
New Horizons in Structural Biology of Membrane Proteins: Experimental Evaluation of the Role of Conformational Dynamics and Intrinsic Flexibility



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Nuclear Magnetic Resonance
- High magnetic sensitivity: the 19F isotope, in contrast to proton (1H), the gold standard, has a high relative sensitivity of ~83% and a 100% natural abundance (note that relative sensitivity for detection in NMR experiments at a constant number of nuclei is roughly proportional to the cube of their gyromagnetic ratios [7], (γF19/γH1)3 = 0.943 = 0.83).
- The lack of endogenous fluorine and thereby the absence of a background signal.
- Sensitivity to local environment: 19F exhibits large chemical shifts dispersion (CSD) which spans 2000 ppm in comparison to a meager 13 ppm for 1H; although chemical shifts, arising solely from local van der Waals electrostatic and solvent interactions, typically vary between 2.5 (CF3) and 20 ppms (mono-fluoro-aromatics) [2], they may still be enough to characterize motional and structural properties of IMPs in different environments such as lipid vesicles, detergents or organic solvents.
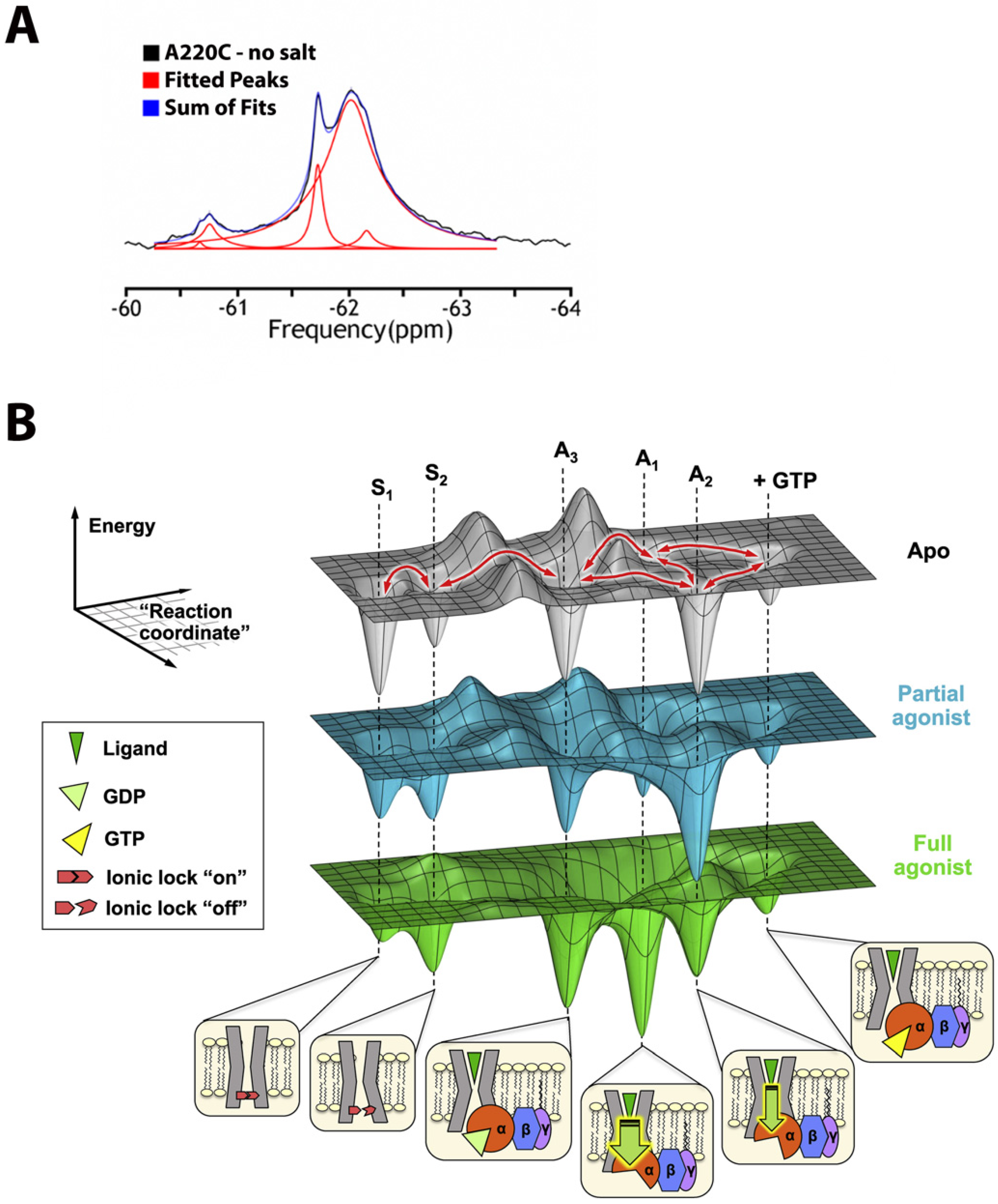
- 1D spectroscopy, used to avoid unfavorable relaxation associated with multidimensional NMR methods, is usually sufficient for the separation of the peaks in 19F spectrum (Figure 1A) and works even for potentially dynamic states that are characterized by broad lines. Thus, different states can be resolved, and their corresponding population quantified.
- Lastly, the lack of protein deuteration significantly improves the ease and efficacy of sample preparations.

