
p53 Signaling on Microenvironment and Its Contribution to Tissue Chemoresistance

 ,
,
Abstract
1. Introduction
2. Keeping the Tissue in Check: Extrinsic Function of Wild-Type p53
3. Turn of the Resistant Tide: Mutant p53’s Nonautonomous Gain-of-Function
3.1. A Darker Side to SASP
3.2. Hypoxia: p53 as Friend or Foe
3.3. CSCs and EMT: Gearing Up for TME-Driven Chemoresistance
3.4. ECM Remodeling and Integrin Expression: Survival of the Stiffest
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Venkatesan, S.; Swanton, C.; Taylor, B.S.; Costello, J.F. Treatment-Induced Mutagenesis and Selective Pressures Sculpt Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a026617. [Google Scholar] [CrossRef] [PubMed]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.J.; Potdar, S.; Kyttälä, A.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M.; et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer 2019, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Minn, A.J. Exosome Transfer from Stromal to Breast Cancer Cells Regulates Therapy Resistance Pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Kreger, B.T.; Johansen, E.R.; Cerione, R.A.; Antonyak, M.A. The Enrichment of Survivin in Exosomes from Breast Cancer Cells Treated with Paclitaxel Promotes Cell Survival and Chemoresistance. Cancers 2016, 8, 111. [Google Scholar] [CrossRef]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 Paracrine Network Links Cancer Chemoresistance and Metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Cox, T.R. The matrix in cancer. Nat. Rev. Cancer. 2021, 21, 217–238. [Google Scholar] [CrossRef]
- Pietilä, E.A.; Gonzalez-Molina, J.; Moyano-Galceran, L.; Jamalzadeh, S.; Zhang, K.; Lehtinen, L.; Turunen, S.P.; Martins, T.A.; Gultekin, O.; Lamminen, T.; et al. Co-evolution of matrisome and adaptive adhesion dynamics drives ovarian cancer chemoresistance. Nat. Commun. 2021, 12, 3904. [Google Scholar] [CrossRef]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.; Hernández, A.D.R. Matrix stiffness induces epithelial–mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef]
- Shin, J.-W.; Mooney, D.J. Extracellular matrix stiffness causes systematic variations in proliferation and chemosensitivity in myeloid leukemias. Proc. Natl. Acad. Sci. USA 2016, 113, 12126–12131. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharife, H.; Kreisel, T.; Mogilevsky, M.; Bar-Lev, L.; Grunewald, M.; Aizenshtein, E.; Karni, R.; Paldor, I.; Shlomi, T.; et al. Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell Metab. 2019, 30, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Delou, J.; Souza, A.S.; Souza, L.; Borges, H.L. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells 2019, 8, 1013. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Dou, B.; Tan, H.; Feng, Y.; Wang, N.; Wang, D. Tumor microenvironment-driven non-cell-autonomous resistance to antineoplastic treatment. Mol. Cancer 2019, 18, 69. [Google Scholar] [CrossRef]
- Nwabo Kamdje, A.H.; Bassi, G.; Pacelli, L.; Malpeli, G.; Amati, E.; Nichele, I.; Krampera, M. Role of stromal cell-mediated Notch signaling in CLL resistance to chemotherapy. Blood Cancer J. 2012, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef]
- Li, B.; Jia, R.; Li, W.; Zhou, Y.; Guo, D.; Teng, Q.; Ji, C. PAK1 Mediates Bone Marrow Stromal Cell-Induced Drug Resistance in Acute Myeloid Leukemia via ERK1/2 Signaling Pathway. Front. Cell Dev. Biol. 2021, 8, 686695. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef]
