recombinant M-CSF (R&D Systems, Minneapolis, MN, USA). The culture medium was changed after 3 days, and non-adherent cells were discarded. On day 7, the presence of mature bone marrow-derived Mϕ was evaluated by expression of CD11b and F4/80 using fluorophore-conjugated antibodies and flow cytometry. On average, more than 98% of the cells were CD11b+ and F4/80+, corresponding to Mϕ.
Resident naïve peritoneal macrophages were isolated by peritoneal cavity lavage as described above. Cell suspensions were immediately centrifuged for 10 min at 350× g, resuspended in RPMI1640 with L-glutamine and penicillin/streptomycin and supplemented with 10% FBS, and seeded on a 12-well plate at a concentration of 5 × 105 cells per well. Cells were then allowed to attach for 1 h at 37 °C in a CO2 incubator. Non-adherent cells were removed, and fresh medium was added. Cells were incubated for an additional 16 h, cellular viability was determined by the Trypan-blue exclusion test (90% viability was considered acceptable), and purity of the cell population was determined by flow cytometry using F4/80 antibodies. A 90–95% population of macrophages was considered acceptable.
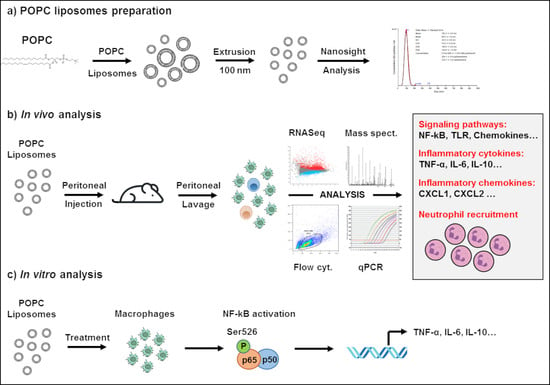
2.3. Liposome Preparation and Treatment
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) in chloroform (Avanti Polar Lipids, Alabaster, AL, USA) was dried under a nitrogen stream. Liposomes were formed by rehydrating the dried lipid film (400 μg) in 50 mM endotoxin-free Tris Buffer pH 7.4 (120 μL) and vortexing every 5 min for 30 min. In POPC liposome uptake experiments, liposomes were formed by rehydrating the dried lipid film (400 μg) in 50 mM endotoxin-free Tris Buffer pH 7.4 (120 µL) containing either 0.01 mg/mL AlexaFluor647-labeled dextran (10,000 MW; Invitrogen, Carlsbad, CA, USA) or 0.5 mg/mL AlexaFluor488-labeled dextran (10,000 MW; Invitrogen) and vortexing every 5 min for 30 min. The preparation was extruded by 15 passages through a 100 nm membrane filter using a mini-extruder apparatus (Avanti Polar Lipids). Liposomes were then centrifuged at 100,000× g for 60 min at 4 °C and resuspended in sterile PBS at a concentration of 1 mg/mL. The size distribution and concentration for each liposome preparation were verified by Nanoparticle Tracking Analysis (NTA, Nanosight NS300, Malvern Panalytical, Malvern, UK). The level of endotoxin contamination was determined using the ToxinSensor™ Chromogenic LAL Endotoxin Assay Kit (GenScript). The concentration of endotoxin found in POPC liposome preparation was generally less than 0.01 EU/mL, which corresponds to less than 0.002 ng/mL of endotoxin contamination.
2.4. RNA Extraction, cDNA Production and Quantitative Real-Time PCR (qPCR)
Levels of mRNA were measured by quantitative real-time PCR (qPCR). Peritoneal cells and bone marrow-derived macrophages were resuspended in TRIzol reagent following treatment and mixed thoroughly. RNA was purified according to the manufacturer’s protocol and treated with DNase I (DNA-free kit, Ambion, Austin, TX, USA) to remove any DNA contamination. DNA-free RNA was then reverse transcribed to cDNA using the High Capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Newly synthesized cDNA was further diluted and stored at −20 °C. The cDNA levels of genes were measured by quantitative real-time PCR (qPCR) using the QuantiTect SYBR Green PCR kit (Qiagen, Valencia, CA, USA) with QuantiTect validated primer sets (Tnfa: QT00104006; Il6: QT00098875; Il10: QT00106169; Cxcl1 (KC): QT00115647; Cxcl2 (MIP2-α): QT00113253, all from Qiagen). All PCR reactions were performed using the StepOnePlus Real-Time PCR System (Applied Biosystems). Melting curve analysis was performed for each primer set to ensure amplification specificity. Corresponding standard curves were added in each PCR reaction. The housekeeping gene GAPDH (QT01658692, Qiagen, Valencia, CA, USA) was used to normalize data to cDNA inputs. The results are expressed as copy numbers of the target gene per copy numbers of GAPDH.