3. Cryo-Electron Microscopy
4. Serial X-ray Crystallography
5. Förster Resonance Energy Transfer
- Single-Molecule FRET
- Total Internal Reflection Fluorescence
6. Electron Paramagnetic Resonance
7. Hydrogen-Deuterium Exchange
8. Antibody Fragments
9. Computational Approaches
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lau, C.; Hunter, M.J.; Stewart, A.; Perozo, E.; Vandenberg, J.I. Never at rest: Insights into the conformational dynamics of ion channels from cryo-electron microscopy. J. Physiol. 2018, 596, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Picard, L.P.; Prosser, R.S. Advances in the study of GPCRs by (19)F NMR. Curr. Opin. Struct. Biol. 2021, 69, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, R.; Vinogradova, O. Solution NMR: A powerful tool for structural and functional studies of membrane proteins in reconstituted environments. J. Biol. Chem. 2019, 294, 15914–15931. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.; Riek, R.; Wider, G.; Wüthrich, K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 1997, 94, 12366–12371. [Google Scholar] [CrossRef] [PubMed]
- Schutz, S.; Sprangers, R. Methyl TROSY spectroscopy: A versatile NMR approach to study challenging biological systems. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 116, 56–84. [Google Scholar] [CrossRef] [PubMed]
- Mooney, E.F.; Winson, P.H. Fluorine-19 Nuclear Magnetic Resonance Spectroscopy. Annu. Rep. NMR Spectrosc. 1968, 1, 243–311. [Google Scholar]
- Canet, D. NMR: Concepts and Methods; Wiley: New York, NY, USA, 1991. [Google Scholar]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef]
- Huang, S.K.; Pandey, A.; Tran, D.P.; Villanueva, N.L.; Kitao, A.; Sunahara, R.K.; Sljoka, A.; Prosser, R.P. Delineating the conformational landscape of the adenosine A2A receptor during G protein coupling. Cell 2021, 184, 1884–1894.e14. [Google Scholar] [CrossRef]
- Sljoka, A. Probing Allosteric Mechanism with Long-Range Rigidity Transmission Across Protein Networks. Methods Mol. Biol. 2021, 2253, 61–75. [Google Scholar]
- Di Pietrantonio, C.; Pandey, A.; Gould, J.; Hasabins, A.; Prosser, R.S. Understanding Protein Function Through an Ensemble Description: Characterization of Functional States by (19)F NMR. Methods Enzymol. 2019, 615, 103–130. [Google Scholar]
- Huang, Y.; Wang, X.; Lv, G.; Razavi, A.M.; Huysmans, G.H.M.; Weinstein, H.; Bracken, C.; Eliezer, D.; Boudker, O. Use of paramagnetic (19)F NMR to monitor domain movement in a glutamate transporter homolog. Nat. Chem. Biol. 2020, 16, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Meiboom, S.; Gill, D. Modified SpinEcho Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 28, 688–691. [Google Scholar] [CrossRef]
- Zhuravleva, A.; Korzhnev, D.M. Protein folding by NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 100, 52–77. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural Insights into the Dynamic Process. of beta2-Adrenergic Receptor Signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef]
- Boeszoermenyi, A.; Chhabra, S.; Dubey, A.; Radeva, D.L.; Burdzhiev, N.T.; Chanev, C.D.; Petrov, O.I.; Gelev, V.M.; Zhang, M.; Anklin, C.; et al. Aromatic (19)F-(13)C TROSY: A background-free approach to probe biomolecular structure, function, and dynamics. Nat. Methods 2019, 16, 333–340. [Google Scholar] [CrossRef]
- Cellitti, S.E.; Jones, D.H.; Lagpacan, L.; Hao, X.; Zhang, Q.; Hu, H.; Brittain, S.M.; Brinker, A.; Caldwell, J.; Bursulaya, B.; et al. In vivo incorporation of unnatural amino acids to probe structure, dynamics, and ligand binding in a large protein by nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 2008, 130, 9268–9281. [Google Scholar] [CrossRef]
- Didenko, T.; Liu, J.J.; Horst, R.; Stevens, R.C.; Wüthrich, K. Fluorine-19 NMR of integral membrane proteins illustrated with studies of GPCRs. Curr. Opin. Struct. Biol. 2013, 23, 740–747. [Google Scholar] [CrossRef]
- Yang, F.; Yu, X.; Liu, C.; Qu, C.-X.; Gong, Z.; Liu, H.-D.; Li, F.-H.; Wang, H.-M.; He, D.-F.; Yi, F.; et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat. Commun. 2015, 6, 8202. [Google Scholar] [CrossRef]
- Kuhlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef]
- Hite, R.K.; MacKinnon, R. Structural Titration of Slo2.2, a Na+-Dependent K+ Channel. Cell 2017, 168, 390–399.e11. [Google Scholar] [CrossRef]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, Y. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Armache, J.P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 525, 552. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Bulkley, D.; Guo, Y.; Zhang, W.; Guo, Z.; Huynh, W.; Wu, S.; Meltzer, S.; Cheng, T.; Jan, L.Y.; et al. Electron. cryo-microscopy structure of the mechanotransduction channel NOMPC. Nature 2017, 547, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Hirschi, M.; Herzik, M.A., Jr.; Wie, J.; Suo, Y.; Borschel, W.F.; Ren, D.; Lander, G.C.; Lee, S.-Y. Cryo-electron microscopy structure of the lysosomal calcium-permeable channel TRPML3. Nature 2017, 550, 411–414. [Google Scholar] [CrossRef]
- Chen, Q.; She, J.; Zeng, W.; Guo, J.; Xu, H.; Bai, X.C.; Jiang, Y. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 2017, 550, 415–418. [Google Scholar] [CrossRef]
- Zhang, K.; Julius, D.; Cheng, Y. Structural snapshots of TRPV1 reveal mechanism of polymodal functionality. Cell 2021, 184, 5138–5150.e12. [Google Scholar] [CrossRef]
- Steinberg, X.; Kasimova, M.A.; Cabezas-Bratesco, D.; Galpin, J.D.; Ladron-de-Guevara, E.; Villa, F.; Carnevale, V.; Islas, L.; Ahern, C.A.; Brauchi, S.E. Conformational dynamics in TRPV1 channels reported by an encoded coumarin amino acid. eLife 2017, 6, e28626. [Google Scholar] [CrossRef]
- Chen, I.; Pant, S.; Wu, Q.; Cater, R.J.; Sobti, M.; Vandenberg, R.J.; Stewart, A.G.; Tajkhorshid, E.; Font, J.; Ryan, R.M. Glutamate transporters have a chloride channel with two hydrophobic gates. Nature 2021, 591, 327–331. [Google Scholar] [CrossRef]