- Maia, J.; Caja, S.; Moraes, M.C.S.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, S.; Zhang, R.; Sohrabi, A.; Yu, Q.; Liu, S.; Ehsanipour, A.; Liang, J.; Bierman, R.D.; Nathanson, D.A.; et al. Bioengineered scaffolds for 3D culture demonstrate extracellular matrix-mediated mechanisms of chemotherapy resistance in glioblastoma. Matrix Biol. 2020, 85–86, 128–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to Stromal Fibroblasts Induces Resistance of Lung Cancer to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Sheng, J.; Gao, D.; Li, F.; Durrans, A.; Ryu, S.; Lee, S.B.; Narula, N.; Rafii, S.; Elemento, O.; et al. Transcriptome Analysis of Individual Stromal Cell Populations Identifies Stroma-Tumor Crosstalk in Mouse Lung Cancer Model. Cell Rep. 2015, 10, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Ouahoud, S.; Voorneveld, P.W.; van der Burg, L.R.A.; De Jonge-Muller, E.S.M.; Schoonderwoerd, M.J.A.; Paauwe, M.; De Vos, T.; De Wit, S.; van Pelt, G.W.; Mesker, W.E.; et al. Bidirectional tumor/stroma crosstalk promotes metastasis in mesenchymal colorectal cancer. Oncogene 2020, 39, 2453–2466. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stanger, B.Z. The tumor as organizer model. Science 2019, 363, 1038–1039. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Rivlin, N.; Koifman, G.; Rotter, V. p53 orchestrates between normal differentiation and cancer. Semin. Cancer Biol. 2015, 32, 10–17. [Google Scholar] [CrossRef]
- Humpton, T.J.; Vousden, K.H. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026146. [Google Scholar] [CrossRef]
- Shetzer, Y.; Molchadsky, A.; Rotter, V. Oncogenic Mutant p53 Gain of Function Nourishes the Vicious Cycle of Tumor Development and Cancer Stem-Cell Formation. Cold Spring Harb. Perspect. Med. 2016, 6, a026203. [Google Scholar] [CrossRef]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Wheeler, D.A. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Hoyos, L.F.; Penson, A.; Kannan, R.; Cho, H.; Pan, C.-H.; Singh, R.K.; Apken, L.H.; Hobbs, G.A.; Luo, R.; Lecomte, N.; et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 2020, 38, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Vijayakumaran, R.; Miranda, P.J.; Burgess, A.; Lim, E.; Haupt, Y. The role of MDM2 and MDM4 in breast cancer development and prevention. J. Mol. Cell Biol. 2017, 9, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; van den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.-C.; Prats, A.-C. The p53 isoform, Δ133p53α, stimulates angiogenesis and tumour progression. Oncogene 2012, 32, 2150–2160. [Google Scholar] [CrossRef]
- Horikawa, I.; Fujita, K.; Jenkins, L.M.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.; Appella, E.; Harris, C.C. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53-mediated senescence. Nat. Commun. 2014, 5, 4706. [Google Scholar] [CrossRef]
- Beck, J.; Turnquist, C.; Horikawa, I.; Harris, C.C. Targeting cellular senescence in cancer and aging: Roles of p53 and its isoforms. Carcinogenesis 2020, 41, 1017–1029. [Google Scholar] [CrossRef]
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Roux, P. TP53 drives invasion through expression of its Δ133p53β variant. eLife 2016, 15, e14734. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-Cell-Autonomous Tumor Suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Molchadsky, A.; Rotter, V. p53 and its mutants on the slippery road from stemness to carcinogenesis. Carcinogenesis 2017, 38, 347–358. [Google Scholar] [CrossRef]
- Pavlakis, E.; Stiewe, T. p53’s Extended Reach: The Mutant p53 Secretome. Biomolecules 2020, 10, 307. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, E.; Neumann, M.; Stiewe, T. Extracellular Vesicles: Messengers of p53 in Tumor–Stroma Communication and Cancer Metastasis. Int. J. Mol. Sci. 2020, 21, 9648. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant p53-a potential player in shaping the tumor-stroma crosstalk. J. Mol. Cell Biol. 2019, 11, 600–604. [Google Scholar] [CrossRef]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Gimenez, G.; Bowie, S.; Campbell, H.; Reily-Bell, A.L.; Roth, I.; Ray, S.; Drummond, C.J.; et al. The Δ133p53β isoform promotes an immunosuppressive environment leading to aggressive prostate cancer. Cell Death Dis. 2019, 10, 631. [Google Scholar] [CrossRef]