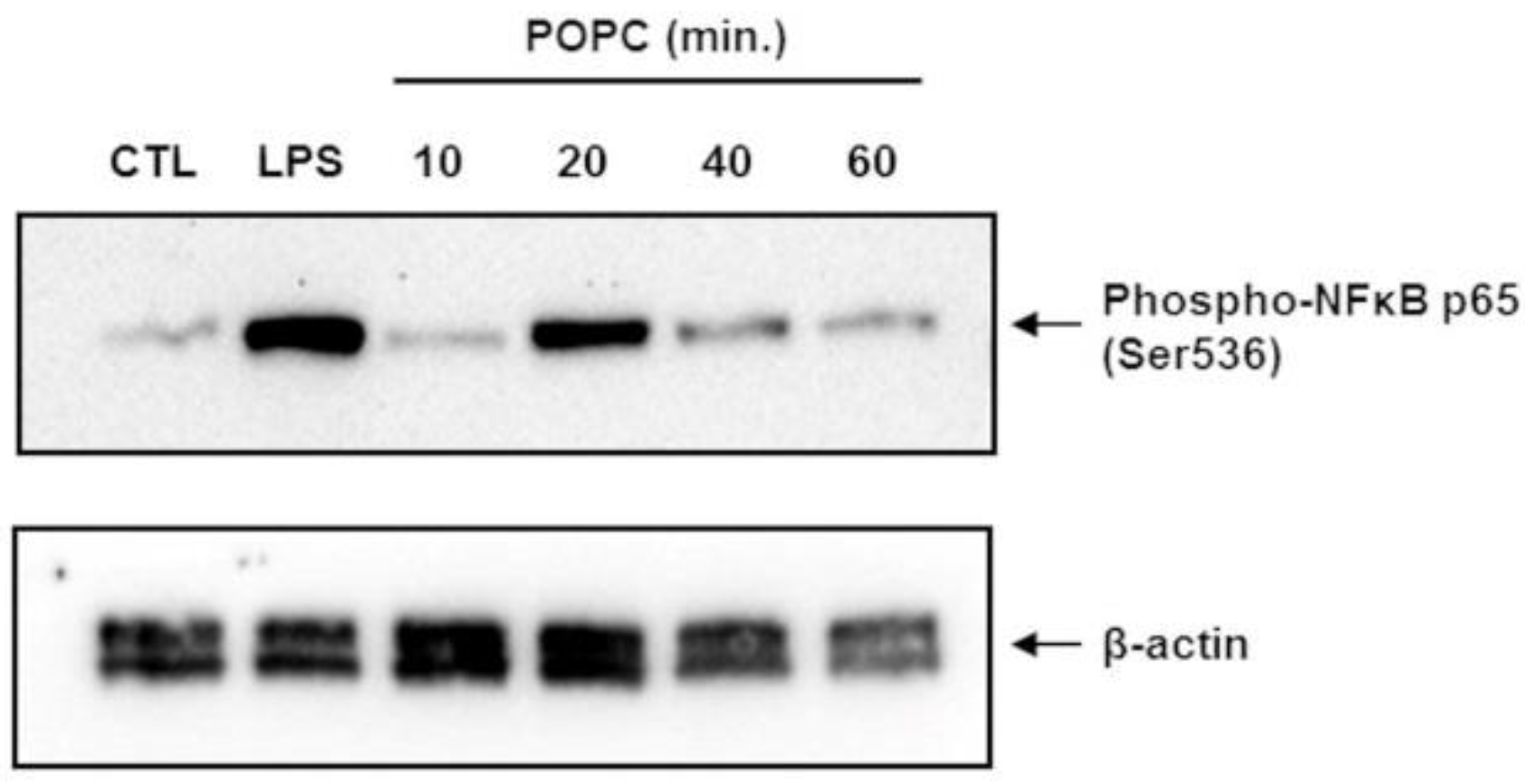
2.5. Immunoblot Analysis
J774 cells treated with PBS (vehicle control), LPS (100 ng/mL, 10 min.), or POPC (10, 20, 40, and 60 min.) were lysed in the RIPA lysis buffer containing 0.01M sodium fluoride as serine/threonine phosphatases inhibitor, vortexed for 30 s and cell homogenates were incubated for 30 min at 4 °C. Samples were then centrifuged at 10,000× g for 8 min at 4 °C. The supernatant was collected, and a 10 µL aliquot was used to determine protein concentration using a BCA protein assay (Pierce Biotechnology, Rockford, IL, USA). Cell homogenates were mixed with NuPAGE LDS sample buffer (Life Technologies, Carlsbad, CA, USA), and 60 µg of total protein was resolved by SDS-PAGE using NuPAGE 4–12% Bis-Tris gels (Life Technologies, Carlsbad, CA). Proteins were then transferred to nitrocellulose membranes and blocked with 5% BSA diluted in Tris-buffered saline (TBS) for 1 h at 23 °C. Blots were probed with a rabbit anti-phospho-NF-kB p65 mAb (1/2000, clone 93H1; Cell Signaling Technology, Danvers, MA, USA) in 5% BSA-TBS and incubated overnight at 4 °C followed by three 15 min washes with TBS supplemented with 0.1% Tween 20 (TBST) at 23 °C. Blots were then incubated with HRP-conjugated goat anti-rabbit IgG antibodies (1:3000—Santa Cruz Biotechnology, Dallas, TX, USA) in 5% BSA-TBS for 1 h at 23 °C. After three 15 min washes in TBST, bands were detected by chemiluminescence using SuperSignal reagents (Pierce Biotechnology). As a loading control, blots were probed with mouse anti- β-actin monoclonal antibodies (1:3000—Thermo Scientific, Waltham, MA, USA) in 5% BSA-TBS for 16 h at 4 °C. Goat anti-mouse HRP-conjugated IgG