- Cater, R.J.; Ryan, R.M.; Vandenberg, R.J. The Split Personality of Glutamate Transporters: A Chloride Channel and a Transporter. Neurochem. Res. 2016, 41, 593–599. [Google Scholar] [CrossRef]
- Arkhipova, V.; Guskov, A.; Slotboom, D.J. Structural ensemble of a glutamate transporter homologue in lipid nanodisc environment. Nat. Commun. 2020, 11, 998. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Elferich, J.; Dehghani-Ghahnaviyeh, S.; Zhao, Z.; Meadows, M.; von Gersdorff, H.; Tajkhorshid, E.; Gouaux, E. Molecular mechanism of prestin electromotive signal amplification. Cell 2021, 184, 4669–4679.e13. [Google Scholar] [CrossRef]
- Bavi, N.; Clark, M.D.; Contreras, G.F.; Shen, R.; Reddy, B.G.; Milewski, W.; Perozo, E. The conformational cycle of prestin underlies outer-hair cell electromotility. Nature 2021, 600, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Butan, C.; Song, Q.; Bai, J.-P.; Tan, W.J.Y.; Navaratnam, D.; Santos-Sacchi, J. Single particle cryo-EM structure of the outer hair cell motor protein prestin. Nat. Commun. 2022, 13, 290. [Google Scholar] [CrossRef] [PubMed]
- Santos-Sacchi, J.; Shen, W.; Zheng, J.; Dallos, P. Effects of membrane potential and tension on prestin, the outer hair cell lateral membrane motor protein. J. Physiol. 2001, 531, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, S.; Deddebm, D.; Nunez, R.V.; Matoba, K.; Takagi, J.; Biertümpfel, C.; Mizuno, N. Structural insights into integrin alpha5beta1 opening by fibronectin ligand. Sci. Adv. 2021, 7, eabe9716. [Google Scholar] [CrossRef]
- Klebl, D.P.; White, H.D.; Sobott, F.; Muench, S.P. On-grid and in-flow mixing for time-resolved cryo-EM. Acta Crystallogr. D Struct. Biol. 2021, 77, 1233–1240. [Google Scholar] [CrossRef]
- Kaledhonkar, S.; Fu, Z.; White, H.; Frank, J. Time-Resolved Cryo-electron Microscopy Using a Microfluidic Chip. Methods Mol. Biol. 2018, 1764, 59–71. [Google Scholar]
- Maeots, M.E.; Lee, B.; Nans, A.; Jeong, S.-G.; Esfahani, M.M.N.; Ding, S.; Smith, D.J.; Lee, C.-S.; Lee, S.S.; Peter, M.; et al. Modular microfluidics enables kinetic insight from time-resolved cryo-EM. Nat. Commun. 2020, 11, 3465. [Google Scholar] [CrossRef]
- Dandey, V.P.; Budell, W.C.; Wei, H.; Bobe, D.; Maruthi, K.; Kopylov, M.; Eng, E.T.; Kahn, P.A.; Hinshaw, J.E.; Kundu, N.; et al. Time-resolved cryo-EM using Spotiton. Nat. Methods 2020, 17, 897–900. [Google Scholar] [CrossRef]
- Shaikh, T.R.; Barnard, D.; Meng, X.; Wagenknecht, T. Implementation of a flash-photolysis system for time-resolved cryo-electron microscopy. J. Struct. Biol. 2009, 165, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Yoder, N.; Jalali-Yazdi, F.; Noreng, S.; Houser, A.; Baconguis, I.; Gouaux, E. Light-coupled cryo-plunger for time-resolved cryo-EM. J. Struct. Biol. 2020, 212, 107624. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.; Skopintsev, P.; James, D.; Dworkowski, F.; Panepucci, E.; Kekilli, D.; Furrer, A.; Brünle, S.; Mous, S.; Ozerov, D.; et al. Proton uptake mechanism in bacteriorhodopsin captured by serial synchrotron crystallography. Science 2019, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Martin-Garcia, J.M. Protein Dynamics and Time Resolved Protein Crystallography at Synchrotron Radiation Sources: Past, Present and Future. Crystals 2021, 11, 521. [Google Scholar] [CrossRef]
- Wickstrand, C.; Nogly, P.; Nango, E.; Iwata, S.; Standfuss, J.; Neutze, R. Bacteriorhodopsin: Structural Insights Revealed Using X-ray Lasers and Synchrotron Radiation. Annu. Rev. Biochem. 2019, 88, 59–83. [Google Scholar] [CrossRef]
- Baxter, R.H.; Ponomarenko, N.; Srajer, V.; Pahl, R.; Moffat, K.; Norris, J.R. Time-resolved crystallographic studies of light-induced structural changes in the photosynthetic reaction center. Proc. Natl. Acad. Sci. USA 2004, 101, 5982–5987. [Google Scholar] [CrossRef]
- Wohri, A.B.; Katona, G.; Johansson, L.C.; Fritz, E.; Malmerberg, E.; Andersson, M.; Vincent, J.; Eklund, M.; Cammarata, M.; Wulff, M.; et al. Light-induced structural changes in a photosynthetic reaction center caught by Laue diffraction. Science 2010, 328, 630–633. [Google Scholar] [CrossRef]
- Neutze, R.; Wouts, R.; van der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 752–757. [Google Scholar] [CrossRef]
- Aquila, A.; Hunter, M.S.; Doak, R.B.; Kirian, R.A.; Fromme, P.; White, T.A.; Andreasson, J.; Arnlund, D.; Bajt, S.; Barends, T.R.M.; et al. Time-resolved protein nanocrystallography using an X-ray free-electron laser. Opt. Express 2012, 20, 2706–2716. [Google Scholar] [CrossRef]
- Kern, J.; Alonso-Mori, R.; Tran, R.; Hattne, J.; Gildea, R.J.; Echols, N.; Glöckner, C.; Hellmich, J.; Laksmono, H.; Sierra, R.G.; et al. Simultaneous femtosecond X-ray spectroscopy and diffraction of photosystem II at room temperature. Science 2013, 340, 491–495. [Google Scholar] [CrossRef]
- Kupitz, C.; Basu, S.; Grotjohann, I.; Fromme, R.; Zatsepin, N.A.; Rendek, K.N.; Hunter, M.S.; Shoeman, R.L.; White, T.A.; Wang, D.; et al. Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature 2014, 513, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.; Tran, R.; Alonso-Mori, R.; Koroidov, S.; Echols, N.; Hattne, J.; Ibrahim, M.; Gul, S.; Laksmono, H.; Sierra, R.G.; et al. Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy. Nat. Commun. 2014, 5, 4371. [Google Scholar] [CrossRef] [PubMed]
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native structure of photosystem II at 1.95 A resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Ibrahim, M.; Chatterjee, R.; Gul, S.; Fuller, F.; Koroidov, S.; Brewster, A.S.; Tran, R.; Alonso-Mori, R.; Kroll, T.; et al. Structure of photosystem II and substrate binding at room temperature. Nature 2016, 540, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Sauter, N.K.; Echols, N.; Adams, P.D.; Zwart, P.H.; Kern, J.; Brewster, A.S.; Koroidov, S.; Alonso-Mori, R.; Zouni, A.; Messinger, J.; et al. No observable conformational changes in PSII. Nature 2016, 533, E1–E2. [Google Scholar] [CrossRef] [PubMed]
