
Peripheral Groups of Dicationic Pyrazinoporphyrins Regulate Lipid Membrane Binding

 , ,
, ,  and
and 
Abstract
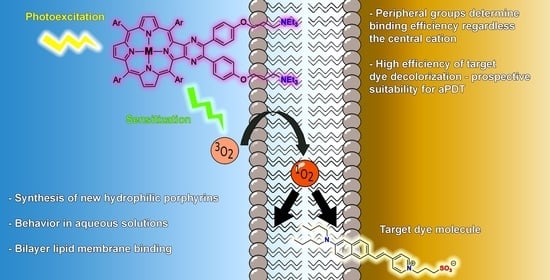
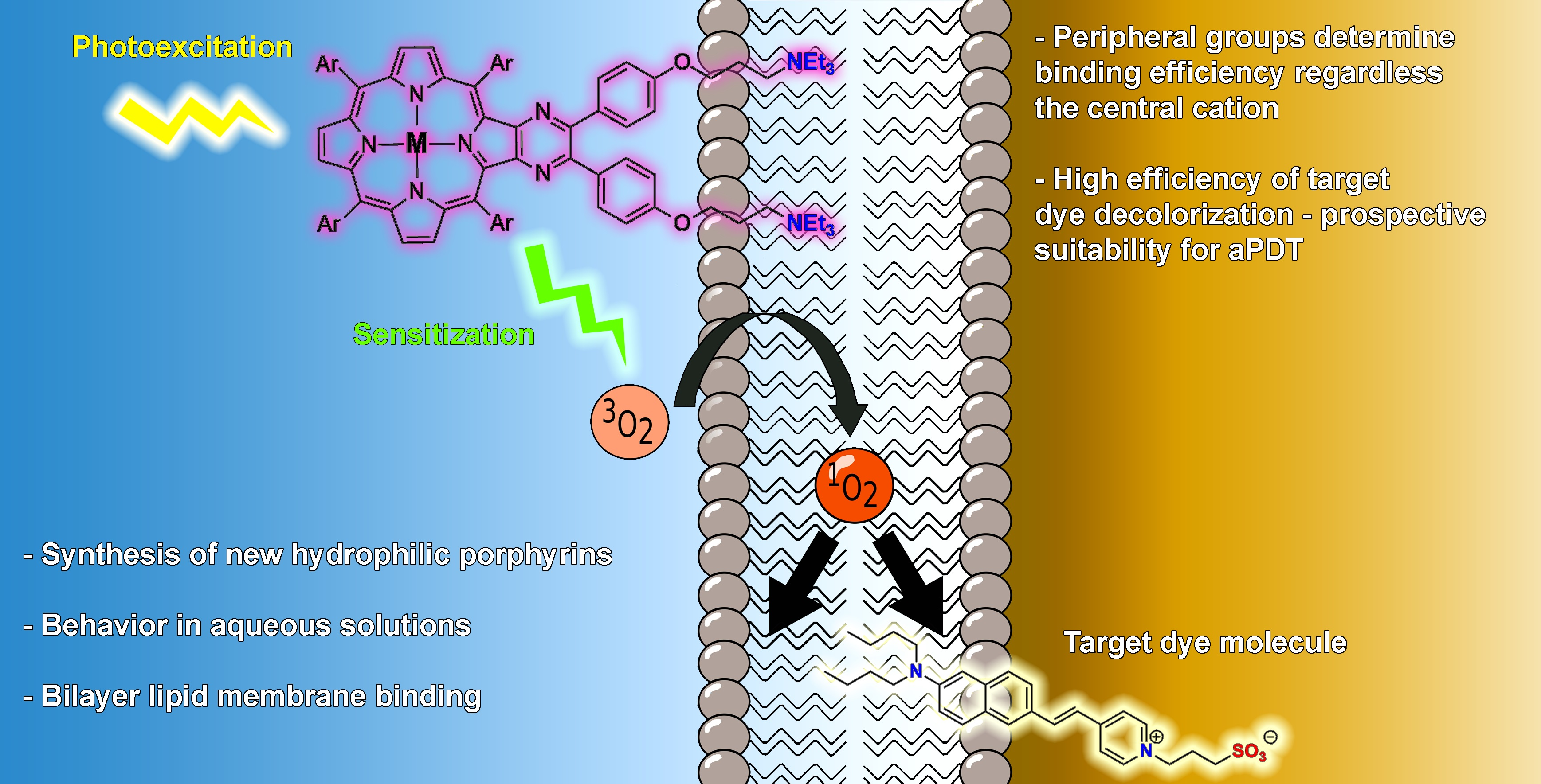
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results and Discussion
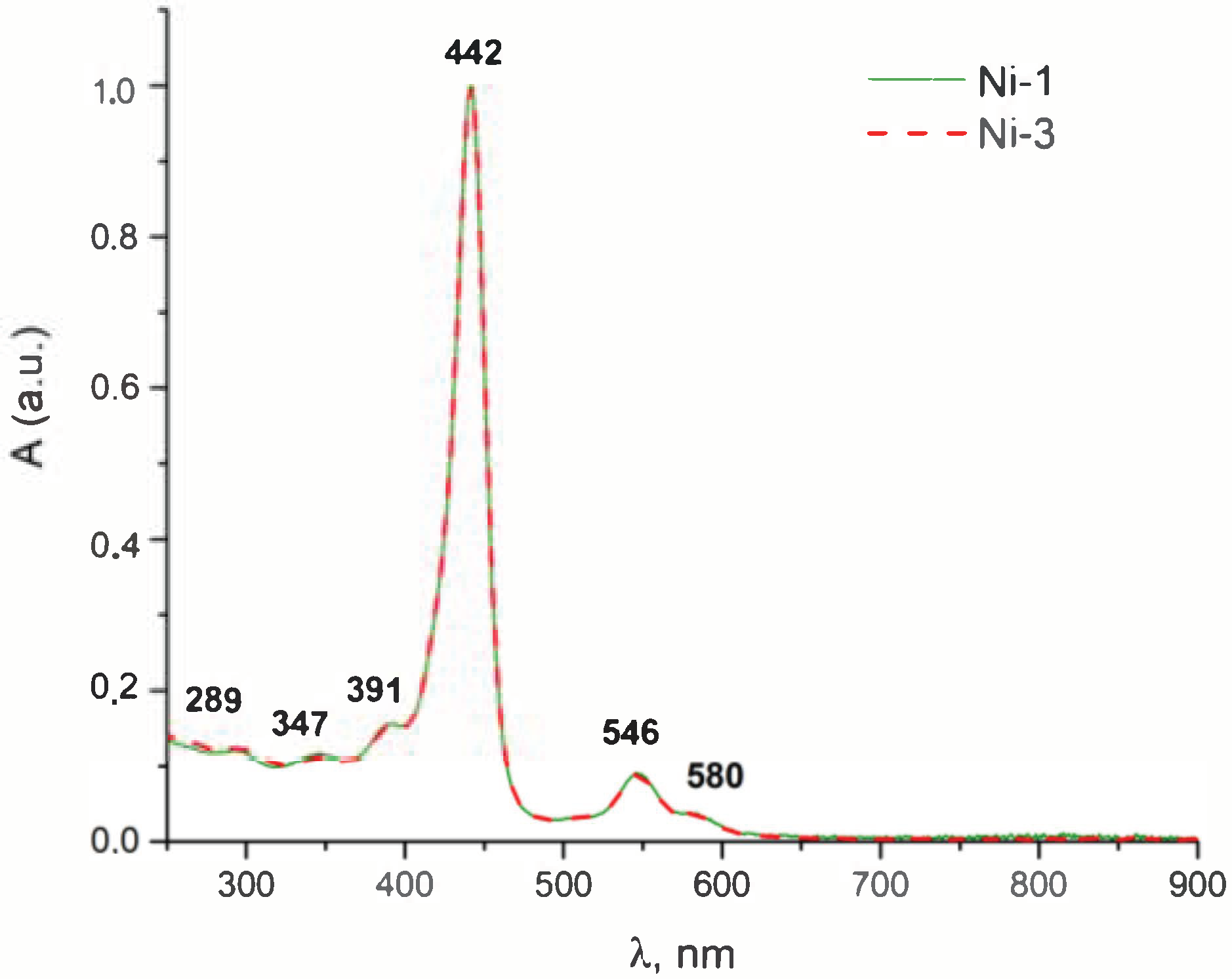
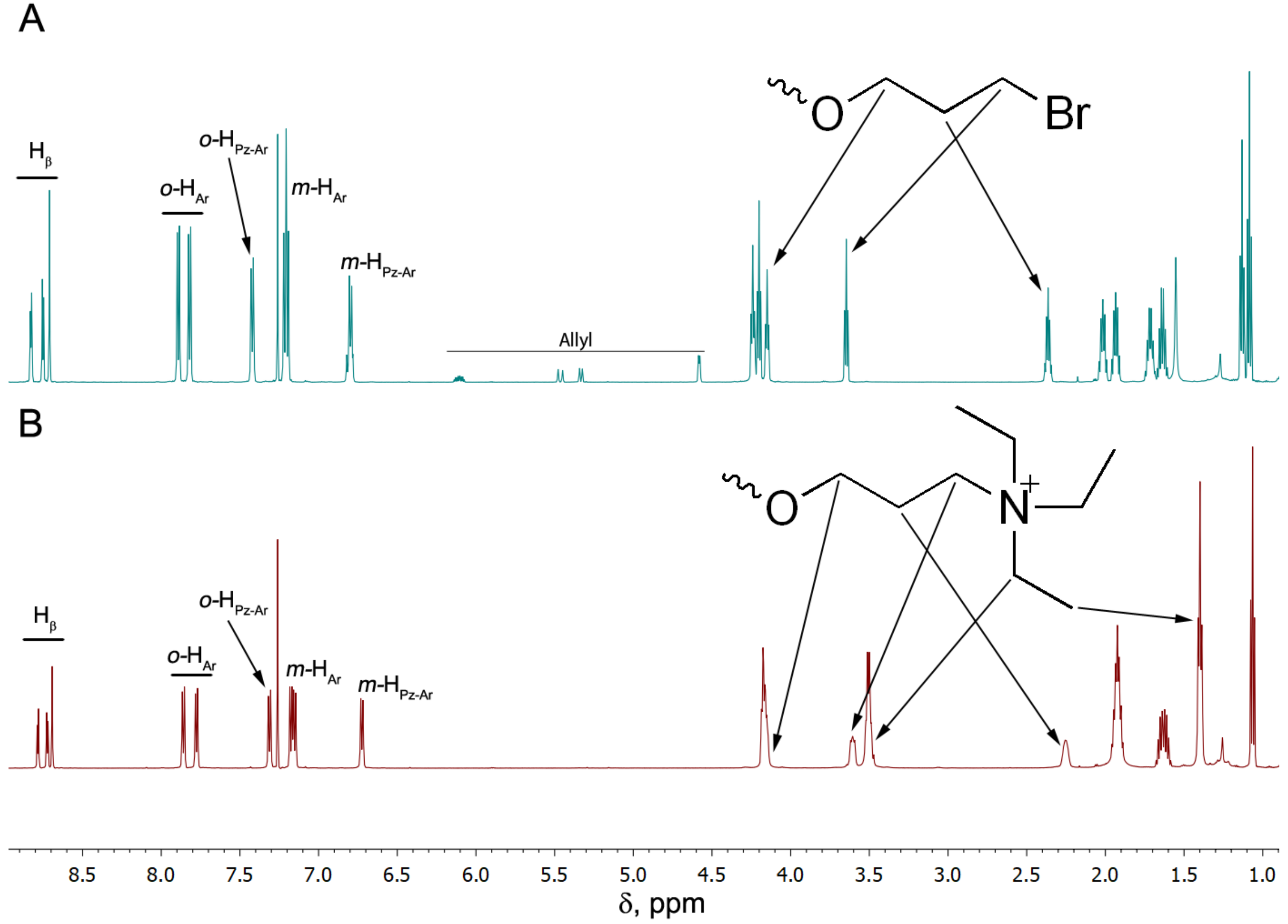
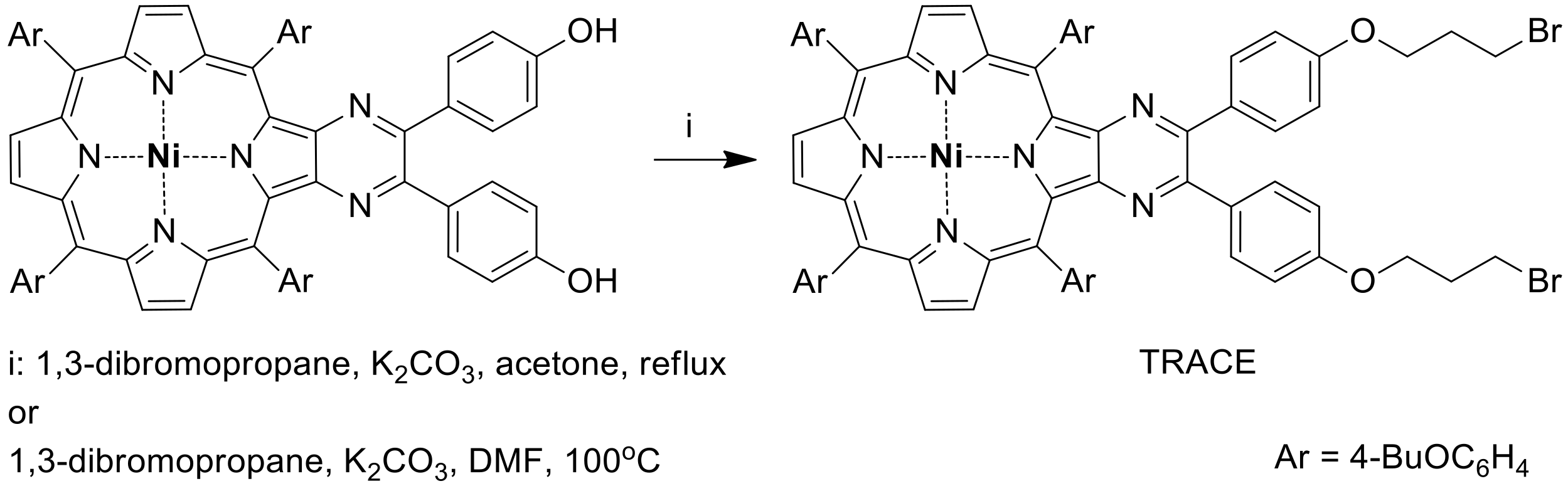
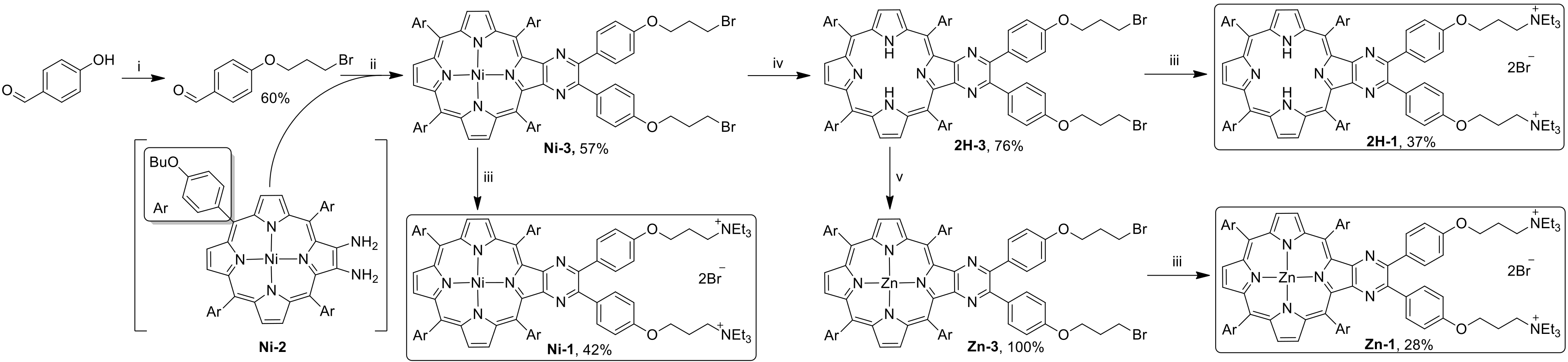
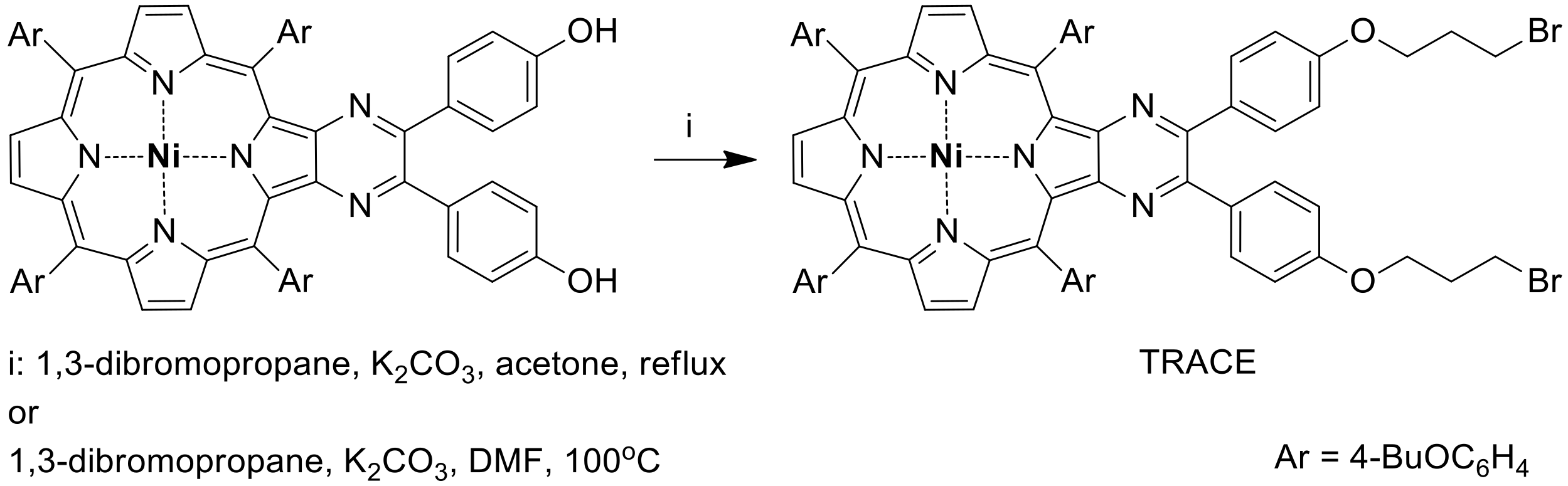
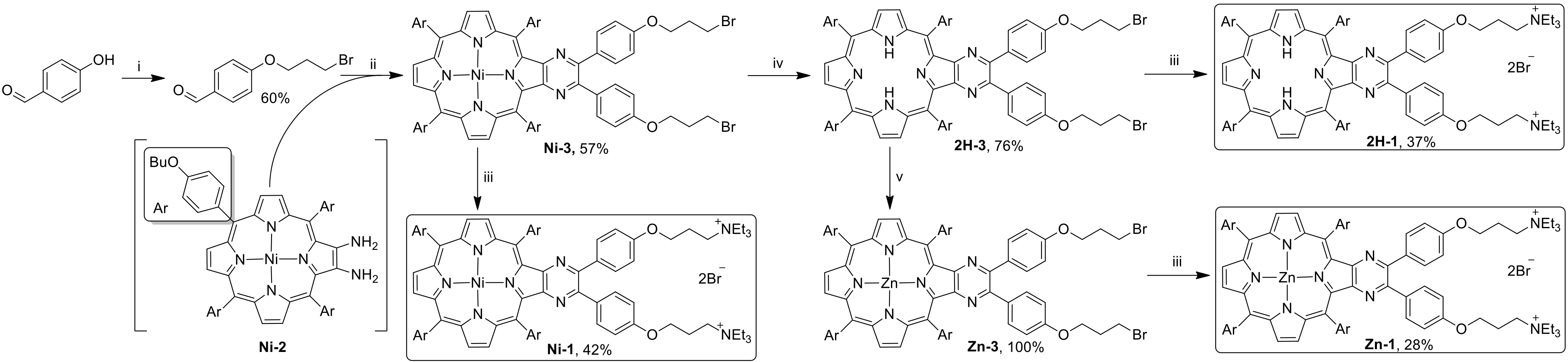
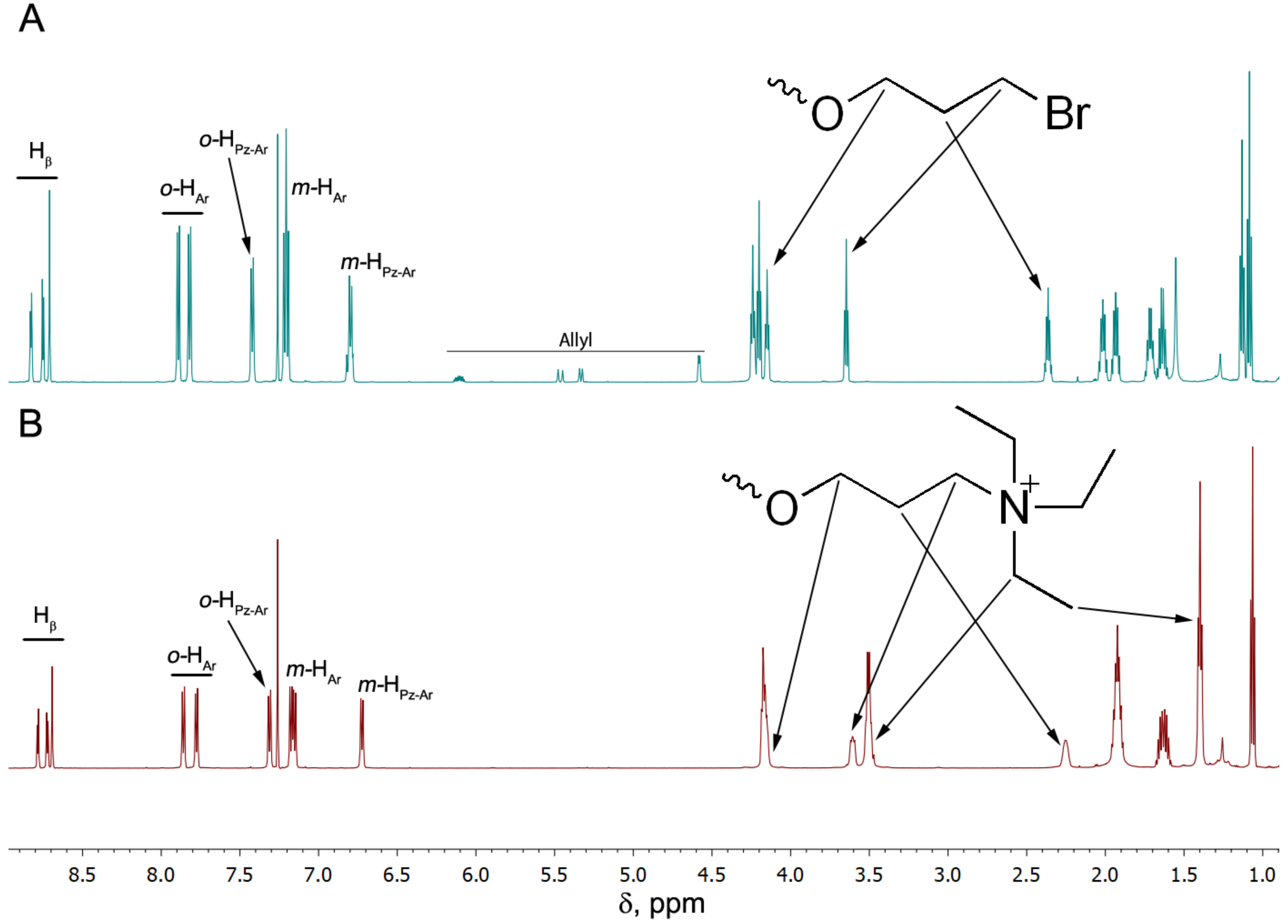
3.1. Synthesis
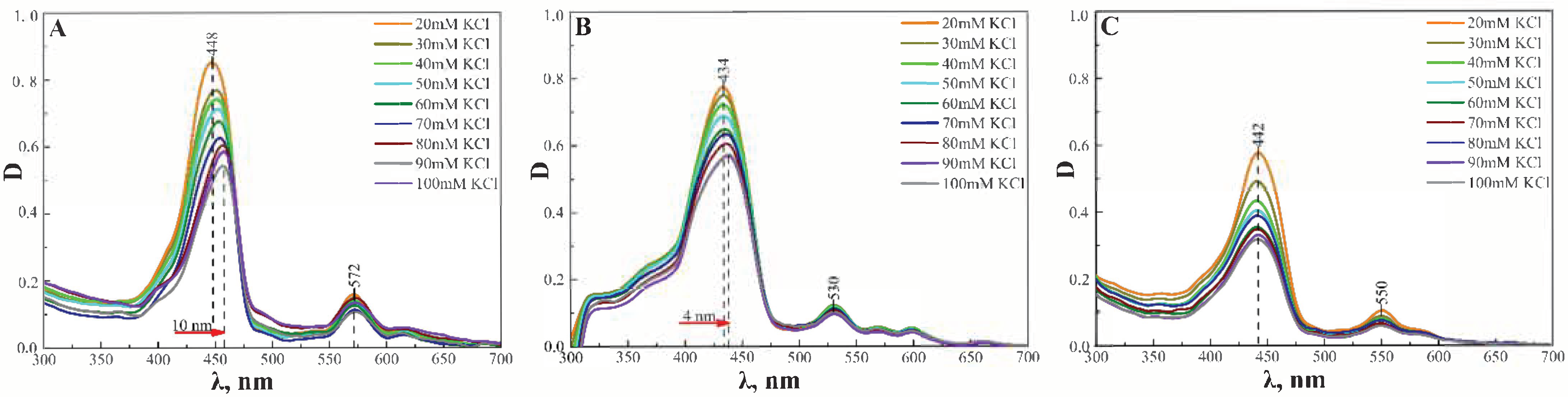
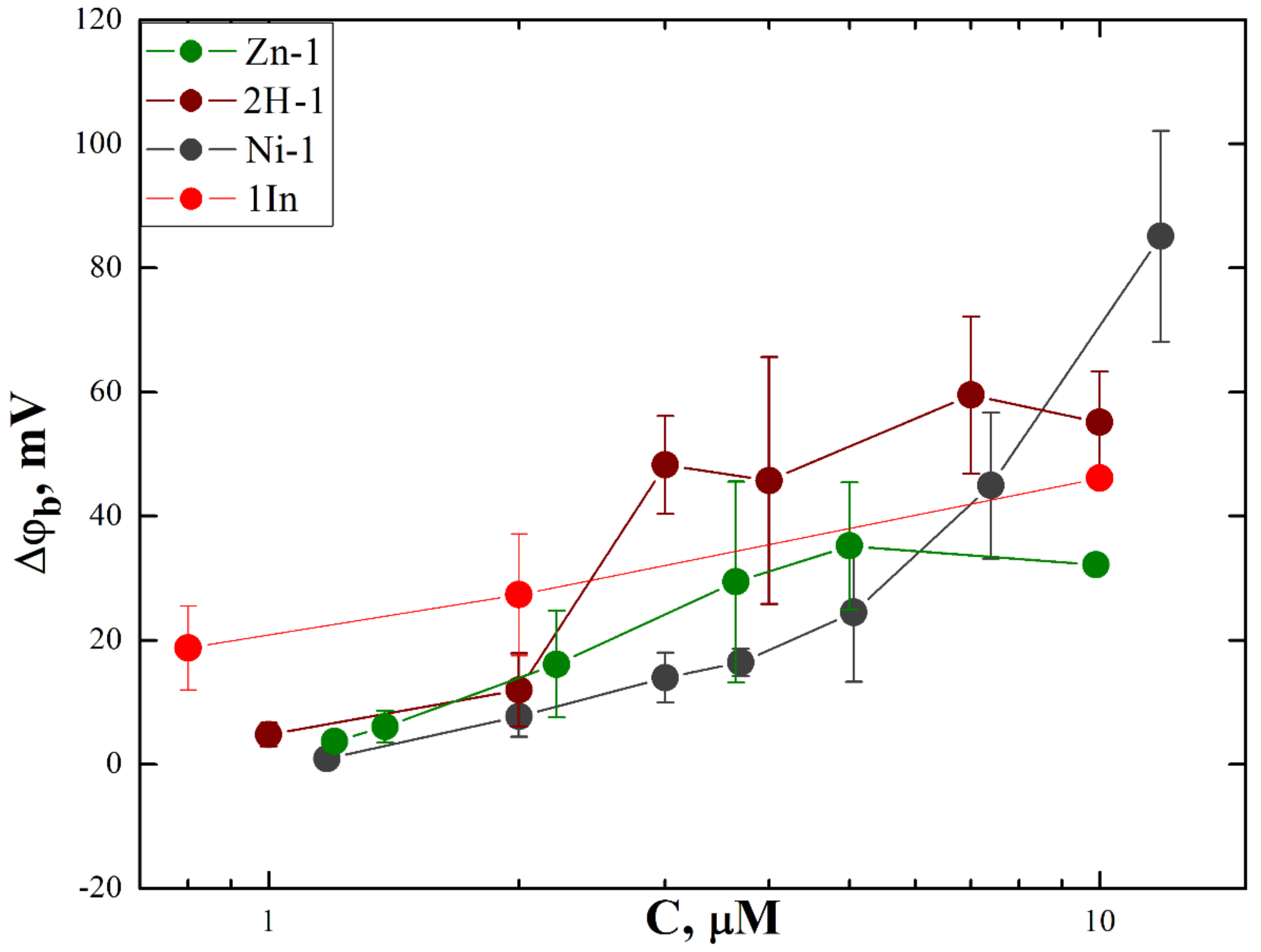
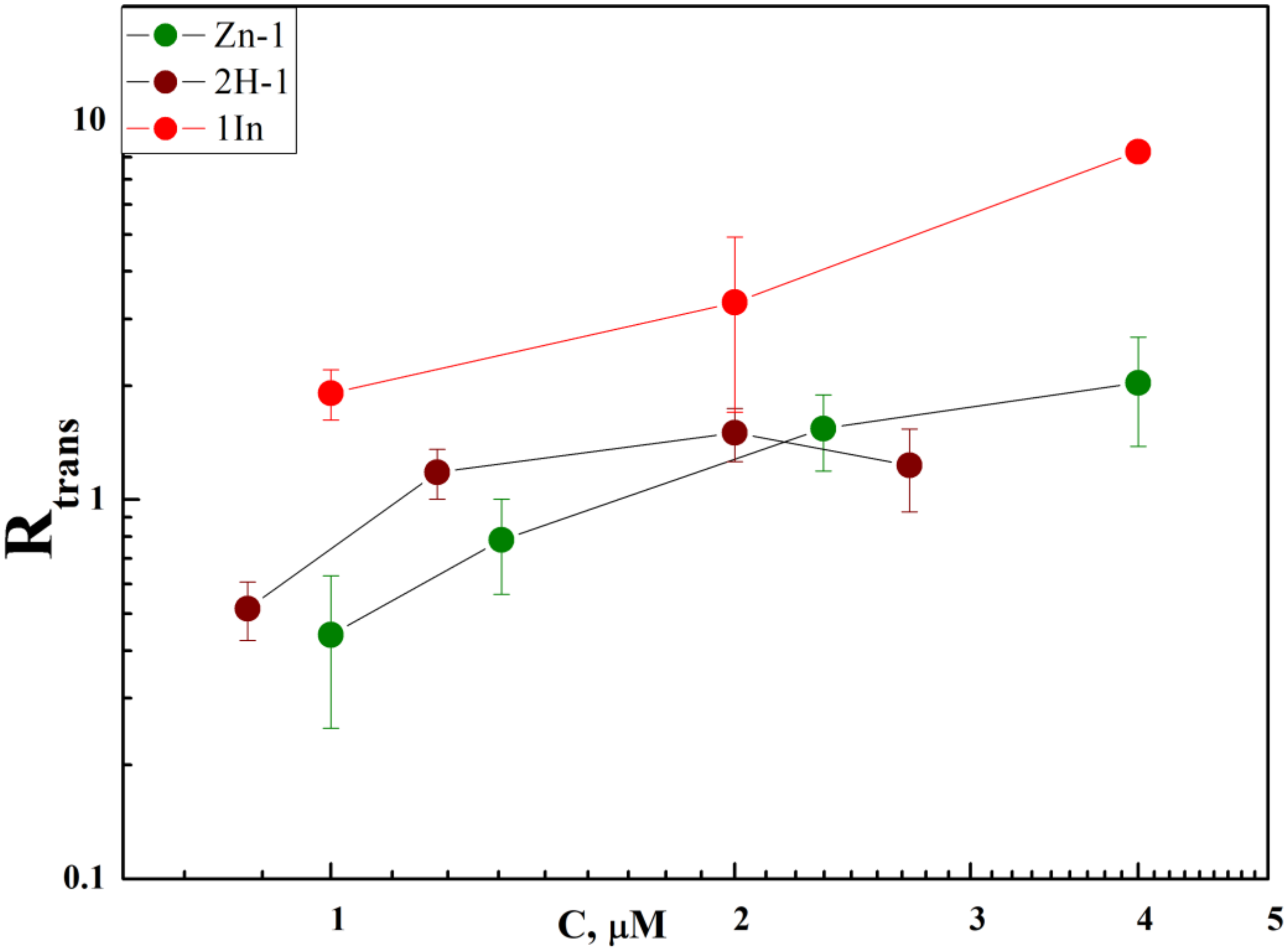
3.2. Membrane Binding and Photodynamic Efficiency
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef] [PubMed]
- Nyokong, T.; Ahsen, V. Photosensitizers in Medicine, Environment, and Security; Nyokong, T., Ahsen, V., Eds.; Springer: Dordrecht, The Netherlands, 2012; ISBN 978-90-481-3870-8. [Google Scholar]