- Kazantseva, M.; Eiholzer, R.A.; Mehta, S.; Taha, A.; Bowie, S.; Roth, I.; Braithwaite, A.W. Elevation of the TP53 isoform Δ133p53β in glioblastomas: An alternative to mutant p53 in promoting tumor development: Δ133p53β isoform in brain cancer. J. Pathol. 2018, 246, 77–88. [Google Scholar] [CrossRef]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496. [Google Scholar] [CrossRef]
- Bezzi, M.; Seitzer, N.; Ishikawa, T.; Reschke, M.; Chen, M.; Wang, G.; Mitchell, C.; Ng, C.; Katon, J.; Lunardi, A.; et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat. Med. 2018, 24, 165–175. [Google Scholar] [CrossRef]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef]
- Shi, D.; Jiang, P. A Different Facet of p53 Function: Regulation of Immunity and Inflammation During Tumor Development. Front. Cell Dev. Biol. 2021, 9, 762651. [Google Scholar] [CrossRef]
- Arandkar, S.; Furth, N.; Elisha, Y.; Nataraj, N.B.; van der Kuip, H.; Yarden, Y.; Aulitzky, W.; Ulitsky, I.; Geiger, B.; Oren, M. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc. Natl. Acad. Sci. USA 2018, 115, 6410–6415. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Clos, A.L.; Castillo-Carranza, D.; Sengupta, U.; Guerrero-Muñoz, M.; Kelly, B.; Wagner, R.; Kayed, R. Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem. Biophys. Res. Commun. 2013, 430, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Rangel, L.P.; Costa, D.C.; Vieira, T.C.; Silva, J.L. The aggregation of mutant p53 produces prion-like properties in cancer. Prion 2014, 8, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Forget, K.J.; Tremblay, G.; Roucou, X. p53 Aggregates Penetrate Cells and Induce the Co-Aggregation of Intracellular p53. PLoS ONE 2013, 8, e69242. [Google Scholar] [CrossRef]
- Silva, J.L.; De Moura Gallo, C.V.; Costa, D.C.F.; Rangel, L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef]
- Pedrote, M.M.; Motta, M.F.; Ferretti, G.D.; Norberto, D.R.; Spohr, T.C.; Lima, F.R.S.; Gratton, E.; Silva, J.L.; de Oliveira, G.A. Oncogenic Gain of Function in Glioblastoma Is Linked to Mutant p53 Amyloid Oligomers. iScience 2020, 23, 100820. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef]
- Rohwer, N.; Dame, C.l.; Haugstetter, A.; Wiedenmann, B.; Detjen, K.; Schmitt, C.A.; Cramer, T. Hypoxia-Inducible Factor 1α Determines Gastric Cancer Chemosensitivity via Modulation of p53 and NF-κB. Fox D, editor. PLoS ONE 2010, 10, e12038. [Google Scholar]
- Wang, P.; Guan, D.; Zhang, X.; Liu, F.; Wang, W. Modeling the regulation of p53 activation by HIF-1 upon hypoxia. FEBS Lett. 2019, 593, 2596–2611. [Google Scholar] [CrossRef]
- Li, H.; Rokavec, M.; Jiang, L.; Horst, D.; Hermeking, H. Antagonistic Effects of p53 and HIF1A on microRNA-34a Regulation of PPP1R11 and STAT3 and Hypoxia-induced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells. Gastroenterology 2017, 153, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintás-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-Mediated Senescence Impairs the Apoptotic Response to Chemotherapy and Clinical Outcome in Breast Cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Kiaris, H.; Chatzistamou, I.; Trimis, G.; Frangou-Plemmenou, M.; Pafiti-Kondi, A.; Kalofoutis, A. Evidence for Nonautonomous Effect of p53 Tumor Suppressor in Carcinogenesis. Cancer Res. 2005, 65, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Marrero, L.; Rodriguez, P.; Del Valle, L.; Ochoa, A.; Cui, Y. Trp53 Inactivation in the Tumor Microenvironment Promotes Tumor Progression by Expanding the Immunosuppressive Lymphoid-like Stromal Network. Cancer Res. 2013, 73, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, P.; Li, W.; Ren, G.; Roberts, A.I.; Cao, W.; Zhang, X.; Su, J.; Chen, X.; Chen, Q.; et al. p53 regulates mesenchymal stem cell-mediated tumor suppression in a tumor microenvironment through immune modulation. Oncogene 2013, 33, 3830–3838. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Huang, H.-Y.; Pak, J.; Cheng, J.; Zhang, Z.-T.; Shapiro, E.; Pellicer, A.; Sun, T.-T.; Wu, X.-R. p53 deficiency provokes urothelial proliferation and synergizes with activated Ha-ras in promoting urothelial tumorigenesis. Oncogene 2004, 23, 687–696. [Google Scholar] [CrossRef]
- Hill, R.; Song, Y.; Cardiff, R.D.; Van Dyke, T. Selective Evolution of Stromal Mesenchyme with p53 Loss in Response to Epithelial Tumorigenesis. Cell 2005, 123, 1001–1011. [Google Scholar] [CrossRef]
- Dudley, A.C.; Shih, S.-C.; Cliffe, A.R.; Hida, K.; Klagsbrun, M. Attenuated p53 activation in tumour-associated stromal cells accompanies decreased sensitivity to etoposide and vincristine. Br. J. Cancer 2008, 99, 118–125. [Google Scholar] [CrossRef]
- Bar, J.; Feniger-Barish, R.; Lukashchuk, N.; Shaham, H.; Moskovits, N.; Goldfinger, N.; Simansky, D.; Perlman, M.; Papa, M.; Yosepovich, A.; et al. Cancer cells suppress p53 in adjacent fibroblasts. Oncogene 2008, 28, 933–936. [Google Scholar] [CrossRef]
- Yoshii, S.; Hayashi, Y.; Iijima, H.; Inoue, T.; Kimura, K.; Sakatani, A.; Nagai, K.; Fujinaga, T.; Hiyama, S.; Kodama, T.; et al. Exosomal micro RNA s derived from colon cancer cells promote tumor progression by suppressing fibroblast TP 53 expression. Cancer Sci. 2019, 110, 2396–2407. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Molchadsky, A.; Goldstein, I.; Solomon, H.; Tal, P.; Goldfinger, N.; Yang, P.; Porat, Z.; Lozano, G.; Rotter, V. Novel p53 target genes secreted by the liver are involved in non-cell-autonomous regulation. Cell Death Differ. 2016, 23, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, B.; Luz, I.; Krueger, C.; Teo, F.; Lane, D.; Sabapathy, K.; Cooks, T. Cancer Cells Shuttle Extracellular Vesicles Containing Oncogenic Mutant p53 Proteins to the Tumor Microenvironment. Cancers 2021, 13, 2985. [Google Scholar] [CrossRef] [PubMed]
- Solomon, H.; Dinowitz, N.; Pateras, I.S.; Cooks, T.; Shetzer, Y.; Molchadsky, A.; Charni-Natan, M.; Rabani, S.; Koifman, G.; Tarcic, O.; et al. Mutant p53 gain of function underlies high expression levels of colorectal cancer stem cells markers. Oncogene 2018, 37, 1669–1684. [Google Scholar] [CrossRef]
- Novo, D.; Heath, N.; Mitchell, L.; Caligiuri, G.; Macfarlane, A.; Reijmer, D.; Charlton, L.; Knight, J.; Calka, M.; McGhee, E.; et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat. Commun. 2018, 9, 5069. [Google Scholar] [CrossRef]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Acevedo, J.D.B.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef]
- Lee, J.-G.; Ahn, J.-H.; Kim, T.J.; Lee, J.H.; Choi, J.-H. Mutant p53 promotes ovarian cancer cell adhesion to mesothelial cells via integrin β4 and Akt signals. Sci. Rep. 2015, 5, 12642. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2011, 31, 3148–3163. [Google Scholar] [CrossRef]
- Wiley, C.D.; Schaum, N.; Alimirah, F.; Lopez-Dominguez, J.A.; Orjalo, A.V.; Scott, G.; Desprez, P.Y.; Benz, C.; Davalos, A.R.; Campisi, J. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep. 2018, 8, 2410. [Google Scholar] [CrossRef] [PubMed]
- Ara, T.; Nakata, R.; Sheard, M.; Shimada, H.; Buettner, R.; Groshen, S.G.; Ji, L.; Yu, H.; Jove, R.; Seeger, R.C.; et al. Critical Role of STAT3 in IL-6–Mediated Drug Resistance in Human Neuroblastoma. Cancer Res. 2013, 73, 3852–3864. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhan, Y.; Yuan, Z.; Qiu, Y.; Wang, H.; Fan, G.; Wang, J.; Li, W.; Cao, Y.; Shen, X.; et al. Hypoxia Induces Drug Resistance in Colorectal Cancer through the HIF-1α/miR-338-5p/IL-6 Feedback Loop. Mol. Ther. 2019, 27, 1810–1824. [Google Scholar] [CrossRef]