secondary antibody (1:3000—Thermo Scientific) was used for 1 h at 23 °C, followed by chemiluminescence detection. Chemiluminescence data were acquired using the ChemiDoc™ MP system (Bio-Rad, Hercules, CA, USA) and densitometry analyses were performed using the Image Lab™ software (Bio-Rad).
2.6. Global Gene Analysis by RNA Sequencing
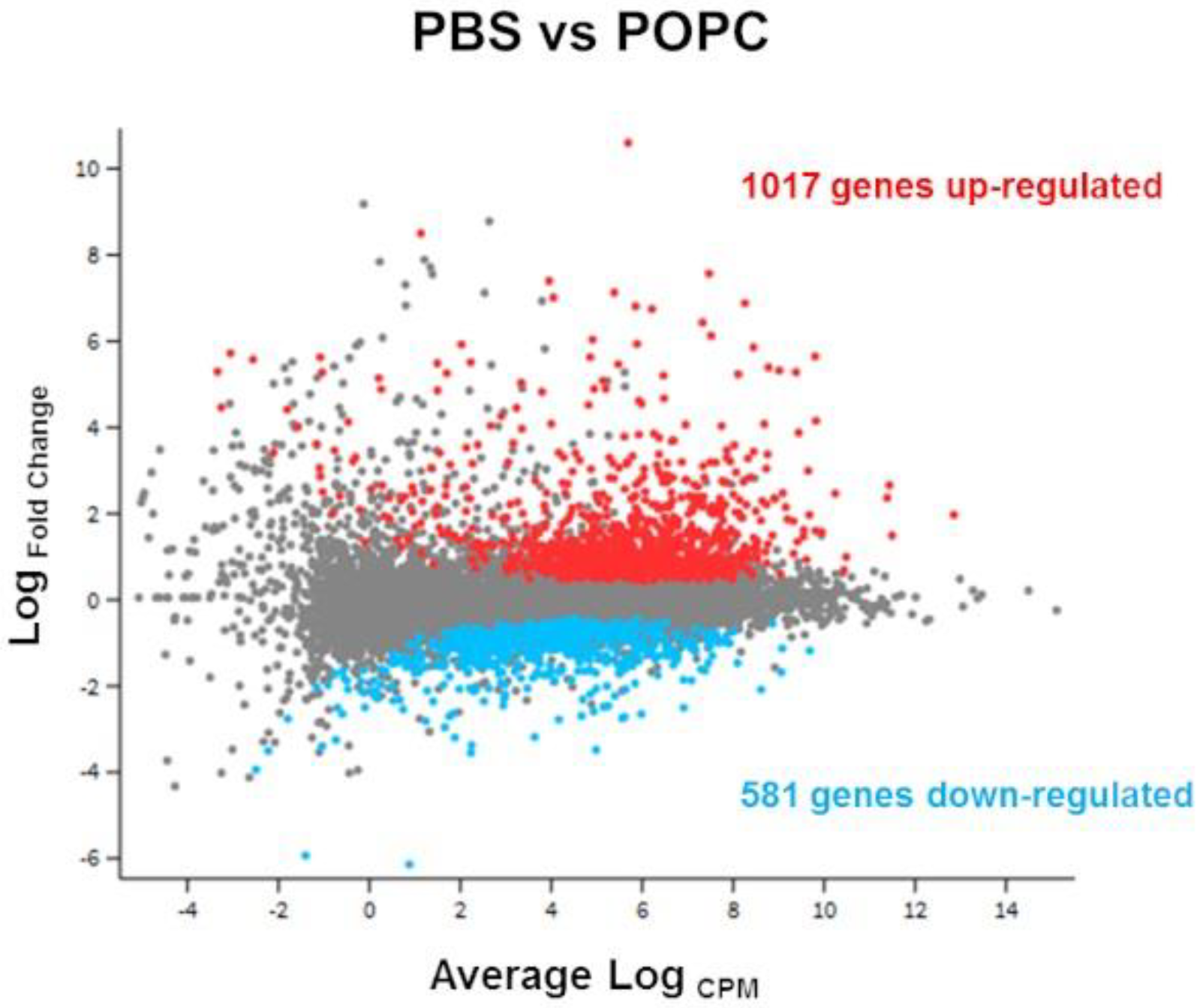
CD-1 mice were injected i.p. with 400 µg of POPC liposomes, and total peritoneal cells were then collected 1 h later by peritoneal lavage and processed for global gene analysis by RNA sequencing. Control animals received an equal volume of PBS. RNA was isolated using the RNeasy Mini Kit according to the manufacturer’s protocol (Qiagen, Valencia, CA, USA), and RNA integrity numbers (RIN) were determined by the Agilent 4200 Tapestation. RNA sequencing libraries were then generated from 1 µg of RNA using the TruSeq® Stranded Total RNA Library Prep Gold Sample Prep Kit following the manufacturer’s instructions (Illumina, San Diego, CA, USA), modifying the shear time to 5 min. RNA libraries were multiplexed and sequenced with 75 base pair (bp) single reads (SR75) to a depth of approximately 40 million reads per sample on an Illumina HiSeq4000 (Illumina, San Diego, CA, USA). Quality control of the raw fastq files was performed using the FastQC software tool. Sequencing reads were aligned to the mouse genome (mm10) using the STAR v2.5.1a aligner. Read quantification was performed with RSEM v1.3.0 and GENCODE annotation (Mus_musculus.GRCm38.68.gtf). The R BioConductor packages edgeR and limma were used to implement the Limma-voom method for differential expression analysis. The experimental design was modeled