- Suga, M.; Akita, F.; Sugahara, M.; Kubo, M.; Nakajima, Y.; Nakane, T.; Yamashita, K.; Umena, Y.; Nakabayashi, M.; Yamane, T.; et al. Light-induced structural changes and the site of O = O bond formation in PSII caught by XFEL. Nature 2017, 543, 131–135. [Google Scholar] [CrossRef]
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso-Mori, R.; et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Yamashita, K.; Nakajima, Y.; Ueno, G.; Li, H.; Yamane, T.; Hirata, K.; Umena, Y.; Yonekura, S.; et al. An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an X-ray free-electron laser. Science 2019, 366, 334–338. [Google Scholar] [CrossRef]
- Ibrahim, M.; Fransson, T.; Chatterjee, R.; Cheah, M.H.; Hussein, R.; Lassalle, L.; Sutherlin, K.D.; Young, I.D.; Fuller, F.D.; Gul, S.; et al. Untangling the sequence of events during the S2 --> S3 transition in photosystem II and implications for the water oxidation mechanism. Proc. Natl. Acad. Sci. USA 2020, 117, 12624–12635. [Google Scholar] [CrossRef]
- Hussein, R.; Ibrahim, M.; Bhowmick, A.; Simon, P.S.; Chatterjee, R.; Lassalle, L.; Doyle, M.; Bogacz, I.; Kim, I.-S.; Cheah, M.H.; et al. Structural dynamics in the water and proton channels of photosystem II during the S2 to S3 transition. Nat. Commun. 2021, 12, 6531. [Google Scholar] [CrossRef]
- Bao, H.; Burnap, R.L. Photoactivation: The Light-Driven Assembly of the Water Oxidation Complex. of Photosystem II. Front. Plant Sci. 2016, 7, 578. [Google Scholar] [CrossRef] [PubMed]
- Kok, B.; Forbush, B.; McGloin, M. Cooperation of charges in photosynthetic O2 evolution-I. A linear four step mechanism. Photochem. Photobiol. 1970, 11, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Dods, R.; Båth, P.; Morozov, D.; Gagnér, V.A.; Arnlund, D.; Luk, H.L.; Kübel, J.; Maj, M.; Vallejos, A.; Wickstrand, C.; et al. Ultrafast structural changes within a photosynthetic reaction centre. Nature 2021, 589, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Lanyi, J.K. Bacteriorhodopsin. Annu. Rev. Physiol. 2004, 66, 665–688. [Google Scholar] [CrossRef]
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.; Tanaka, R.; et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 2016, 354, 1552–1557. [Google Scholar] [CrossRef]
- Nogly, P.; Weinert, T.; James, D.; Carbajo, S.; Ozerov, D.; Furrer, A.; Gashi, D.; Borin, V.; Skopintsev, P.; Jaeger, K.; et al. Retinal isomerization in bacteriorhodopsin captured by a femtosecond X-ray laser. Science 2018, 361, eaat0094. [Google Scholar] [CrossRef]
- Kovacs, G.N.; Colletier, J.P.; Grünbein, M.L.; Yang, Y.; Stensitzki, T.; Batyuk, A.; Carbajo, S.; Doak, R.B.; Ehrenberg, D.; Foucar, L.; et al. Three-dimensional view of ultrafast dynamics in photoexcited bacteriorhodopsin. Nat. Commun. 2019, 10, 3177. [Google Scholar] [CrossRef]
- Skopintsev, P.; Ehrenberg, D.; Weinert, T.; James, D.; Kar, R.K.; Johnson, P.J.M.; Ozerov, D.; Furrer, A.; Martiel, I.; Dworkowski, F.; et al. Femtosecond-to-millisecond structural changes in a light-driven sodium pump. Nature 2020, 583, 314–318. [Google Scholar] [CrossRef]
- Yun, J.H.; Li, X.; Yue, J.; Park, J.-H.; Jin, Z.; Li, C.; Hu, H.; Shim, Y.; Pandey, S.; Carbajo, S.; et al. Early-stage dynamics of chloride ion-pumping rhodopsin revealed by a femtosecond X-ray laser. Proc. Natl. Acad. Sci. USA 2021, 118, e2020486118. [Google Scholar] [CrossRef]
- Oda, K.; Nomura, T.; Nakane, T.; Yamashita, K.; Inoue, K.; Ito, S.; Vierock, J.; Hirata, K.; Maturana, A.D.; Katayama, K.; et al. Time-resolved serial femtosecond crystallography reveals early structural changes in channelrhodopsin. eLife 2021, 10, e62389. [Google Scholar] [CrossRef]
- Shimada, A.; Kubo, M.; Baba, S.; Yamashita, K.; Hirata, K.; Ueno, G.; Nomura, T.; Kimura, T.; Shinzawa-Itoh, K.; Baba, J.; et al. A nanosecond time-resolved XFEL analysis of structural changes associated with CO release from cytochrome c oxidase. Sci. Adv. 2017, 3, e1603042. [Google Scholar] [CrossRef]
- Ishigami, I.; Lewis-Ballester, A.; Echelmeier, A.; Brehm, G.; Zatsepin, N.A.; Grant, T.D.; Coe, J.D.; Lisova, S.; Nelson, G.; Zhang, S.; et al. Snapshot of an oxygen intermediate in the catalytic reaction of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2019, 116, 3572–3577. [Google Scholar] [CrossRef] [PubMed]
- Gisriel, C.; Coe, J.; Letrun, R.; Yefanov, O.M.; Luna-Chavez, C.; Stander, N.E.; Lisova, S.; Mariani, V.; Kuhn, M.; Aplin, S.; et al. Membrane protein megahertz crystallography at the European XFEL. Nat. Commun. 2019, 10, 5021. [Google Scholar] [CrossRef] [PubMed]
- Masters, B.R. Paths to Förster’s resonance energy transfer (FRET) theory. Eur. Phys. J. H 2014, 39, 87–139. [Google Scholar] [CrossRef]
- Forster, T. Energiewanderung und Fluoreszenz. Naturwissenschaften 1946, 33, 166–175. [Google Scholar] [CrossRef]
- Stryer, L.; Haugland, R.P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719–726. [Google Scholar] [CrossRef]
- Moerner, W.E.; Kador, L. Optical detection and spectroscopy of single molecules in a solid. Phys. Rev. Lett. 1989, 62, 2535–2538. [Google Scholar] [CrossRef]
- Orrit, M.; Bernard, J. Single pentacene molecules detected by fluorescence excitation in a p-terphenyl crystal. Phys. Rev. Lett. 1990, 65, 2716–2719. [Google Scholar] [CrossRef]
- Aznauryan, M.; Nettels, D.; Holla, A.; Hofmann, H.; Schuler, B. Single-molecule spectroscopy of cold denaturation and the temperature-induced collapse of unfolded proteins. J. Am. Chem. Soc. 2013, 135, 14040–14043. [Google Scholar] [CrossRef]
- Vancraenenbroeck, R.; Harel, Y.S.; Zheng, W.; Hofmann, H. Polymer effects modulate binding affinities in disordered proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 19506–19512. [Google Scholar] [CrossRef]