- Sreenivasan, L.; Wang, H.; Yap, S.Q.; Leclair, P.; Tam, A.; Lim, C.J. Autocrine IL-6/STAT3 signaling aids development of acquired drug resistance in Group 3 medulloblastoma. Cell Death Dis. 2020, 11, 1035. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef]
- Dong, Y.-L.; Vadla, G.P.; Lu, J.-Y.; Ahmad, V.; Klein, T.J.; Liu, L.-F.; Glazer, P.M.; Xu, T.; Chabu, C.-Y. Cooperation between oncogenic Ras and wild-type p53 stimulates STAT non-cell autonomously to promote tumor radioresistance. Commun. Biol. 2021, 4, 374. [Google Scholar] [CrossRef]
- Parmakhtiar, B.; Burger, R.A.; Kim, J.-H.; Fruehauf, J.P. HIF Inactivation of p53 in Ovarian Cancer Can Be Reversed by Topotecan, Restoring Cisplatin and Paclitaxel Sensitivity. Mol. Cancer Res. 2019, 17, 1675–1686. [Google Scholar] [CrossRef]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Lee, J.-Y. Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. Int. J. Mol. Sci. 2017, 18, 1854. [Google Scholar] [CrossRef]
- Chen, W.-L.; Wang, C.-C.; Lin, Y.-J.; Wu, C.-P.; Hsieh, C.-H. Cycling hypoxia induces chemoresistance through the activation of reactive oxygen species-mediated B-cell lymphoma extra-long pathway in glioblastoma multiforme. J. Transl. Med. 2015, 13, 389. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Lotterman, C.D.; Bao, C.; Hruban, R.H.; Karim, B.; Mendell, J.T.; Huso, D.; Lowenstein, C.J. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6334–6339. [Google Scholar] [CrossRef] [PubMed]
- Sermeus, A.; Michiels, C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011, 2, e164. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Song, X.; Song, B.; Liu, Y.; Wei, L.; Wang, X.; Yu, J. Effects of lentivirus-mediated HIF-1α knockdown on hypoxia-related cisplatin resistance and their dependence on p53 status in fibrosarcoma cells. Cancer Gene Ther. 2008, 15, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Z.-G.; Du, C.-Z.; Wang, H.-W.; Yan, L.; Gu, J. Cancer stem cell marker CD133+ tumour cells and clinical outcome in rectal cancer. Histopathology 2009, 55, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Pallini, R.; Vitiani, L.R.; Banna, G.L.; Signore, M.; Lombardi, D.; Todaro, M.; Stassi, G.; Martini, M.; Maira, G.; LaRocca, L.M.; et al. Cancer Stem Cell Analysis and Clinical Outcome in Patients with Glioblastoma Multiforme. Clin. Cancer Res. 2008, 14, 8205–8212. [Google Scholar] [CrossRef]
- Levina, V.; Marrangoni, A.M.; Demarco, R.; Gorelik, E.; Lokshin, A.E. Drug-Selected Human Lung Cancer Stem Cells: Cytokine Network, Tumorigenic and Metastatic Properties. PLoS ONE 2008, 3, e3077. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Warner, K.A.; Dong, Z.; Imai, A.; Nör, C.; Ward, B.B.; Nör, J.E. Endothelial Interleukin-6 Defines the Tumorigenic Potential of Primary Human Cancer Stem Cells: Endothelial IL-6 and Cancer Stem Cells. Stem Cells 2014, 32, 2845–2857. [Google Scholar] [CrossRef]
- Li, N.; Babaei-Jadidi, R.; Lorenzi, F.; Spencer-Dene, B.; Clarke, P.; Domingo, E.; Tulchinsky, E.; Vries, R.G.J.; Kerr, D.; Pan, Y.; et al. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncology 2019, 8, 13. [Google Scholar] [CrossRef]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal. 2019, 12, eaau8544. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Zarkoob, H.; Taube, J.; Singh, S.K.; Mani, S.; Kohandel, M. Investigating the Link between Molecular Subtypes of Glioblastoma, Epithelial-Mesenchymal Transition, and CD133 Cell Surface Protein. PLoS ONE 2013, 8, e64169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, H.; Sun, L.; Zhan, P.; Chen, M.; Zhang, F.; Ran, Y.; Wan, J. LGR5, a novel functional glioma stem cell marker, promotes EMT by activating the Wnt/β-catenin pathway and predicts poor survival of glioma patients. J. Exp. Clin. Cancer Res. 2018, 37, 225. [Google Scholar] [CrossRef] [PubMed]