upon treatment (~0 +treatment). Significance was defined by using an adjusted p-value cut-off of 0.05 after multiple testing corrections using a moderated t-statistic in Limma. Functional enrichment of the differentially expressed genes was performed using the Bioconductor GSVA package for implementing GSEA. The Signaling Pathway Impact Analysis (SPIA) and overrepresentation analysis were performed by using the Bioconductor package and the WebGestalt toolkit, respectively. The data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession number GSE115489.
2.7. Flow Cytometry Analysis of Peritoneal Cells
Peritoneal cells were collected by lavage of the peritoneum as described above. Cells were centrifuged for 10 min at 300× g, resuspended in PBS without Ca2+/Mg2+ supplemented with 0.5% BSA (FACS staining buffer, FSB), and counted. Peritoneal cells (5 × 105 cells/tube) were then incubated for 15 min with 0.5 µg of FcγR blocking antibodies (Fc block; BD Biosciences, San Jose, CA, USA), followed by antibody staining for 30 min in the dark at 4 °C. Cells were then washed, centrifuged, and resuspended in FSB for analysis. Each anti-mouse antibody was added at 0.5 µg/tube and included FITC-conjugated anti-Ly6G (clone 1A8, Biolegend, San Diego, CA, USA), PE-conjugated anti-CD11b (clone M1/70, eBioscience), PerCP-conjugated anti-CD19 (clone 1D3, BD Bioscience), APC-conjugated anti-F4/80 (clone BM8, eBioscience). Propidium iodide was also used to assess cell viability. Flow cytometry was performed using a FACSCanto II flow cytometer with FACSDiva software (BD Biosciences, San Jose, CA, USA). The data were analyzed using FlowJo software v.10.1 (Tree Star, Ashland, OR, USA).