- Jiang, L.; Gan, C.R.R.; Gao, J.; Loh, X.J. A Perspective on the Trends and Challenges Facing Porphyrin-Based Anti-Microbial Materials. Small 2016, 12, 3609–3644. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowski, J.M.; Pucelik, B.; Regiel-Futyra, A.; Brindell, M.; Mazuryk, O.; Kyzioł, A.; Stochel, G.; Macyk, W.; Arnaut, L.G. Engineering of Relevant Photodynamic Processes through Structural Modifications of Metallotetrapyrrolic Photosensitizers. Coord. Chem. Rev. 2016, 325, 67–101. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, R.; Zaat, S.A.J.; Breukink, E.; Heger, M. Antibacterial Photodynamic Therapy: Overview of a Promising Approach to Fight Antibiotic-Resistant Bacterial Infections. J. Clin. Transl. Res. 2015, 1, 140. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Faustino, M.A.F.; Tomé, J.P.C. Photodynamic Inactivation of Bacteria: Finding the Effective Targets. Future Med. Chem. 2015, 7, 1221–1224. [Google Scholar] [CrossRef]
- Miretti, M.; Clementi, R.; Tempesti, T.C.; Baumgartner, M.T. Photodynamic Inactivation of Multiresistant Bacteria (KPC) Using Zinc(II)Phthalocyanines. Bioorganic Med. Chem. Lett. 2017, 27, 4341–4344. [Google Scholar] [CrossRef]