- Ziberi, S.; Zuccarini, M.; Carluccio, M.; Giuliani, P.; Ricci-Vitiani, L.; Pallini, R.; Caciagli, F.; Di Iorio, P.; Ciccarelli, R. Upregulation of Epithelial-To-Mesenchymal Transition Markers and P2X7 Receptors Is Associated to Increased Invasiveness Caused by P2X7 Receptor Stimulation in Human Glioblastoma Stem Cells. Cells 2019, 9, 85. [Google Scholar] [CrossRef]
- Bai, Z.; Wang, J.; Wang, T.; Li, Y.; Zhao, X.; Wu, G.; Zhang, Z. The MiR-495/Annexin A3/P53 Axis Inhibits the Invasion and EMT of Colorectal Cancer Cells. Cell Physiol. Biochem. 2017, 44, 1882–1895. [Google Scholar] [CrossRef]
- Cho, S.-Y.; Park, C.; Na, D.; Han, J.Y.; Lee, J.; Park, O.-K.; Zhang, C.; Sung, C.O.; Moon, H.E.; Kim, Y.; et al. High prevalence of TP53 mutations is associated with poor survival and an EMT signature in gliosarcoma patients. Exp. Mol. Med. 2017, 49, e317. [Google Scholar] [CrossRef]
- Aloni-Grinstein, R.; Shetzer, Y.; Kaufman, T.; Rotter, V. p53: The barrier to cancer stem cell formation. FEBS Lett. 2014, 588, 2580–2589. [Google Scholar] [CrossRef]
- Ghatak, D.; Das Ghosh, D.; Roychoudhury, S. Cancer Stemness: p53 at the Wheel. Front. Oncol. 2021, 10, 2910. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2017, 553, 96–100. [Google Scholar] [CrossRef]
- Wei, S.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.; et al. Matrix stiffness drives epithelial–mesenchymal transition and tumour metastasis through a TWIST1–G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Nandi, S.; Hindi, E.S.; Wainwright, D.A.; Han, Y.; Lesniak, M.S. Short Hairpin RNA-Mediated Fibronectin Knockdown Delays Tumor Growth in a Mouse Glioma Model. Neoplasia 2010, 12, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.H.; Lu, C.; James, E.R.; Hegab, R.; Allen, S.C.; Suggs, L.J.; Brock, A. Phenotypic Basis for Matrix Stiffness-Dependent Chemoresistance of Breast Cancer Cells to Doxorubicin. Front. Oncol. 2018, 8, 337. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Cao, T.; Guo, K.; Zhou, Y.; Liu, H.; Pan, Y.; Hou, Q.; Nie, Y.; Fan, D.; Lu, Y.; et al. Regulation of Integrin Subunit Alpha 2 by miR-135b-5p Modulates Chemoresistance in Gastric Cancer. Front. Oncol. 2020, 10, 308. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 Drives Invasion by Promoting Integrin Recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef]
- Shi, F.; Sottile, J. Caveolin-1-dependent β1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci. 2008, 121, 2360–2371. [Google Scholar] [CrossRef]
- Yu, V.Z.; So, S.S.; Lung, M.L. Gain-of-function hot spot mutant p53R248Q regulation of integrin/FAK/ERK signaling in esophageal squamous cell carcinoma. Transl. Oncol. 2021, 14, 100982. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Trinidad, A.G.; Timpson, P.; Morton, J.P.; Zanivan, S.; Berghe, P.V.E.V.D.; Nixon, C.; Karim, A.S.; Caswell, P.T.; Noll, E.J.; et al. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene 2012, 32, 1252–1265. [Google Scholar] [CrossRef]
- Lakoduk, A.M.; Roudot, P.; Mettlen, M.; Grossman, H.M.; Schmid, S.L.; Chen, P.-H. Mutant p53 amplifies a dynamin-1/APPL1 endosome feedback loop that regulates recycling and migration. J. Cell Biol. 2019, 218, 1928–1942. [Google Scholar] [CrossRef]
- Phatak, V.; von Grabowiecki, Y.; Janus, J.; Officer, L.; Behan, C.; Aschauer, L.; Pinon, L.; Mackay, H.; Zanivan, S.; Norman, J.C.; et al. Mutant p53 promotes RCP-dependent chemoresistance coinciding with increased delivery of P-glycoprotein to the plasma membrane. Cell Death Dis. 2021, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Xiao, W.; Sun, S.; Sohrabi, A.; Liang, J.; Seidlits, S.K. Extracellular Matrix Proteins Confer Cell Adhesion-Mediated Drug Resistance Through Integrin αv in Glioblastoma Cells. Front. Cell Dev. Biol. 2021, 9, 616580. [Google Scholar] [CrossRef] [PubMed]