2.8. POPC Liposome Uptake
For flow cytometry analysis, J774A.1 macrophages were incubated for different periods of time with POPC liposomes loaded with AlexaFuor647-labeled dextran. Cells were then collected by scraping and processed for flow cytometry analysis. For microscopy analysis, J774A.1 macrophages were plated onto Nunc Lab-Tek II Chamber slides (ThermoFisher Scientific, Waltham, MA, USA) and incubated for different periods of time with POPC liposomes loaded with AlexaFuor488-labeled dextran. Cells were then washed and fixed with 4% paraformaldehyde and the slides were mounted with Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA). Fluorescence was acquired using an inverted fluorescence microscope (100× oil immersion objective, Eclipse TE300, Nikon Instruments, Melville, NY, USA) equipped with an AxioCam HRm (Zeiss, Pleasanton, CA, USA). Image analysis was performed using the Zen microscope software (Zeiss).
2.9. Mass Spectrometry Analysis
CD-1 mice were injected i.p. with 400 µg of POPC liposomes and total peritoneal cells were collected 1 h later by peritoneal lavage, centrifuged, and cell pellets were processed for mass spectrometry analysis by the Biomolecular and Proteomics Mass Spectrometry Facility (University of California, San Diego). Control animals received an equal volume of PBS. Briefly, samples were digested with trypsin and analyzed by HPLC coupled with tandem mass spectrometry (LC-MS/MS) using nano-spray ionization (TripleTOF 5600 hybrid mass spectrometer (AB SCIEX)). MS/MS spectra acquisition, peptide/protein identification, quantification of the data, and statistical calculations were performed using the peptide-feature-based PEAKS Studio X (Bioinformatics Solutions Inc., Canada) [
12].
2.10. Statistical Analysis
All data were analyzed using GraphPad Prism software (GraphPad Prism Software, San Diego, CA, USA). Significance was analyzed using one-way ANOVA followed by Tukey’s Multiple Comparison Test, two-way ANOVA followed by Bonferroni post-tests, or unpaired Student’s t-test. A p < 0.05 value was considered statistically significant.
4. Discussion
The lipid composition of eukaryote cellular membranes is complex, in which glycerophospholipids are the major component, with the additional presence of sphingolipids and sterols [
13]. Indeed, the lipid organization of all subcellular membranes, including the plasmalemma, is different and specific for each subcellular compartment [
14]. Moreover, phospholipid membranes are key components of the export of vesicles into the extracellular environment, named exosomes or extracellular vesicles, which play a role in intracellular communication [
15,
16]. Finally, phospholipids are fundamental forming blocks for the assembly of LNP, which has displayed great utility for the delivery of nucleic acids to cells directed at changing gene expression as well as for the development of mRNA vaccines [
1,
2,
3,
4,
5,
6]. The great advantage of LNP is that nucleic acids can be encapsulated at high concentrations into the vesicle lumen and protected from the activity of circulating lytic agents.
Particles containing phospholipid membranes, including LNP, exosomes and apoptotic bodies, are primarily engulfed by phagocytic cells. In this regard, very little attention has been devoted to the possible contribution of phospholipids within these particles to modify cellular functions. The earlier assumption was that phospholipids were inert components [
9]. However, other studies have shown that phospholipids could be involved in cellular responses [
10]. Certainly, we showed that liposomes made of palmitoyl oleoyl phosphatidylserine (POPS) altered gene expression in macrophages in vivo. Thus, peritoneal injection of POPS liposomes increased the peritoneal expression of cytokines and chemokines that attract neutrophils into the cavity and neutralize bacterial infections [
11]. These observations open the possibility that phospholipids are not just bystanders but rather active players in modulating cellular functions.