- Schuler, B.; Hofmann, H. Single-molecule spectroscopy of protein folding dynamics—Expanding scope and timescales. Curr. Opin. Struct. Biol. 2013, 23, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Eaton, W.A. Protein folding transition path times from single molecule FRET. Curr. Opin. Struct. Biol. 2018, 48, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Ngo, T.T.; Zhang, Q.; Zhou, R.; Yodh, J.G.; Ha, T. Asymmetric unwrapping of nucleosomes under tension directed by DNA local flexibility. Cell 2015, 160, 1135–1144. [Google Scholar] [CrossRef]
- Guin, D.; Gruebele, M. Chaperones Hsc70 and Hsp70 Bind to the Protein PGK Differently inside Living Cells. J. Phys. Chem. B 2020, 124, 3629–3635. [Google Scholar] [CrossRef]
- Ha, T.; Ogletree, E.D.F.; Chemla, D.S.; Selvin, P.R.; Weiss, S. Probing the interaction between two single molecules: Fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA 1996, 93, 6264–6268. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B. Single-molecule FRET of protein structure and dynamics—A primer. J. Nanobiotechnol. 2013, 11 (Suppl. 1), S2. [Google Scholar] [CrossRef] [PubMed]
- Mazal, H.; Haran, G. Single-molecule FRET methods to study the dynamics of proteins at work. Curr. Opin. Biomed. Eng. 2019, 12, 8–17. [Google Scholar] [CrossRef]
- Han, L.; Zhu, Y.; Liu, M.; Zhou, Y.; Lu, G.; Lan, L.; Wang, X.; Zhao, Y.; Zhang, X.C. Molecular mechanism of substrate recognition and transport by the AtSWEET13 sugar transporter. Proc. Natl. Acad. Sci. USA 2017, 114, 10089–10094. [Google Scholar] [CrossRef]
- Haney, C.M.; Wissner, R.F.; Petersson, E.J. Multiply labeling proteins for studies of folding and stability. Curr. Opin. Chem. Biol. 2015, 28, 123–130. [Google Scholar] [CrossRef]
- Prive, G.G. Detergents for the stabilization and crystallization of membrane proteins. Methods 2007, 41, 388–397. [Google Scholar] [CrossRef]
- Zhao, Y.; Terry, D.; Shi, L.; Weinstein, H.; Blanchard, S.C.; Javitch, J.A. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature 2010, 465, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, R.; Nguyen, K.; Vinogradova, O. Nanodiscs and Solution NMR: Preparation, application and challenges. Nanotechnol. Rev. 2017, 6, 111–126. [Google Scholar] [CrossRef]
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. Science 2018, 359, eaan1133. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.; He, L.; Zhao, Y.; Zhang, X.C. Single-molecule fluorescence studies on the conformational change of the ABC transporter MsbA. Biophys. Rep. 2018, 4, 153–165. [Google Scholar] [CrossRef]
- Ural-Blimke, Y.; Flayhan, A.; Strauss, J.; Rantos, V.; Bartels, K.; Nielsen, R.; Pardon, E.; Steyaert, J.; Kosinski, J.; Quistgaard, E.M.; et al. Structure of Prototypic Peptide Transporter DtpA from E. coli in Complex. with Valganciclovir Provides Insights into Drug Binding of Human PepT1. J. Am. Chem. Soc. 2019, 141, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Popovic, K.; Holyoake, J.; Pomès, R.; Privé, G.G. Structure of saposin A lipoprotein discs. Proc. Natl. Acad. Sci. USA 2012, 109, 2908–2912. [Google Scholar] [CrossRef] [PubMed]
- Lasitza-Male, T.; Jungwirth, J.; Wiggers, F.; Rosenblum, G.; Hofmann, H.; Löw, C. Membrane Chemistry Tunes the Structure of a Peptide Transporter. Angew. Chem. Int. Ed. Engl. 2020, 59, 19121–19128. [Google Scholar] [CrossRef]
- Kanner, B.I.; Zomot, E. Sodium-Coupled neurotransmitter transporters. Chem. Rev. 2008, 108, 1654–1668. [Google Scholar] [CrossRef]
- Terry, D.S.; Kolster, R.A.; Quick, M.; LeVine, M.V.; Khelashvili, G.; Zhou, Z.; Weinstein, H.; Javitch, J.A.; Blanchard, S.C. A partially-open inward-facing intermediate conformation of LeuT is associated with Na+ release and substrate transport. Nat. Commun. 2018, 9, 230. [Google Scholar] [CrossRef]
- LeVine, M.V.; Terry, D.S.; Khelashvili, G.; Siegel, Z.S.; Quick, M.; Javitch, J.A.; Blanchard, S.C.; Weinstein, H. The allosteric mechanism of substrate-specific transport in SLC6 is mediated by a volumetric sensor. Proc. Natl. Acad. Sci. USA 2019, 116, 15947–15956. [Google Scholar] [CrossRef]
- Kalstrup, T.; Blunck, R. Dynamics of internal pore opening in K(V) channels probed by a fluorescent unnatural amino acid. Proc. Natl. Acad. Sci. USA 2013, 110, 8272–8277. [Google Scholar] [CrossRef] [PubMed]
- Dolino, D.M.; Cooper, D.; Ramaswamy, S.; Jaurich, H.; Landes, C.F.; Jayaraman, V. Structural dynamics of the glycine-binding domain of the N-methyl-D-aspartate receptor. J. Biol. Chem. 2015, 290, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Belov, V.N.; Wurm, C.A.; Boyarskiy, V.P.; Jakobs, S.; Hell, S.W. Rhodamines NN: A novel class of caged fluorescent dyes. Angew. Chem. Int. Ed. Engl. 2010, 49, 3520–3523. [Google Scholar] [CrossRef] [PubMed]
- Jazi, A.A.; Ploetz, E.; Arizki, M.; Dhandayuthapani, B.; Waclawska, I.; Krämer, R.; Ziegler, C.; Cordes, T. Caging and Photoactivation in Single-Molecule Forster Resonance Energy Transfer Experiments. Biochemistry 2017, 56, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Juette, M.F.; Jockusch, S.; Wasserman, M.R.; Zhou, Z.; Altman, R.B.; Blanchard, S.C. Ultra-stable organic fluorophores for single-molecule research. Chem. Soc. Rev. 2014, 43, 1044–1056. [Google Scholar] [CrossRef]
- Zheng, Q.; Jockusch, S.; Zhou, Z.; Altman, R.B.; Zhao, H.; Asher, W.; Holsey, M.; Mathiasen, S.; Geggier, P.; Javitch, J.A.; et al. Electronic tuning of self-healing fluorophores for live-cell and single-molecule imaging. Chem. Sci. 2017, 8, 755–762. [Google Scholar] [CrossRef]
- Stone, T.J.; Buckman, T.; Nordio, P.L.; McConnell, H.M. Spin-labeled biomolecules. Proc. Natl. Acad. Sci. USA 1965, 54, 1010–1017. [Google Scholar] [CrossRef]
- Weil, J.B.; Bolton, J.R. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications; Wiley-Interscience: Hoboken, NJ, USA, 2007. [Google Scholar]