- Bon, G.; Di Carlo, S.E.; Folgiero, V.; Avetrani, P.; Lazzari, C.; D’Orazi, G.; Brizzi, M.F.; Sacchi, A.; Soddu, S.; Blandino, G.; et al. Negative Regulation of β4 Integrin Transcription by Homeodomain-Interacting Protein Kinase 2 and p53 Impairs Tumor Progression. Cancer Res. 2009, 69, 5978–5986. [Google Scholar] [CrossRef]
- Brannon, A.; Drouillard, D.; Steele, N.; Schoettle, S.; Abel, E.V.; Crawford, H.C.; di Magliano, M.P. Beta 1 integrin signaling mediates pancreatic ductal adenocarcinoma resistance to MEK inhibition. Sci. Rep. 2020, 10, 11133. [Google Scholar] [CrossRef] [PubMed]
- Fanucchi, S.; Veale, R.B. Delayed caspase-8 activation and enhanced integrin β1-activated FAK underpins anoikis in oesophageal carcinoma cells harbouring mt p53-R175H. Cell Biol. Int. 2011, 35, 819–826. [Google Scholar] [CrossRef]
- Yang, J.; Bahcecioglu, G.; Zorlutuna, P. The Extracellular Matrix and Vesicles Modulate the Breast Tumor Microenvironment. Bioengineering 2020, 7, 124. [Google Scholar] [CrossRef]
- Janouskova, H.; Maglott, A.; Leger, D.Y.; Bossert, C.; Noulet, F.; Guerin, E.; Guenot, D.; Pinel, S.; Chastagner, P.; Plenat, F.; et al. Integrin α5β1 Plays a Critical Role in Resistance to Temozolomide by Interfering with the p53 Pathway in High-Grade Glioma. Cancer Res. 2012, 72, 3463–3470. [Google Scholar] [CrossRef]
- Janouskova, H.; Ray, A.-M.; Noulet, F.; Lelong-Rebel, I.; Choulier, L.; Schaffner, F.; Lehmann, M.; Martin, S.; Teisinger, J.; Dontenwill, M. Activation of p53 pathway by Nutlin-3a inhibits the expression of the therapeutic target α5 integrin in colon cancer cells. Cancer Lett. 2013, 336, 307–318. [Google Scholar] [CrossRef]
- Khwaja, F.W.; Svoboda, P.; Reed, M.; Pohl, J.; Pyrzynska, B.; van Meir, E.G. Proteomic identification of the wt-p53-regulated tumor cell secretome. Oncogene 2006, 25, 7650–7661. [Google Scholar] [CrossRef][Green Version]
- Zhu, H.; Evans, B.; O’Neill, P.; Ren, X.; Xu, Z.; Hait, W.N.; Yang, J.-M. A role for p53 in the regulation of extracellular matrix metalloproteinase inducer in human cancer cells. Cancer Biol. Ther. 2009, 8, 1722–1728. [Google Scholar] [CrossRef]
- Australian Pancreatic Genome Initiative (APGI); Vennin, C.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat Commun. 2019, 10, 3637. [Google Scholar] [CrossRef] [PubMed]
- Biasoli, D.; Sobrinho, M.F.; Da Fonseca, A.C.C.; De Matos, D.G.; Romao, L.; Maciel, R.D.M.; Rehen, S.; Moura-Neto, V.; Borges, H.L.; Lima, F.R.S. Glioblastoma cells inhibit astrocytic p53-expression favoring cancer malignancy. Oncogenesis 2014, 3, e123. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Wang, J.; Chen, M.; Chen, F.; Tu, W. Exosomal microRNA146a derived from mesenchymal stem cells increases the sensitivity of ovarian cancer cells to docetaxel and taxane via a LAMC2mediated PI3K/Akt axis. Int. J. Mol. Med. 2020, 5, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.; Day, B.W.; Boyd, A.W.; Guillemin, G.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef]
- Silva, J.L.; Lima, C.; Rangel, L.P.; Ferretti, G.D.S.; Pauli, F.; Ribeiro, R.C.B.; Silva, T.D.B.D.; Da Silva, F.C.; Ferreira, V.F. Recent Synthetic Approaches towards Small Molecule Reactivators of p53. Biomololecules 2020, 10, 635. [Google Scholar] [CrossRef]
- Jiang, L.; Zawacka-Pankau, J. The p53/MDM2/MDMX-targeted therapies—a clinical synopsis. Cell Death Dis. 2020, 11, 237. [Google Scholar] [CrossRef]
- Guo, G.; Yu, M.; Xiao, W.; Celis, E.; Cui, Y. Local Activation of p53 in the Tumor Microenvironment Overcomes Immune Suppression and Enhances Antitumor Immunity. Cancer Res. 2017, 77, 2292–2305. [Google Scholar] [CrossRef]