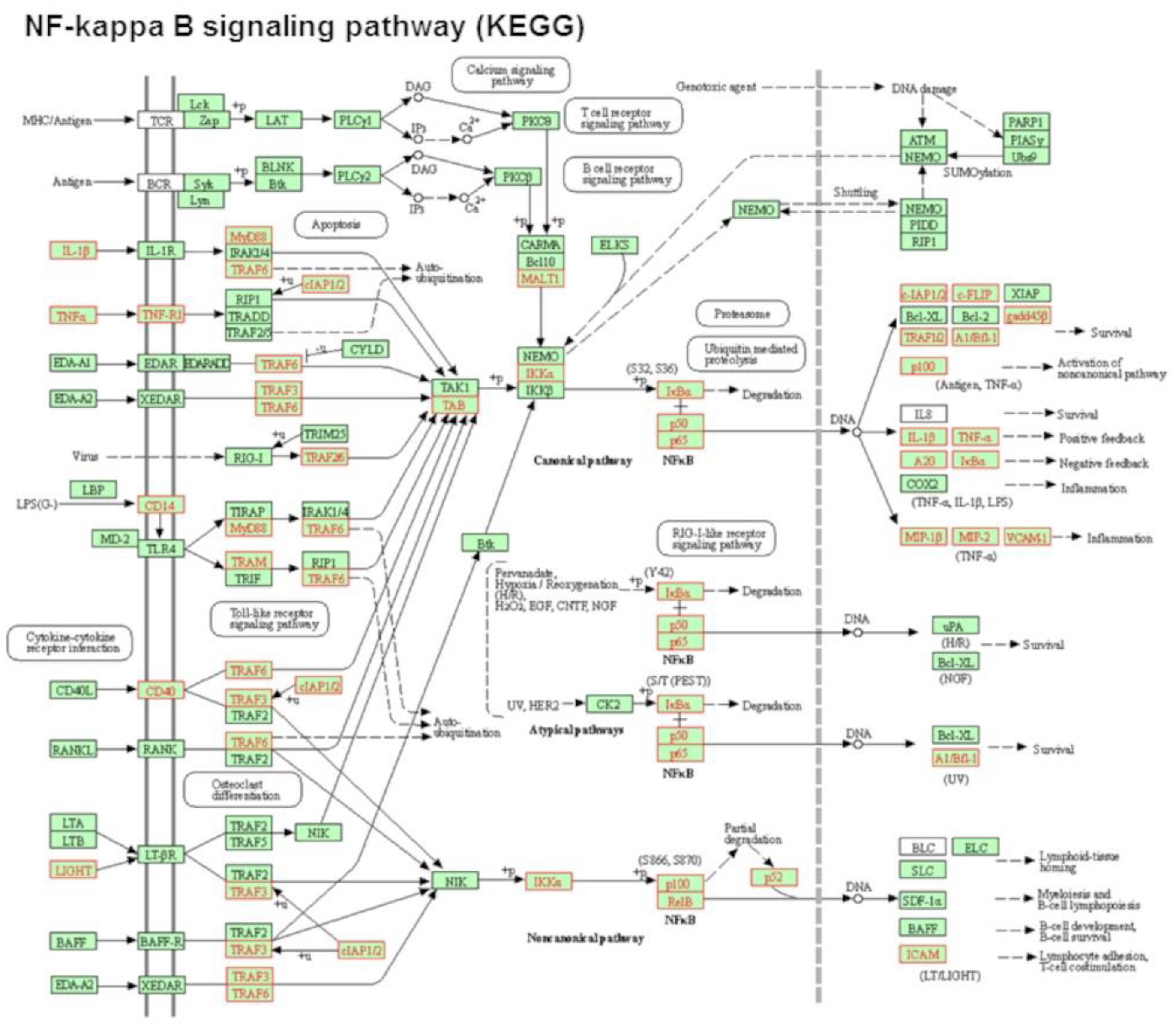
In the present study, we have expanded our prior investigations on the effect of phospholipids on macrophage function by focusing on palmitoyl oleoyl phosphatidylcholine (POPC) liposomes. Phosphatidylcholine is the most abundant phospholipid in cellular membranes and exosomes. We found that this glycerophospholipid increased the expression of a variety of genes in vivo, particularly those involved in the NF-κB activation pathway, resulting in the expression of cytokines (TNF-α, IL-6 and IL-10) and chemokines (Cxcl1 and Cxcl2). These observations were validated in primary and cultured macrophage lines in ex vivo conditions. We are highly confident that the increase in expression of these inflammatory genes was not due to the endotoxin contamination of POPC liposome preparations since any potential contamination was tested using a very sensitive assay system. In addition, the biological activity of LPS within liposomes is highly reduced due to the entrapment of lipid A, which is the active component of endotoxin, within the lipid bilayer [
17]. Since phosphatidylcholine is a scaffold component of the cassette encapsulating the mRNA encoding for the spike protein of the SARS-CoV-2 virus, it is possible that this early activation of innate immune response by phosphatidylcholine may contribute to the success of the COVID-19 vaccine, perhaps acting as an adjuvant.