- Sahu, I.D.; Lorigan, G.A. Site-Directed Spin Labeling EPR for Studying Membrane Proteins. BioMed Res. Int. 2018, 2018, 3248289. [Google Scholar] [CrossRef]
- Fleissner, M.R.; Brustad, E.M.; Kálai, T.; Altenbach, C.; Cascio, D.; Peters, F.B.; Hideg, K.; Peuker, S.; Schultz, P.G.; Hubbell, W.L. Site-directed spin labeling of a genetically encoded unnatural amino acid. Proc. Natl. Acad. Sci. USA 2009, 106, 21637–21642. [Google Scholar] [CrossRef]
- Chiang, Y.W.; Zheng, T.-Y.; Kao, C.-J.; Horng, J.-C. Determination of interspin distance distributions by cw-ESR is a single linear inverse problem. Biophys. J. 2009, 97, 930–936. [Google Scholar] [CrossRef][Green Version]
- Borbat, P.P.; McHaourab, H.S.; Freed, J.H. Protein structure determination using long-distance constraints from double-quantum coherence ESR: Study of T4 lysozyme. J. Am. Chem. Soc. 2002, 124, 5304–5314. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G.; Polyhach, Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 2007, 9, 1895–1910. [Google Scholar] [CrossRef] [PubMed]
- Pannier, M.; Veit, S.; Godt, A.; Jeschke, G.; Spiess, H.W. Dead-time free measurement of dipole-dipole interactions between electron spins. J. Magn. Reson. 2000, 142, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.; Bowman, A.; Sozudogru, E.; El-Mkami, H.; Owen-Hughes, T.; Norman, D.G. EPR distance measurements in deuterated proteins. J. Magn. Reson. 2010, 207, 164–167. [Google Scholar] [CrossRef]
- Chiang, Y.W.; Borbat, P.P.; Freed, J.H. The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. J. Magn. Reson. 2005, 172, 279–295. [Google Scholar] [CrossRef]
- Zhou, N.; Fan, L.; Ho, H.-T.; Nowicka-Sans, B.; Sun, Y.; Zhu, Y.; Hu, Y.; McAuliffe, B.; Rose, B.; Fang, H.; et al. Increased sensitivity of HIV variants selected by attachment inhibitors to broadly neutralizing antibodies. Virology 2010, 402, 256–261. [Google Scholar] [CrossRef]
- Schmidt, T.; Wälti, M.A.; Baber, J.L.; Hustedt, E.J.; Clore, G.M. Long Distance Measurements up to 160 A in the GroEL Tetradecamer Using Q-Band DEER EPR Spectroscopy. Angew. Chem. Int. Ed. Engl. 2016, 55, 15905–15909. [Google Scholar] [CrossRef]
- Lerch, M.T.; Matt, R.A.; Masureel, M.; Elgeti, M.; Kumar, K.K.; Hilger, D.; Foys, B.; Kobilka, B.K.; Hubbell, W.L. Viewing rare conformations of the beta2 adrenergic receptor with pressure-resolved DEER spectroscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 31824–31831. [Google Scholar] [CrossRef]
- Georgieva, E.R.; Borbat, P.P.; Ginter, C.; Freed, J.H.; Boudker, O. Conformational ensemble of the sodium-coupled aspartate transporter. Nat. Struct. Mol. Biol. 2013, 20, 215–221. [Google Scholar] [CrossRef]
- Hanelt, I.; Wunnicke, D.; Bordignon, E.; Steinhoff, H.-J.; Slotboom, D.J. Conformational heterogeneity of the aspartate transporter Glt(Ph.). Nat. Struct. Mol. Biol. 2013, 20, 210–214. [Google Scholar] [CrossRef]
- Basak, S.; Schmandt, N.; Gicheru, Y.; Chakrapani, S. Crystal structure and dynamics of a lipid-induced potential desensitized-state of a pentameric ligand-gated channel. eLife 2017, 6, e23886. [Google Scholar] [CrossRef]
- Mishra, S.; Verhalen, B.; Stein, R.A.; Wen, P.-C.; Tajkhorshid, E.; Mchaourab, H.S. Conformational dynamics of the nucleotide binding domains and the power stroke of a heterodimeric ABC transporter. eLife 2014, 3, e02740. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Sikora, A.; Cafiso, D.S. Ligand Induced Conformational Changes of a Membrane Transporter in E. coli Cells Observed with DEER/PELDOR. J. Am. Chem. Soc. 2016, 138, 1844–1847. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. A Bioresistant Nitroxide Spin Label. for In-Cell EPR Spectroscopy: In Vitro and In Oocytes Protein Structural Dynamics Studies. Angew. Chem. Int. Ed. Engl. 2018, 57, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chance, M.R. Protein Footprinting Comes of Age: Mass Spectrometry for Biophysical Structure Assessment. Mol. Cell. Proteom. 2017, 16, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Katta, V.; Chait, B.T. Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1991, 5, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Smith, D.L. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 1993, 2, 522–531. [Google Scholar] [CrossRef]
- Hanai, R.; Wang, J.C. Protein footprinting by the combined use of reversible and irreversible lysine modifications. Proc. Natl. Acad. Sci. USA 1994, 91, 11904–11908. [Google Scholar] [CrossRef]
- Eisinger, M.L.; Dörrbaum, A.R.; Michel, H.; Padan, E.; Langer, J.D. Ligand-induced conformational dynamics of the Escherichia coli Na+/H+ antiporter NhaA revealed by hydrogen/deuterium exchange mass spectrometry. Proc. Natl. Acad. Sci. USA 2017, 114, 11691–11696. [Google Scholar] [CrossRef]
- Vahidi, S.; Bi, Y.; Dunn, S.D.; Konermann, L. Load-dependent destabilization of the gamma-rotor shaft in FOF1 ATP synthase revealed by hydrogen/deuterium-exchange mass spectrometry. Proc. Natl. Acad. Sci. USA 2016, 113, 2412–2417. [Google Scholar] [CrossRef]
- Krishnamurthy, H.; Gouaux, E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 2012, 481, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Focke, P.J.; Wang, X.; Larsson, H.P. Neurotransmitter transporters: Structure meets function. Structure 2013, 21, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Merkle, P.S.; Gotfryd, K.; Cuendet, M.A.; Leth-Espensen, K.Z.; Gether, U.; Loland, C.J.; Rand, K.D. Substrate-modulated unwinding of transmembrane helices in the NSS transporter LeuT. Sci. Adv. 2018, 4, eaar6179. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, S.; Deredge, D.J.; Nagarajan, A.; Forrest, L.R.; Wintrode, P.L.; Singh, S.K. Conformational dynamics of a neurotransmitter:sodium symporter in a lipid bilayer. Proc. Natl. Acad. Sci. USA 2017, 114, E1786–E1795. [Google Scholar] [CrossRef] [PubMed]