- Jiménez-Munguía, I.; Fedorov, A.K.; Abdulaeva, I.A.; Birin, K.P.; Ermakov, Y.A.; Batishchev, O.V.; Gorbunova, Y.G.; Sokolov, V.S. Lipid Membrane Adsorption Determines Photodynamic Efficiency of β-Imidazolyl-Substituted Porphyrins. Biomolecules 2019, 9, 853. [Google Scholar] [CrossRef]
- Sokolov, V.S.; Batishchev, O.V.; Akimov, S.A.; Galimzyanov, T.R.; Konstantinova, A.N.; Malingriaux, E.; Gorbunova, Y.G.; Knyazev, D.G.; Pohl, P. Residence Time of Singlet Oxygen in Membranes. Sci. Rep. 2018, 8, 14000. [Google Scholar] [CrossRef]
- Konstantinova, A.N.; Sokolov, V.S.; Jiménez-Munguía, I.; Finogenova, O.A.; Ermakov, Y.A.; Gorbunova, Y.G. Adsorption and Photodynamic Efficiency of Meso-Tetrakis(p-Sulfonatophenyl)Porphyrin on the Surface of Bilayer Lipid Membranes. J. Photochem. Photobiol. B Biol. 2018, 189, 74–80. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Birin, K.P.; Michalak, J.; Romieu, A.; Stern, C.; Bessmertnykh-Lemeune, A.; Guilard, R.; Gorbunova, Y.G.; Tsivadze, A.Y. On the Synthesis of Functionalized Porphyrins and Porphyrin Conjugates via β-Aminoporphyrins. New J. Chem. 2016, 40, 5758–5774. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Birin, K.P.; Gorbunova, Y.G.; Tsivadze, A.Y.; Bessmertnykh-Lemeune, A. Post-Synthetic Methods for Functionalization of Imidazole-Fused Porphyrins. J. Porphyr. Phthalocyanines 2018, 22, 619–631. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Birin, K.P.; Bessmertnykh-Lemeune, A.; Tsivadze, A.Y.; Gorbunova, Y.G. Heterocycle-Appended Porphyrins: Synthesis and Challenges. Coord. Chem. Rev. 2020, 407, 213108. [Google Scholar] [CrossRef]
- Birin, K.P.; Poddubnaya, A.I.; Abdulaeva, I.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Revisiting 2,3-Diaminoporphyrins: Key Synthons for Heterocycle-Appended Porphyrins. Dyes Pigment. 2018, 156, 243–249. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Birin, K.P.; Polivanovskaia, D.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Functionalized Heterocycle-Appended Porphyrins: Catalysis Matters. RSC Adv. 2020, 10, 42388–42399. [Google Scholar] [CrossRef]
- Birin, K.P.; Abdulaeva, I.A.; Polivanovskaia, D.A.; Martynov, A.G.; Shokurov, A.V.; Gorbunova, Y.G.; Tsivadze, A.Y. Imidazoporphyrins with Appended Polycyclic Aromatic Hydrocarbons: To Conjugate or Not to Conjugate? Dyes Pigment. 