Exposure of macrophages to POPC liposomes resulted in the specific up-regulation of 146 genes, including
Mfsd2a,
Tril,
Tnfsf4 (
Table S1), and the down-regulation of 69 genes (e.g.,
Epha2,
Tmem86a,
Cln8;
Table S2) in comparison with POPS liposomes. Since POPC and POPS contain the same acyl groups, the differences in gene expression modulation by these two phospholipids is likely due to a direct effect of the phospholipid head.
The potential role of phospholipids in modulating cellular physiology is complex due to the independent potential activity of the phospholipid head, acyl groups, or a breakdown of products. We observed that palmitoyl oleoyl phosphatidylglycerol (POPG) or palmitoyl oleoyl phosphatidylethanolamine (POPE) liposomes were harboring the same acyl group as POPC and did not trigger macrophage activation indicated by the absence of TNF-α following stimulation (
Figure S1), thus reaffirming the importance of the phospholipid head in the expression of inflammatory genes by PC. In addition, the observation that neither POPG nor POPE, which were prepared under the same conditions as POPC, induced TNF-α expression further discards the possibility that our findings are due to endotoxin contamination. A potential phosphatidylcholine degradation component is lyso-phosphatidylcholine (LysoPC), produced by the activity of phospholipase A1, which could directly activate a cellular signaling pathway [
18]. Prior studies have shown that the exogenous addition of LysoPC to immune cells resulted in the expression of inflammatory molecules such as IFN-γ and TNF-α [
19,
20]. The transcriptome analysis of macrophages exposed to POPC liposomes revealed that the increased expression of the gene encoding for the major facilitator superfamily domain containing 2A (
Mfsd2a), which is a sodium-dependent LysoPC transporter mainly present on the plasma membrane, but it could also be localized in intracellular membranes such as ER [
21]. Mfsd2a transports LysoPC carrying long-chain fatty acids with more than 14-carbon acyl groups. Moreover, the zwitterionic charge of the PC head is critical for Mfsd2a transport [
22]. Mfsd2a is upregulated during the activation of CD8
+ T cells as well as other inflammatory conditions [
23,
24]. Particularly, its expression could be altered by TNF-α [
25]. It is possible that the uptake of POPC liposomes by macrophages increases the expression of Mfsd2a to activate the transport of long-chain fatty acids produced after the degradation of the liposomes from internal vesicular compartments into the cytosol.
Since POPC liposomes are likely to be targeted to the lysosome, a large excess of these vesicles may overload this compartment, triggering lysosomal stress. Lysosomal stress will activate organelle-specific responses, such as biogenesis, exocytosis and autophagy [
26,
27,
28]. Although none of these events were identified within the signal pathways that were modulated by exposure to POPC liposomes, we cannot discard the possibility that an excess of POPC liposomes may result in lysosomal stress. Additionally, phosphatidylcholine could be transferred to other subcellular compartments changing the lipid environment and modulating the activity of resident proteins.
Another gene that was upregulated by incubation with POPC liposomes was
Tril, which encodes the TLR4 interactor with leucine-rich repeats (Tril). Tril is a modulator of TLR4 signaling, enhancing the activation of the inflammatory cascade [
29]. In addition, Tril was also found to modulate the activity of TLR3, which is localized in endosomes [
30]. Since POPC liposomes are likely captured by pinocytosis initially accumulating into endosomes, it is possible that the increased expression of Tril may be related to the activation of the innate immune response within this internal compartment.