- Reading, E.; Hall, Z.; Martens, C.; Haghighi, T.; Findlay, H.; Ahdash, Z.; Politis, A.; Booth, P.J. Interrogating Membrane Protein Conformational Dynamics within Native Lipid Compositions. Angew. Chem. Int. Ed. Engl. 2017, 56, 15654–15657. [Google Scholar] [CrossRef]
- Zhu, Y.; Guo, T.; Park, J.E.; Li, X.; Meng, W.; Datta, A.; Bern, M.; Lim, S.K.; Sze, S.K. Elucidating in vivo structural dynamics in integral membrane protein by hydroxyl radical footprinting. Mol. Cell. Proteom. 2009, 8, 1999–2010. [Google Scholar] [CrossRef]
- Zhu, Y.; Serra, A.; Guo, T.; Park, J.E.; Zhong, Q.; Sze, S.Q. Application of Nanosecond Laser Photolysis Protein Footprinting to Study EGFR Activation by EGF in Cells. J. Proteome Res. 2017, 16, 2282–2293. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Xenaki, K.T.; Oliveira, S.; van Bergen En Henegouwen, P.M.P. Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors. Front. Immunol. 2017, 8, 1287. [Google Scholar] [CrossRef]
- Renart, J.; Reiser, J.; Stark, G.R. Transfer of proteins from gels to diazobenzyloxymethyl-paper and detection with antisera: A method for studying antibody specificity and antigen structure. Proc. Natl. Acad. Sci. USA 1979, 76, 3116–3120. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.B. Immunocytochemical methods: A technical overview. J. Oral Pathol. 1987, 16, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Lequin, R.M. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 2005, 51, 2415–2418. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Gupta, C.; Srivastava, A.; Jaiman, D. Antibody fragments for stabilization and crystallization of G protein-coupled receptors and their signaling complexes. Methods Enzymol. 2015, 557, 247–258. [Google Scholar] [PubMed]
- Dutzler, R.; Campbell, E.B.; MacKinnon, R. Gating the selectivity filter in ClC chloride channels. Science 2003, 300, 108–112. [Google Scholar] [CrossRef]
- Ishchenko, A.; Wacker, D.; Kapoor, M.; Zhang, A.; Han, G.W.; Basu, S.; Patel, N.; Messerschmidt, M.; Weierstall, U.; Liu, W.; et al. Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc. Natl. Acad. Sci. USA 2017, 114, 8223–8228. [Google Scholar] [CrossRef] [PubMed]
- Cormier, A.; Campbell, M.G.; Ito, S.; Wu, S.; Lou, J.; Marks, J.; Baron, J.L.; Nishimura, S.L.; Cheng, Y. Cryo-EM structure of the alphavbeta8 integrin reveals a mechanism for stabilizing integrin extension. Nat. Struct. Mol. Biol. 2018, 25, 698–704. [Google Scholar] [CrossRef]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Beghein, E.; Gettemans, J. Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein-Protein Interaction Analysis, and Protein Function Exploration. Front. Immunol. 2017, 8, 771. [Google Scholar] [CrossRef]
- Puthenveetil, R.; Lee, C.J.; Banerjee, A. Production of Recombinant Transmembrane Proteins from Mammalian Cells for Biochemical and Structural Analyses. Curr. Protoc. Cell Biol. 2020, 87, e106. [Google Scholar] [CrossRef]
- McMahon, C.; Baier, A.S.; Pascolutti, R.; Wegrecki, M.; Zheng, S.; Ong, J.X.; Erlandson, S.C.; Hilger, D.; Rasmussen, S.G.F.; Ring, A.M.; et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 2018, 25, 289–296. [Google Scholar] [CrossRef]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.; Arnold, F.M.; Stohler, P.; Bocquet, N.; Hug, M.N.; Huber, S.; Siegrist, M.; Hetemann, L.; et al. Synthetic single domain antibodies for the conformational trapping of membrane proteins. eLife 2018, 7, e34317. [Google Scholar] [CrossRef]
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [Google Scholar] [CrossRef] [PubMed]
- Hilger, D. The role of structural dynamics in GPCR-mediated signaling. FEBS J. 2021, 288, 2461–2489. [Google Scholar] [CrossRef] [PubMed]
- Ostermeier, C.; Iwata, S.; Ludwig, B.; Michel, H. Fv fragment-mediated crystallization of the membrane protein bacterial cytochrome c oxidase. Nat. Struct. Biol. 1995, 2, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Morais-Cabral, J.H.; Mann, S.; MacKinnon, R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 2001, 411, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Matulef, K.; Annen, A.W.; Nix, J.C.; Valiyaveetil, F.I. Individual Ion Binding Sites in the K+ Channel Play Distinct Roles in C-type Inactivation and in Recovery from Inactivation. Structure 2016, 24, 750–761. [Google Scholar] [CrossRef]
- Ghanouni, P.; Steenhuis, J.J.; Farrens, D.L.; Kobilka, B.K. Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 5997–6002. [Google Scholar] [CrossRef]
- Hunte, C.; Michel, H. Crystallisation of membrane proteins mediated by antibody fragments. Curr. Opin. Struct. Biol. 2002, 12, 503–508. [Google Scholar] [CrossRef]
- Valiyaveetil, F.I.; Sekedat, M.; MacKinnon, R.; Muir, T.W. Structural and functional consequences of an amide-to-ester substitution in the selectivity filter of a potassium channel. J. Am. Chem. Soc. 2006, 128, 11591–11599. [Google Scholar] [CrossRef]
- Rasmussen, S.G.; Choi, H.J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Tosha, T.; Pisliakov, A.V.; Hino, T.; Sugimoto, H.; Nagano, S.; Sugita, Y.; Shiro, Y. Crystal structure of quinol-dependent nitric oxide reductase from Geobacillus stearothermophilus. Nat. Struct. Mol. Biol. 2012, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Strachan, R.T.; Manglik, A.; Pani, B.; Kahsai, A.W.; Kim, T.H.; Wingler, L.M.; Ahn, S.; Chatterjee, A.; Masoudi, A.; et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 2016, 535, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Masureel, M.; Zou, Y.; Picard, L.P.; van der Westhuizen, E.; Mahoney, J.P.; Rodrigues, J.P.G.L.M.; Mildorf, T.J.; Dror, R.O.; Shaw, D.E.; Bouvier, M.; et al. Structural insights into binding specificity, efficacy and bias of a beta2AR partial agonist. Nat. Chem. Biol. 2018, 14, 1059–1066. [Google Scholar] [CrossRef]