2021, 186, 109042. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Birin, K.P.; Sinelshchikova, A.A.; Grigoriev, M.S.; Lyssenko, K.A.; Gorbunova, Y.G.; Tsivadze, A.Y.; Bessmertnykh-Lemeune, A. Imidazoporphyrins as Supramolecular Tectons: Synthesis and Self-Assembly of Zinc 2-(4-Pyridyl)-1 H -Imidazo[4,5-b]Porphyrinate. CrystEngComm 2019, 21, 1488–1498. [Google Scholar] [CrossRef]
- Abdulaeva, I.A.; Polivanovskaia, D.A.; Birin, K.P.; Gorbunova, Y.G.; Tsivadze, A.Y. A Panchromatic Pyrazine-Fused Porphyrin Dimer. Mendeleev Commun. 2020, 30, 162–164. [Google Scholar] [CrossRef]
- Birin, K.P.; Gorbunova, Y.G.; Tsivadze, A.Y. New Approach for Post-Functionalization of Meso-Formylporphyrins. RSC Adv. 2015, 5, 67242–67246. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L. Lin. Purification of Laboratory Chemicals; Elsevier/Butterworth-Heinemann: Oxford, UK, 2009; ISBN 9781856175678. [Google Scholar]
- Zinin, A.I.; Stepanova, E.V.; Jost, U.; Kondakov, N.N.; Shpirt, A.M.; Chizhov, A.O.; Torgov, V.I.; Kononov, L.O. An Efficient Multigram-Scale Synthesis of 4-(ω-Chloroalkoxy)Phenols. Russ. Chem. Bull. 2017, 66, 304–312. [Google Scholar] [CrossRef]