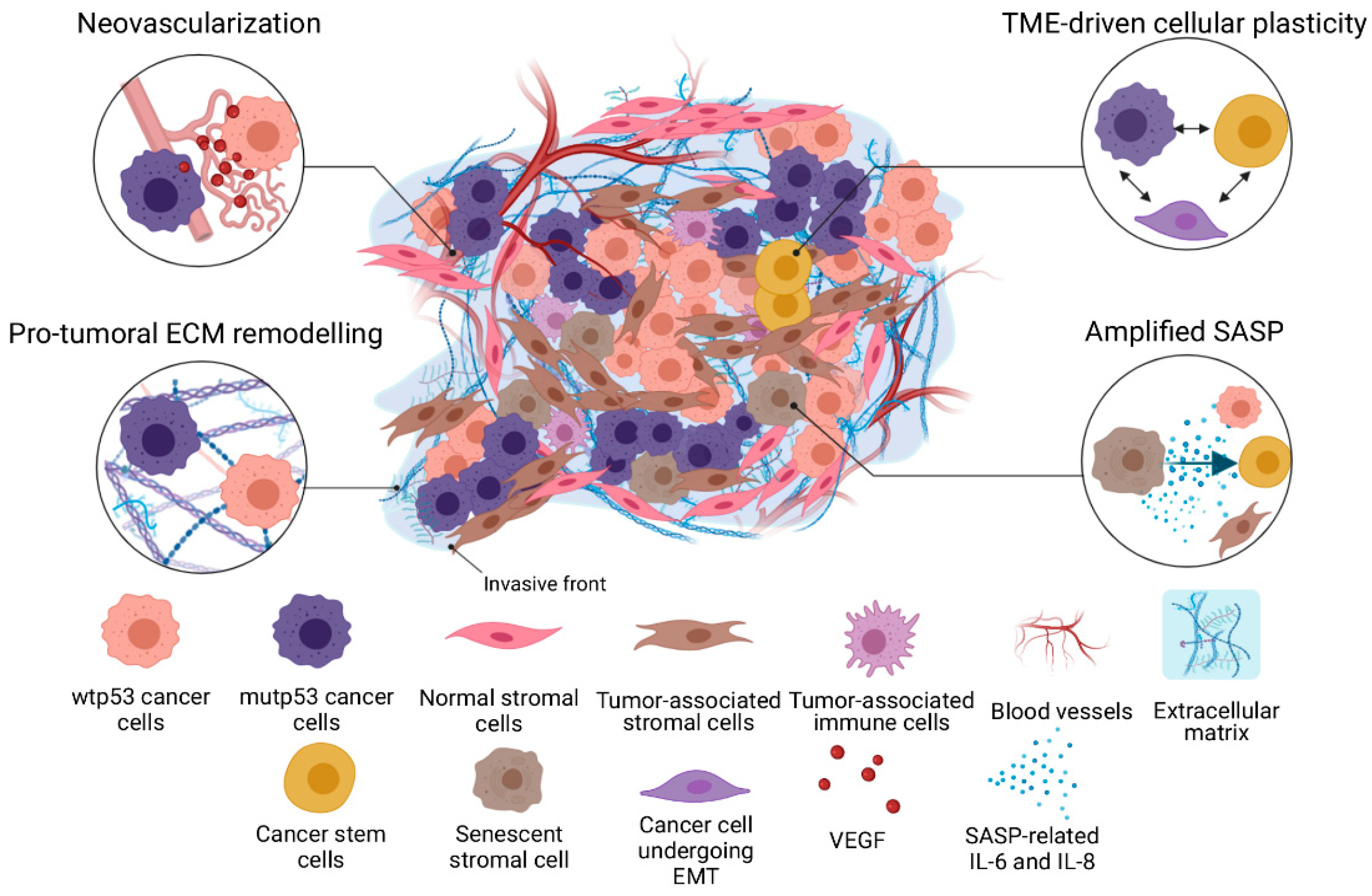{kind=link}
| p53 Mutation | Cancer Type | Change in Microenvironment | Reference |
|---|---|---|---|
| R273H | Pancreatic ductal carcinoma | Release of mutp53-containing | [74] |
| EVs. | |||
| Colon carcinoma | Enhancement of CSC expansion. | [75] | |
| Non-small cell lung carcinoma | Pro-invasive microenvironment and | [76,77] | |
| ECM regulation. | |||
| R175H | Non-small cell lung carcinoma | Pro-invasive microenvironment. | [76] |
| V157F | Pancreatic ductal carcinoma | Release of mutp53-containing | [74] |
| EVs. | |||
| R249S | Pancreatic ductal carcinoma | Release of mutp53-containing | [74] |
| EVs. | |||
| P309S | Colon carcinoma | Enhancement of CSC expansion. | [75] |
| R248W | Colon carcinoma | Enhancement of CSC expansion. | [75] |
| R246I | Non-small cell lung carcinoma | ECM regulation. | [77] |
| R248 | Ovarian cancer | Increased adhesion to mesothelial | [78] |
| cells. |
| p53 Mutation | Integrin Receptor or Subunit | Cancer Cell Line/Type | Resultant Phenotype | Reference |
|---|---|---|---|---|
| R248Q | αVβ3 | KYSE150 (esophageal squamous cell carcinoma) | Upregulation of integrins and downstream activation of ERK signaling. | [118] |
| β4 | OVCAR-3 (high-grade serous ovarian adenocarcinoma) | Upregulation of integrins and downstream activation of PI3K/Akt signaling. | [78] | |
| R273H | α5β1 | H1299 (non-small cell lung carcinoma) | Enhanced integrin and EGFR recycling to the plasma membrane and concomitant activation of MET signaling. | [116,119] |
| H1975 (non-small cell lung carcinoma) | Enhanced integrin and EGFR recycling to the plasma membrane. | [120] | ||
| β1 | A431 (lung squamous cell carcinoma) | Modest cisplatin resistance related to integrin expression. | [121] | |
| αV | GBM6 (primary glioblastoma) | Upregulation of integrin expression resulting in ECM-mediated carmustin resistance. | [122] | |
| β4 | HT29 (colorectal adenocarcinoma) | Loss of wtp53-dependent integrin repression. | [123] | |
| R172H | β1 | Pancreatic ductal adenocarcinoma cells derived from an oncogenic KRAS/mutp53 mouse model | Upregulation of integrin expression resulting in basement membrane-mediated trametinib resistance. | [124] |
| R175H | β1 | SNO (human oesophageal squamous carcinoma) | Upregulation of integrin signaling resulting in sustained FAK activation and resistance to caspase-8 activation. | [125] |
| p53 Mutation | Cancer Type | Drug Chemoresistance | Reference |
|---|---|---|---|
| R273H | Colon carcinoma | 5FU, Cisplatin | [75] |
| Epidermoid carcinoma | Cisplatin | [121] | |
| P309S | Colon carcinoma | 5FU, Cisplatin | [75] |
| R248W | Colon carcinoma | 5FU, Cisplatin | [75] |
| Q136X | Ovarian cancer | Cisplatin, Paclitaxel | [87] |
| G245R | Fibrosarcoma | 5FU, Cisplatin | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souza, L.C.d.M.e.; Faletti, A.; Veríssimo, C.P.; Stelling, M.P.; Borges, H.L. p53 Signaling on Microenvironment and Its Contribution to Tissue Chemoresistance. Membranes 2022, 12, 202. https://doi.org/10.3390/membranes12020202
Souza LCdMe, Faletti A, Veríssimo CP, Stelling MP, Borges HL. p53 Signaling on Microenvironment and Its Contribution to Tissue Chemoresistance. Membranes. 2022; 12(2):202. https://doi.org/10.3390/membranes12020202
Chicago/Turabian StyleSouza, Leonel Cardozo de Menezes e, Anderson Faletti, Carla Pires Veríssimo, Mariana Paranhos Stelling, and Helena Lobo Borges. 2022. "p53 Signaling on Microenvironment and Its Contribution to Tissue Chemoresistance" Membranes 12, no. 2: 202. https://doi.org/10.3390/membranes12020202
APA StyleSouza, L. C. d. M. e., Faletti, A., Veríssimo, C. P., Stelling, M. P., & Borges, H. L. (2022). p53 Signaling on Microenvironment and Its Contribution to Tissue Chemoresistance. Membranes, 12(2), 202. https://doi.org/10.3390/membranes12020202