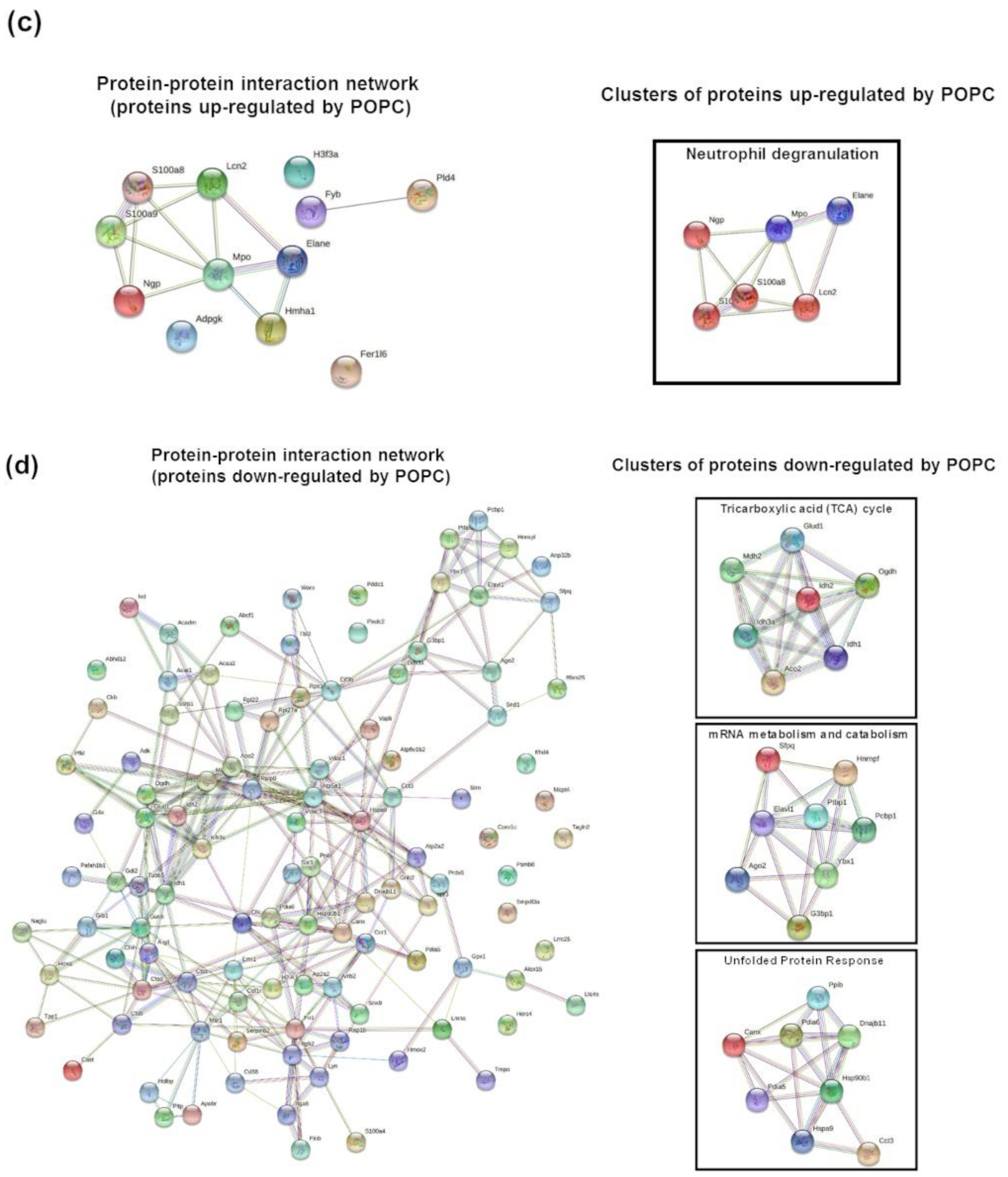
Finally, a parallel proteomic analysis of the same samples used for the transcriptome analysis showed the alteration of a distinct group of proteins without overlapping with the transcriptome profile. This proteomic analysis revealed the presence of proteins of neutrophil granules within the preparation suggesting the presence of these cells in the peritoneum upon injection of POPC liposomes. This possibility was indeed confirmed by flow cytometry analysis of peritoneal cells after exposure to the liposomes. The presence of neutrophils was small (about 12% of total cells) and transient. The differences in the profile between transcriptomic and proteomic analysis were not surprising since cell functions are regulated at both the transcriptional and translational levels. Thus, the dual transcriptome and proteomic analysis could provide a global view of the response to external agents.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}