- Wingler, L.M.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell 2019, 176, 479–490.e12. [Google Scholar] [CrossRef]
- Brauer, P.; Parker, J.L.; Gerondopoulos, A.; Zimmermann, I.; Seeger, M.A.; Barr, F.A.; Newstead, S. Structural basis for pH-dependent retrieval of ER proteins from the Golgi by the KDEL receptor. Science 2019, 363, 1103–1107. [Google Scholar] [CrossRef]
- Sayeed, W.M.; Baenziger, J.E. Structural characterization of the osmosensor ProP. Biochim. Biophys. Acta 2009, 1788, 1108–1115. [Google Scholar] [CrossRef][Green Version]
- Smirnova, I.; Kasho, V.; Sugihara, J.; Kaback, H.R. Trp replacements for tightly interacting Gly-Gly pairs in LacY stabilize an outward-facing conformation. Proc. Natl. Acad. Sci. USA 2013, 110, 8876–8881. [Google Scholar] [CrossRef]
- Smirnova, I.; Kasho, V.; Jiang, X.; Pardon, E.; Steyaert, J.; Kaback, H.R. Outward-facing conformers of LacY stabilized by nanobodies. Proc. Natl. Acad. Sci. USA 2014, 111, 18548–18553. [Google Scholar] [CrossRef]
- Kumar, H.; Finer-Moore, J.S.; Jiang, X.; Smirnova, I.; Kasho, V.; Pardon, E.; Steyaert, J.; Kaback, H.R.; Stroud, R.M. Crystal Structure of a ligand-bound LacY-Nanobody Complex. Proc. Natl. Acad. Sci. USA 2018, 115, 8769–8774. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, R.; Tomshine, J.C.; Tomshine, J.R.; Chevalier, M.; Mahoney, J.P.; Steyaert, J.; Rasmussen, S.G.F.; Sunahara, R.K.; El-Samad, H.; Huang, B.; et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 2013, 495, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Nevoltris, D.; Lombard, B.; Dupuis, E.; Mathis, G.; Chames, P.; Baty, D. Conformational nanobodies reveal tethered epidermal growth factor receptor involved in EGFR/ErbB2 predimers. ACS Nano 2015, 9, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, R.; Martinez-Seoane, J.; Amero, C. Combining Experimental Data and Computational Methods for the Non-Computer Specialist. Molecules 2020, 25, 4783. [Google Scholar] [CrossRef] [PubMed]
- Maximova, T.; Moffatt, R.; Ma, B.; Nussinov, R.; Shehu, A. Principles and Overview of Sampling Methods for Modeling Macromolecular Structure and Dynamics. PLoS Comput. Biol. 2016, 12, e1004619. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–15614. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Bermejo, G.A.; Clore, G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003, 160, 65–73. [Google Scholar] [CrossRef]
- Marsh, J.A.; Forman-Kay, J.D. Structure and disorder in an unfolded state under nondenaturing conditions from ensemble models consistent with a large number of experimental restraints. J. Mol. Biol. 2009, 391, 359–374. [Google Scholar] [CrossRef]
- Boomsma, W.; Frellsen, J.; Harder, T.; Bottaro, S.; Johansson, K.E.; Tian, P.; Stovgaard, K.; Andreetta, C.; Olsson, S.; Valentin, J.B.; et al. PHAISTOS: A framework for Markov chain Monte Carlo simulation and inference of protein structure. J. Comput. Chem. 2013, 34, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Orioli, S.; Frellsen, J.; Harder, T.; Bottaro, S.; Johansson, K.E.; Tian, P.; Stovgaard, K.; Andreetta, C.; Olsson, S.; Valentin, J.B.; et al. How to learn from inconsistencies: Integrating molecular simulations with experimental data. Prog. Mol. Biol. Transl. Sci. 2020, 170, 123–176. [Google Scholar]
- Bonomi, M.; Heller, G.T.; Camilloni, C.; Vendruscolo, M. Principles of protein structural ensemble determination. Curr. Opin. Struct. Biol. 2017, 42, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Leelananda, S.P.; Lindert, S. Iterative Molecular Dynamics-Rosetta Membrane Protein Structure Refinement Guided by Cryo-EM Densities. J. Chem. Theory Comput. 2017, 13, 5131–5145. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, O.P.; Paz, A.; Adelman, J.L.; Colletier, J.-P.; Abramson, J.; Grabe, M. Structure-guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat. Struct. Mol. Biol. 2014, 21, 626–632. [Google Scholar] [CrossRef]
- Zhou, H.X.; Cross, T.A. Modeling the membrane environment has implications for membrane protein structure and function: Influenza A M2 protein. Protein Sci. 2013, 22, 381–394. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.M.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. A method for integrative structure determination of protein-protein complexes. Bioinformatics 2012, 28, 3282–3289. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef]
- Koukos, P.I.; Faro, I.; van Noort, C.W.; Bonvin, A.M.J.J. A Membrane Protein Complex. Docking Benchmark. J. Mol. Biol. 2018, 430, 5246–5256. [Google Scholar] [CrossRef]
- Rudden, L.S.P.; Degiacomi, M.T. Transmembrane Protein Docking with JabberDock. J. Chem. Inf. Model. 2021, 61, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puthenveetil, R.; Christenson, E.T.; Vinogradova, O. New Horizons in Structural Biology of Membrane Proteins: Experimental Evaluation of the Role of Conformational Dynamics and Intrinsic Flexibility. Membranes 2022, 12, 227. https://doi.org/10.3390/membranes12020227
Puthenveetil R, Christenson ET, Vinogradova O. New Horizons in Structural Biology of Membrane Proteins: Experimental Evaluation of the Role of Conformational Dynamics and Intrinsic Flexibility. Membranes. 2022; 12(2):227. https://doi.org/10.3390/membranes12020227
Chicago/Turabian StylePuthenveetil, Robbins, Eric T. Christenson, and Olga Vinogradova. 2022. "New Horizons in Structural Biology of Membrane Proteins: Experimental Evaluation of the Role of Conformational Dynamics and Intrinsic Flexibility" Membranes 12, no. 2: 227. https://doi.org/10.3390/membranes12020227
APA StylePuthenveetil, R., Christenson, E. T., & Vinogradova, O. (2022). New Horizons in Structural Biology of Membrane Proteins: Experimental Evaluation of the Role of Conformational Dynamics and Intrinsic Flexibility. Membranes, 12(2), 227. https://doi.org/10.3390/membranes12020227