- Muller, P.; Rudin, D.O.; Tien, H.T.; Wescott, W.C. Methods for the Formation of Single Bimolecular Lipid Membranes in Aqueous Solution. J. Phys. Chem. 1963, 67, 534–535. [Google Scholar]
- Sokolov, V.; Kuzmin, V. Measurement of the Difference in the Surface Potentials on Bilayer Membranes from the Second Harmonic Og the Capacitance Current. Biophisics 1980, 25, 171–177. [Google Scholar]
- Sokolov, V.S.; Gavrilchik, A.N.; Kulagina, A.O.; Meshkov, I.N.; Pohl, P.; Gorbunova, Y.G. Voltage-Sensitive Styryl Dyes as Singlet Oxygen Targets on the Surface of Bilayer Lipid Membrane. J. Photochem. Photobiol. B Biol. 2016, 161, 162–169. [Google Scholar] [CrossRef]
- Shelby, M.L.; Lestrange, P.J.; Jackson, N.E.; Haldrup, K.; Mara, M.W.; Stickrath, A.B.; Zhu, D.; Lemke, H.T.; Chollet, M.; Hoffman, B.M.; et al. Ultrafast Excited State Relaxation of a Metalloporphyrin Revealed by Femtosecond X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2016, 138, 8752–8764. [Google Scholar] [CrossRef]
- Chirvony, V.S.; Galievsky, V.A.; Terekhov, S.N.; Dzhagarov, B.M.; Ermolenkov, V.V.; Turpin, P. Binding of the Cationic 5-Coordinate Zn(II)-5,10,15,20-Tetrakis(4-N-Methylpyridyl)Porphyrin to DNA and Model Polynucleotides: Ionic-Strength Dependent Intercalation in [Poly(DG-DC)]2. Biospectroscopy 1999, 5, 302–312. [Google Scholar] [CrossRef]
- Lambrechts, S.A.G.; Aalders, M.C.G.; Langeveld-Klerks, D.H.; Khayali, Y.; Lagerberg, J.W.M. Effect of Monovalent and Divalent Cations on the Photoinactivation of Bacteria with Meso-Substituted Cationic Porphyrins. Photochem. Photobiol. 2004, 79, 297. [Google Scholar] [CrossRef]
- Danci, K.-P.S.; Hilario, L.F.; Khoury, R.G.; Mai, K.U.; Nguyen, C.K.; Weddle, K.S.; Shachter, A.S. Synthesis and Aggregation of the Cationic Porphyrincs. J. Heterocycl. Chem. 1997, 34, 749–755. [Google Scholar]
- Dixon, D.W.; Steullet, V. Dimerization of Tetracationic Porphyrins: Ionic Strength Dependence. J. Inorg. Biochem. 1998, 69, 25–32. [Google Scholar] [CrossRef]
- Mosinger, J.; Janošková, M.; Lang, K.; Kubát, P. Light-Induced Aggregation of Cationic Porphyrins. J. Photochem. Photobiol. A Chem. 2006, 181, 283–289. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polivanovskaia, D.A.; Konstantinova, A.N.; Birin, K.P.; Sokolov, V.S.; Batishchev, O.V.; Gorbunova, Y.G. Peripheral Groups of Dicationic Pyrazinoporphyrins Regulate Lipid Membrane Binding. Membranes 2022, 12, 846. https://doi.org/10.3390/membranes12090846
Polivanovskaia DA, Konstantinova AN, Birin KP, Sokolov VS, Batishchev OV, Gorbunova YG. Peripheral Groups of Dicationic Pyrazinoporphyrins Regulate Lipid Membrane Binding. Membranes. 2022; 12(9):846. https://doi.org/10.3390/membranes12090846
Chicago/Turabian StylePolivanovskaia, Daria A., Anna N. Konstantinova, Kirill P. Birin, Valerij S. Sokolov, Oleg V. Batishchev, and Yulia G. Gorbunova. 2022. "Peripheral Groups of Dicationic Pyrazinoporphyrins Regulate Lipid Membrane Binding" Membranes 12, no. 9: 846. https://doi.org/10.3390/membranes12090846
APA StylePolivanovskaia, D. A., Konstantinova, A. N., Birin, K. P., Sokolov, V. S., Batishchev, O. V., & Gorbunova, Y. G. (2022). Peripheral Groups of Dicationic Pyrazinoporphyrins Regulate Lipid Membrane Binding. Membranes, 12(9), 846. https://doi.org/10.3390/membranes12090846